
Development of p-Tau Differentiated Cell Model of Alzheimer’s Disease to Screen Novel Acetylcholinesterase Inhibitors

 , , , ,
, , , ,
Abstract
1. Introduction
2. Results
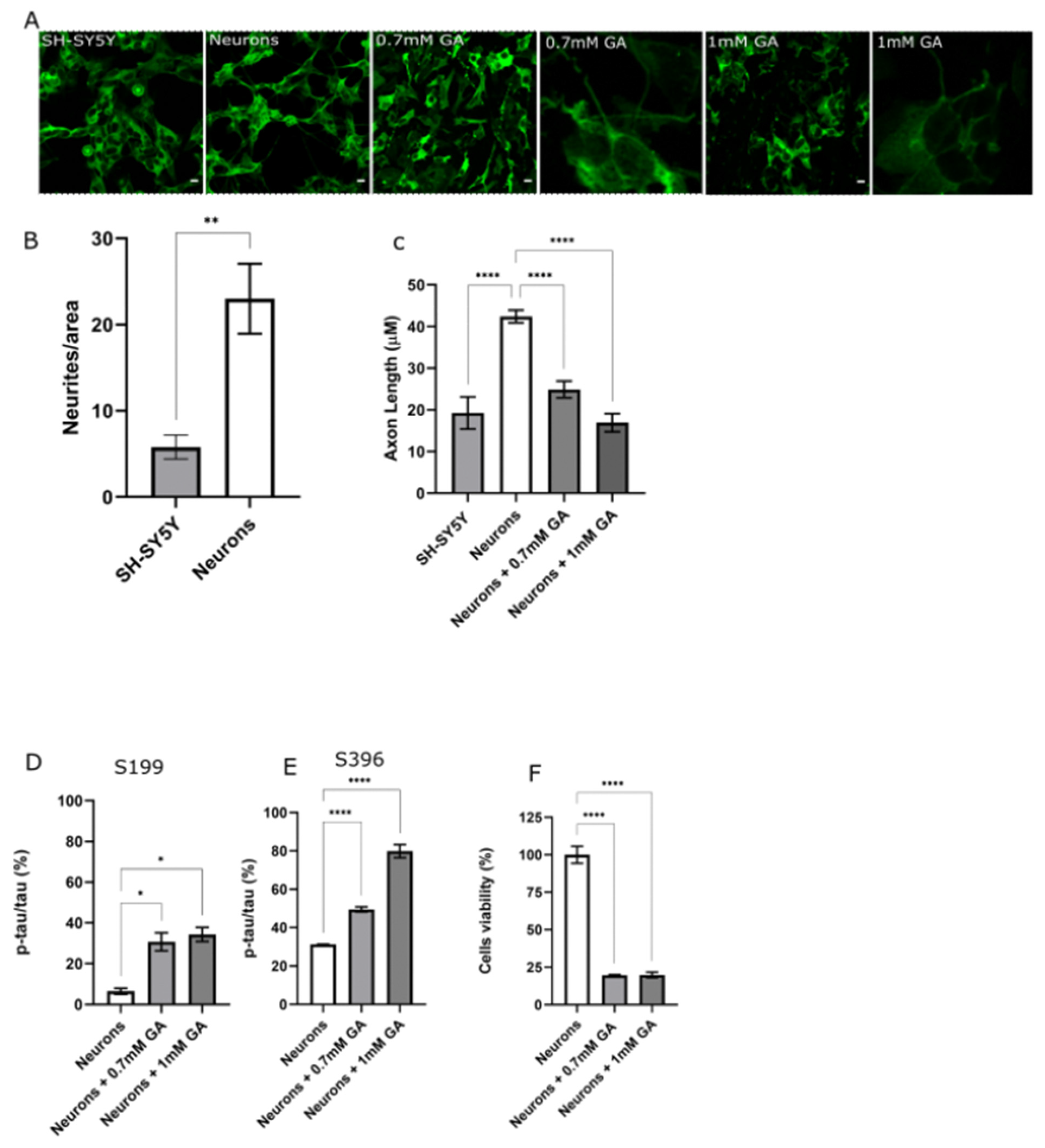
2.1. GA Treatment Induces Abnormal Tau Phosphorylation and Axon Degeneration in SH-SY5Y-Differentiated Neurons
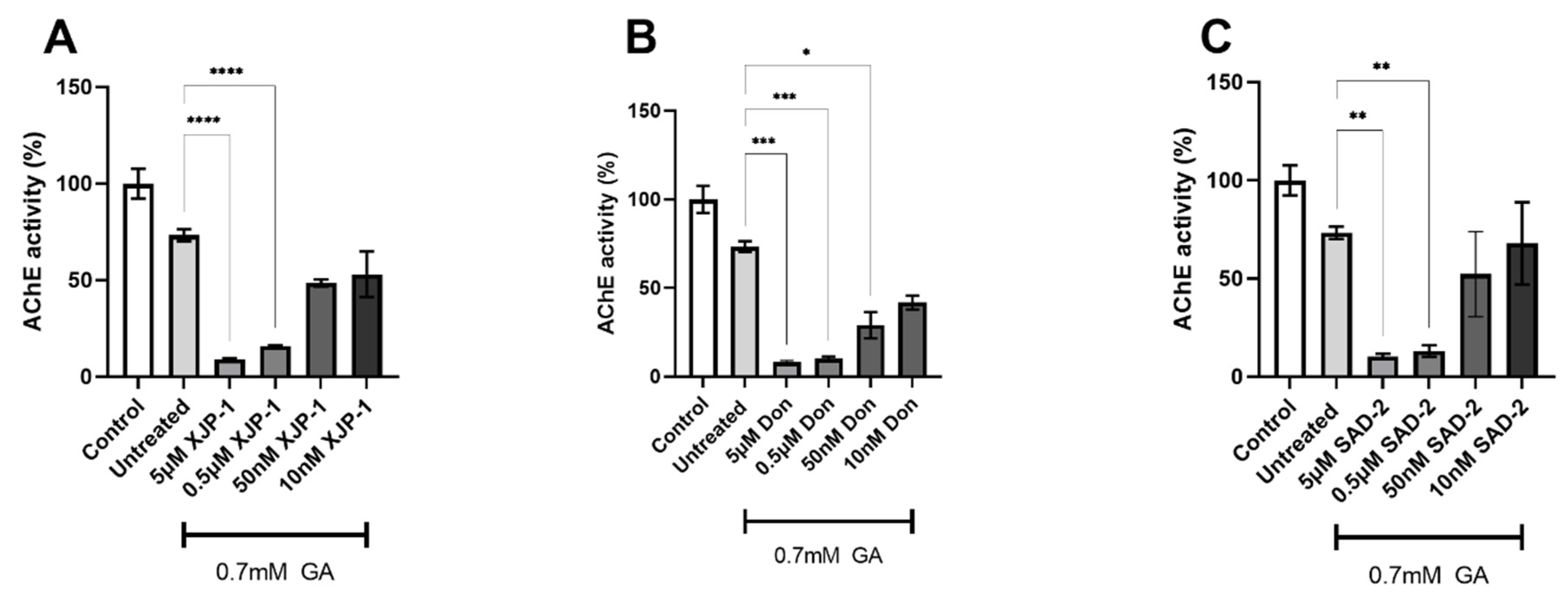
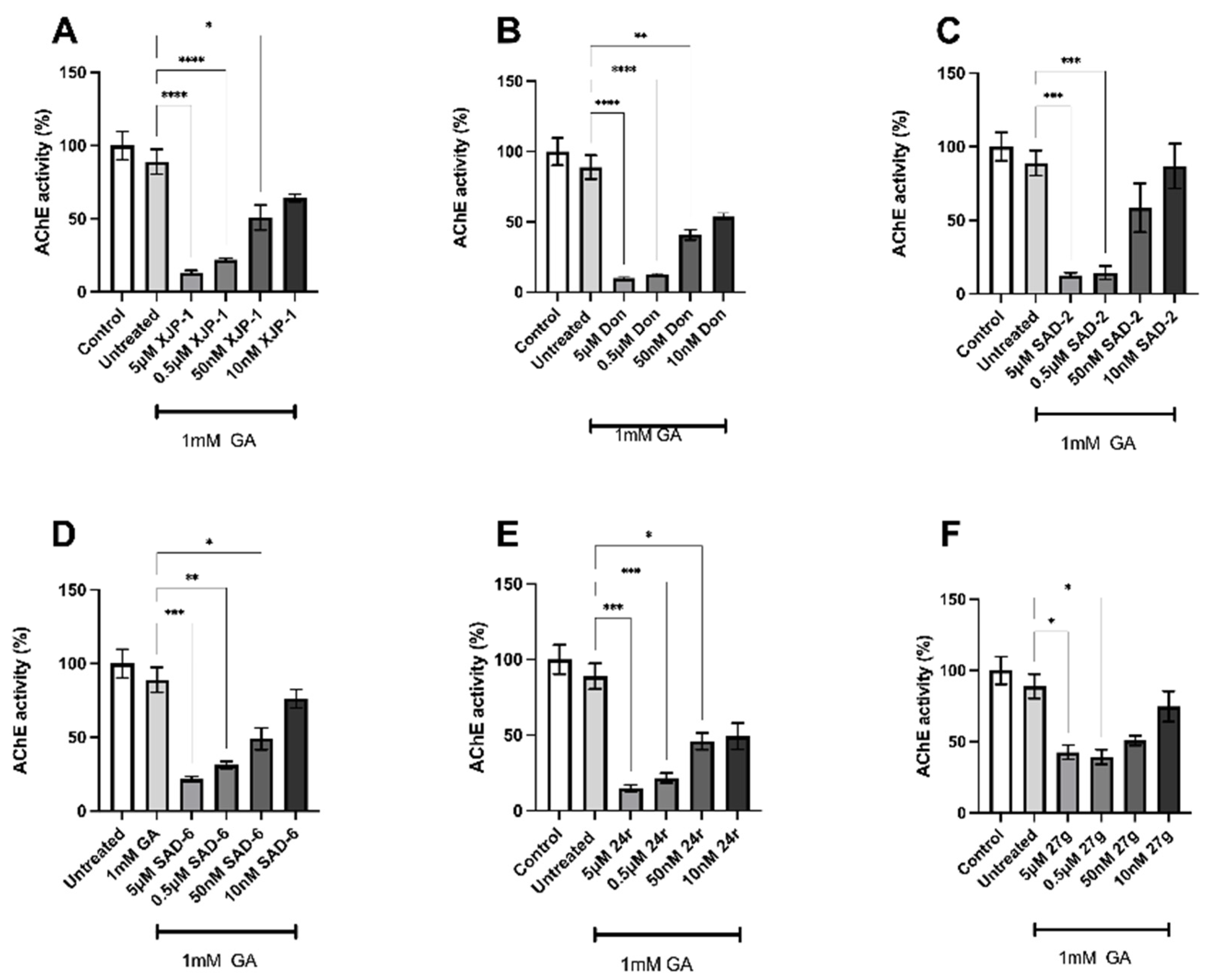
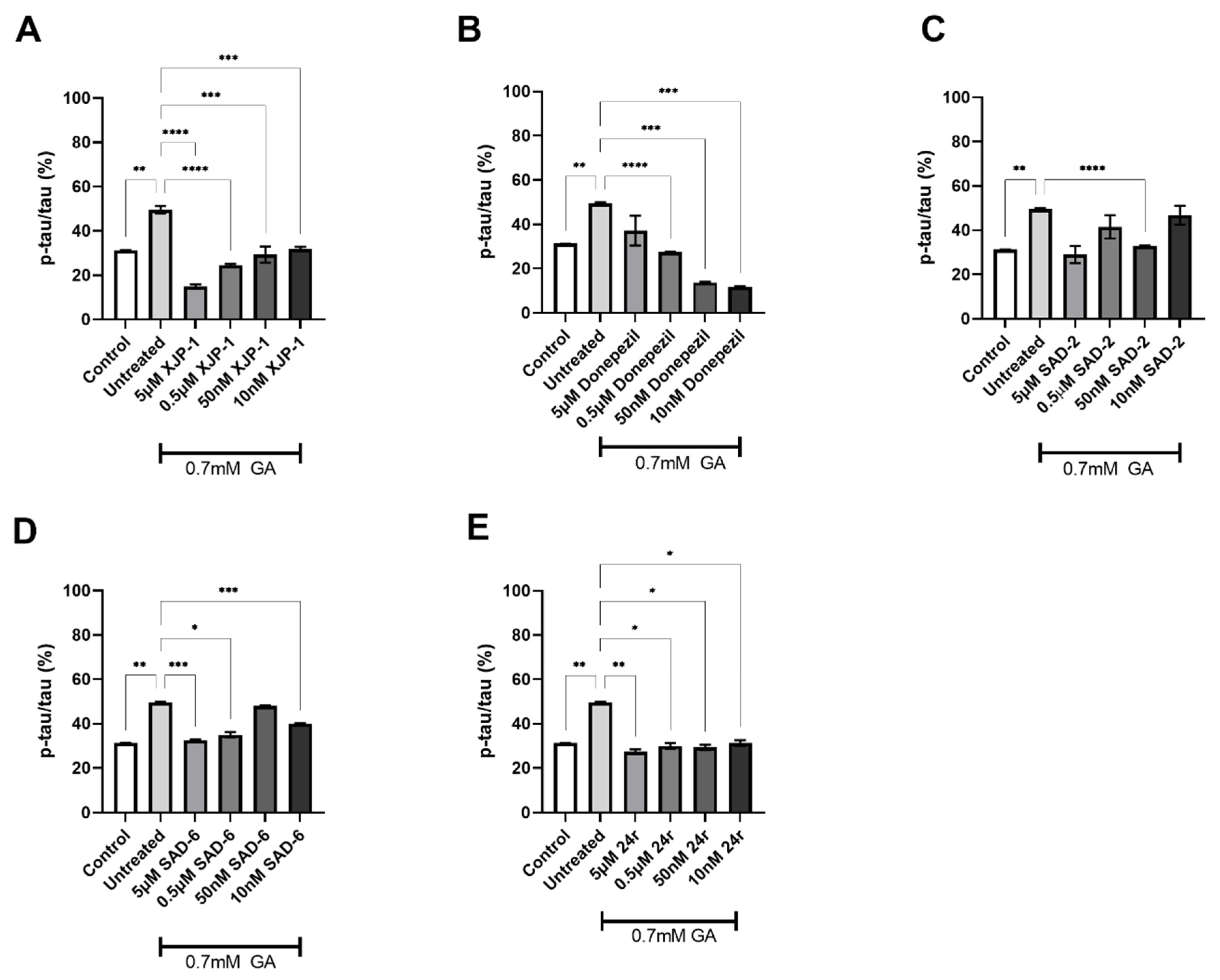
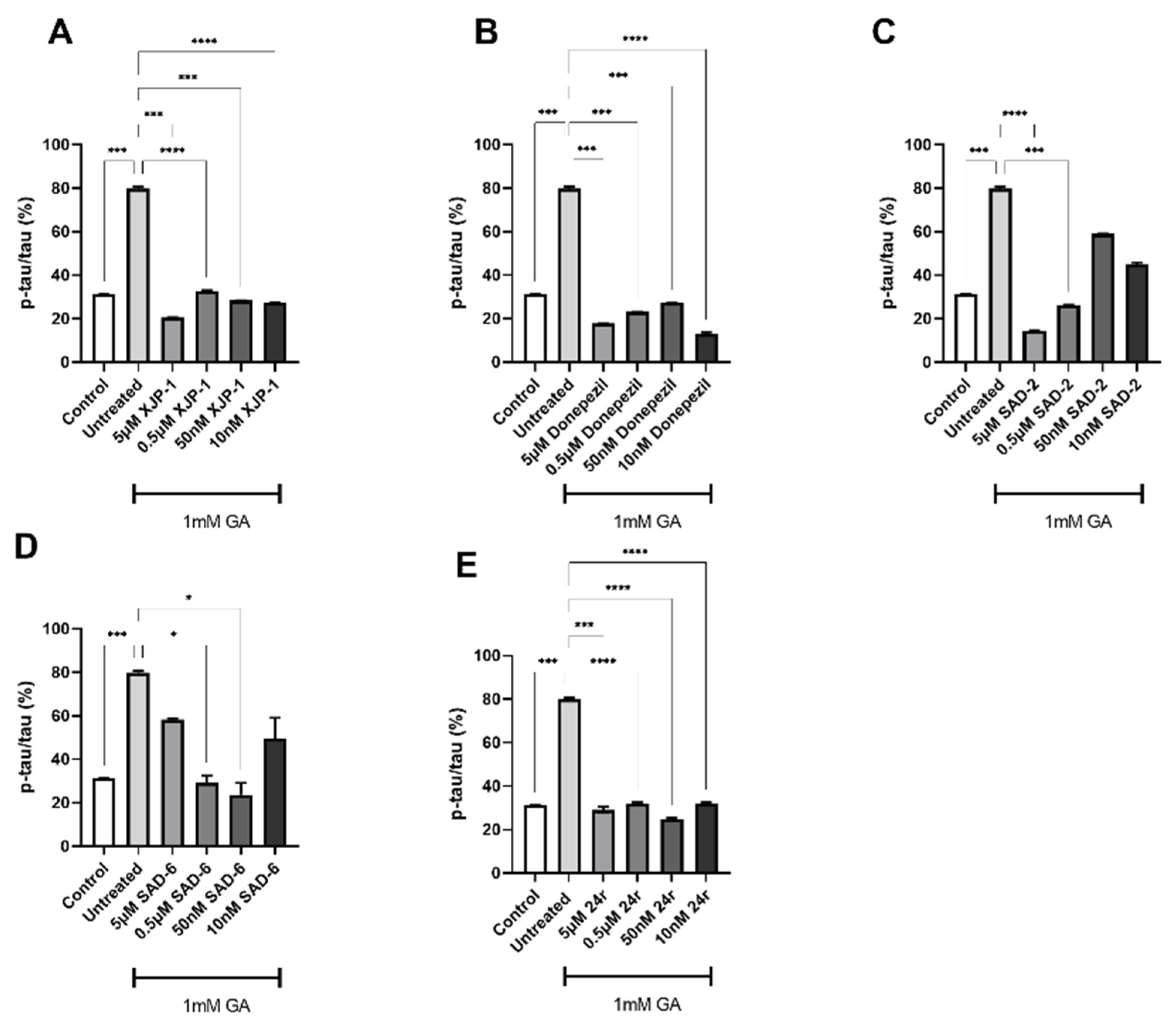
2.2. Novel Compounds Inhibit AChE Enzyme Activity in SH-SY5Y-Differentiated Neurons
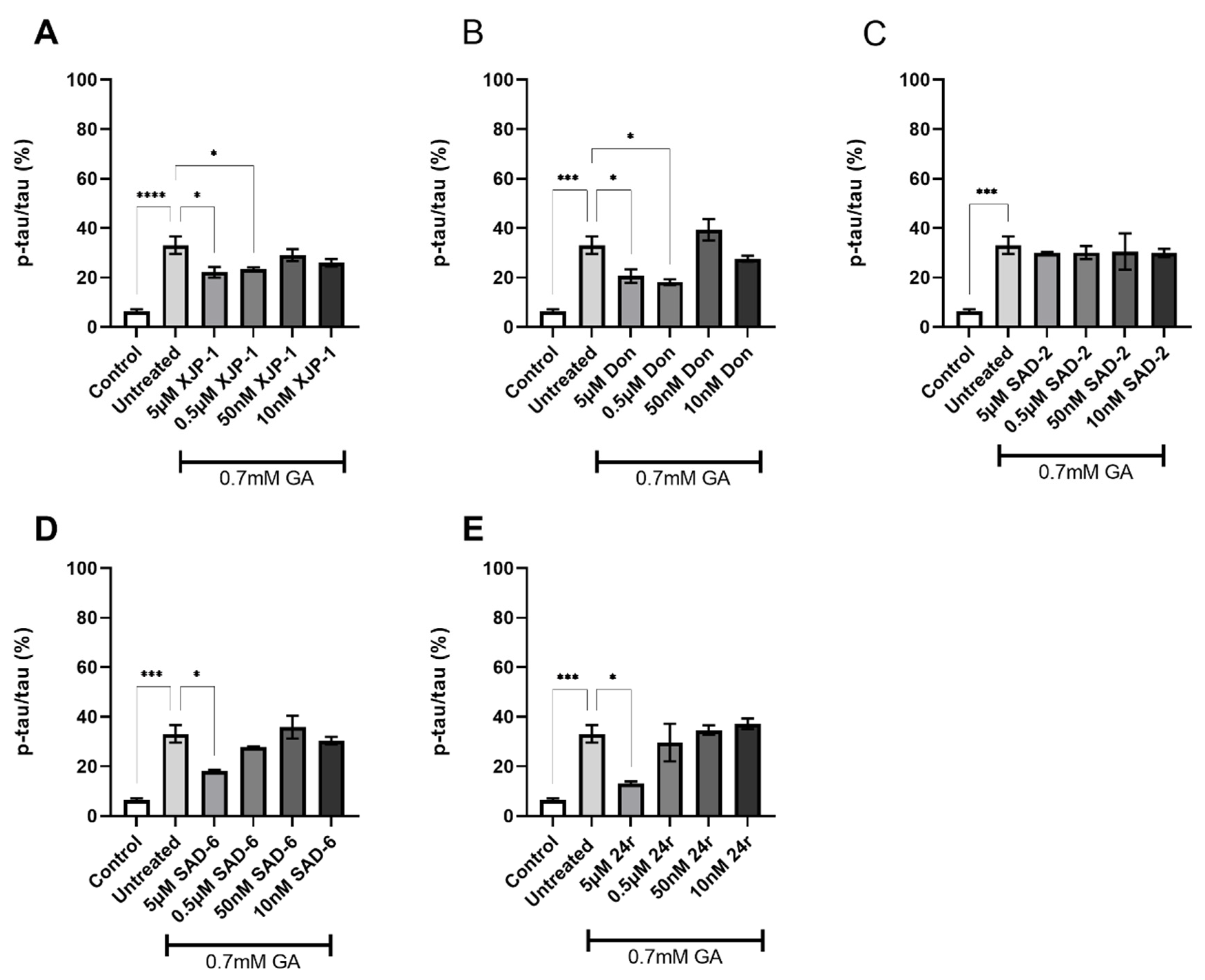
2.3. AChE Inhibition Prevents GA-Induced Tau Phosphorylation on S396 but Not on S199
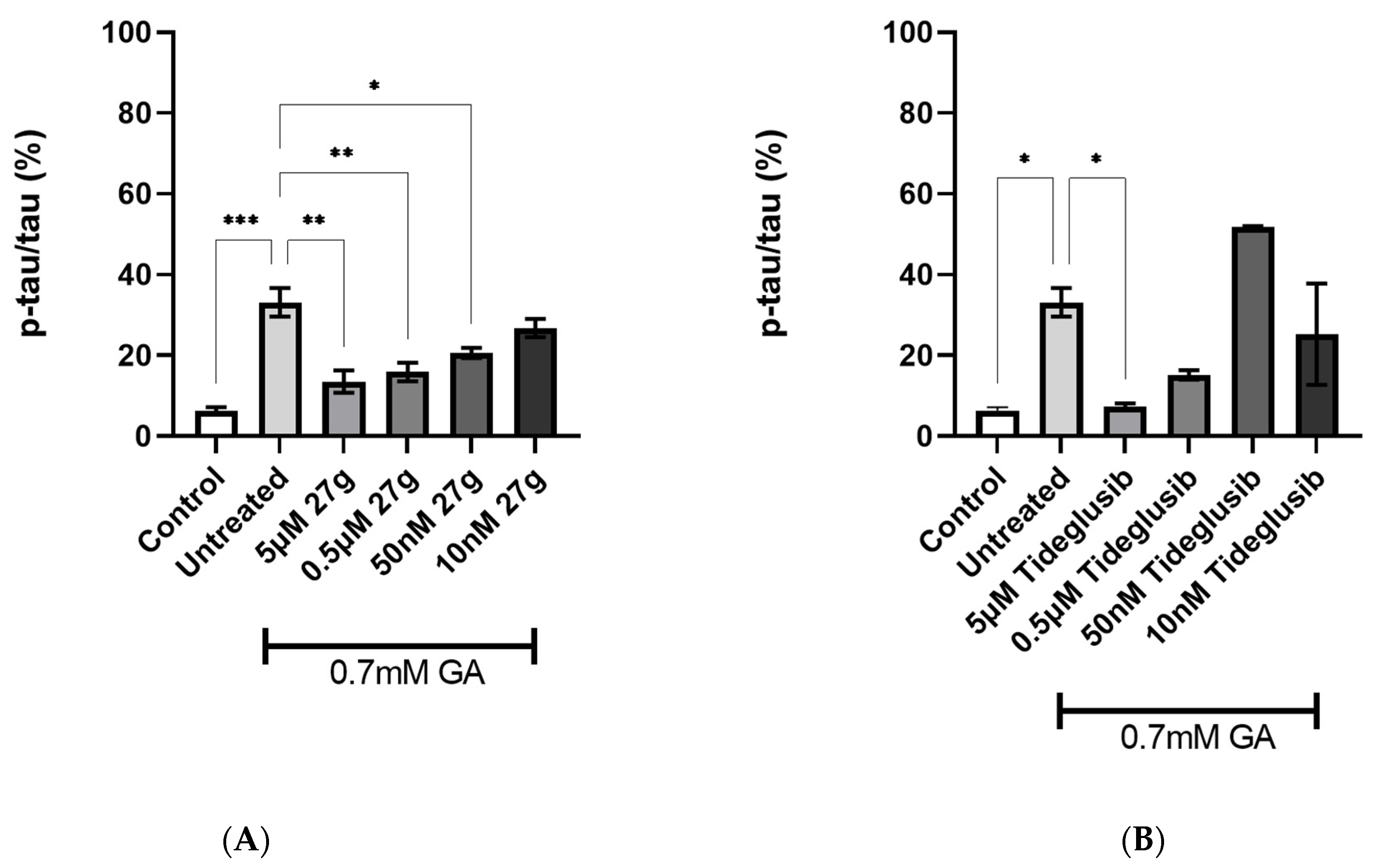
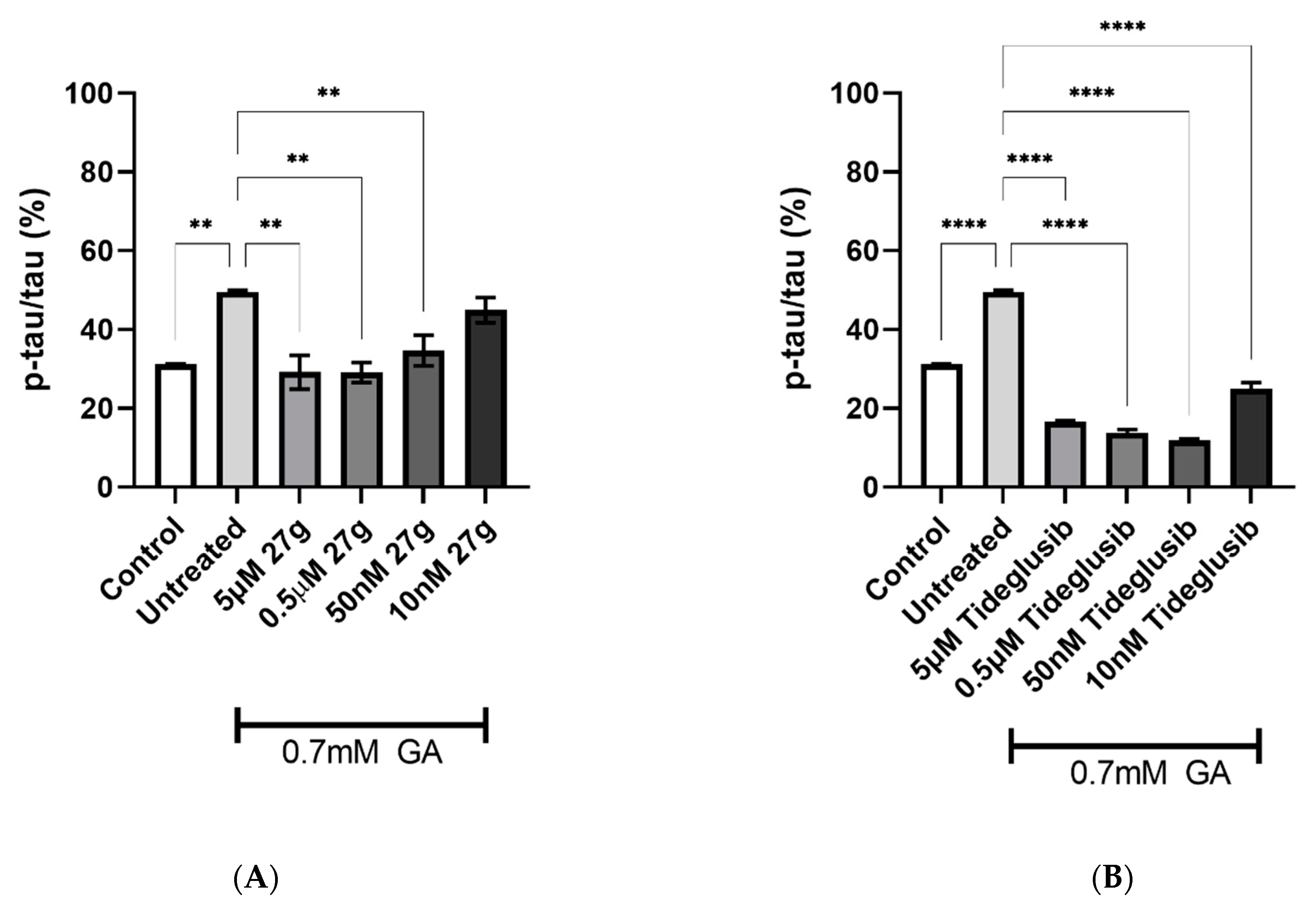
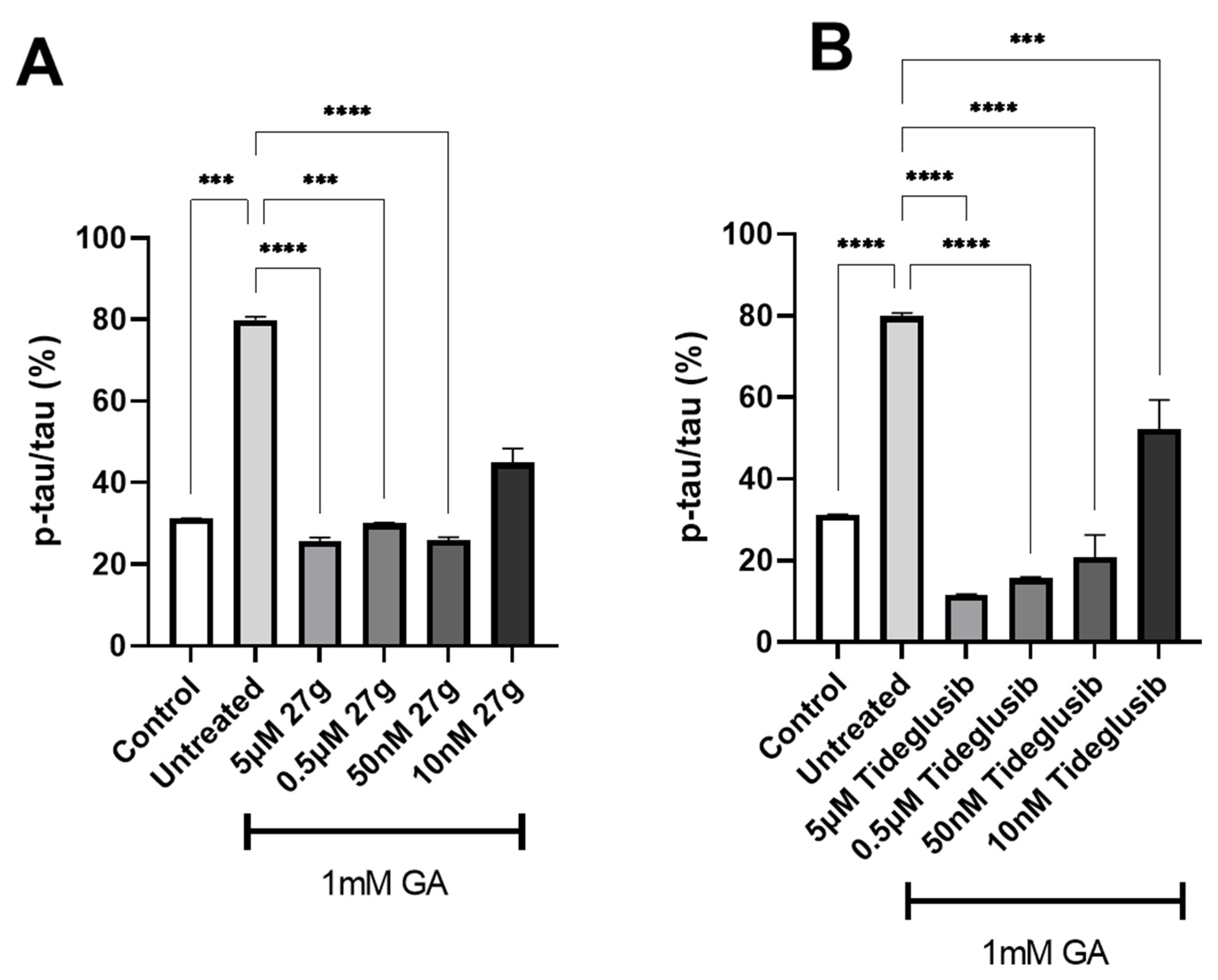
2.4. Dual AChE/GSK-3 Inhibitor 27g Modulates Tau Phosphorylation on Both S199 and S396
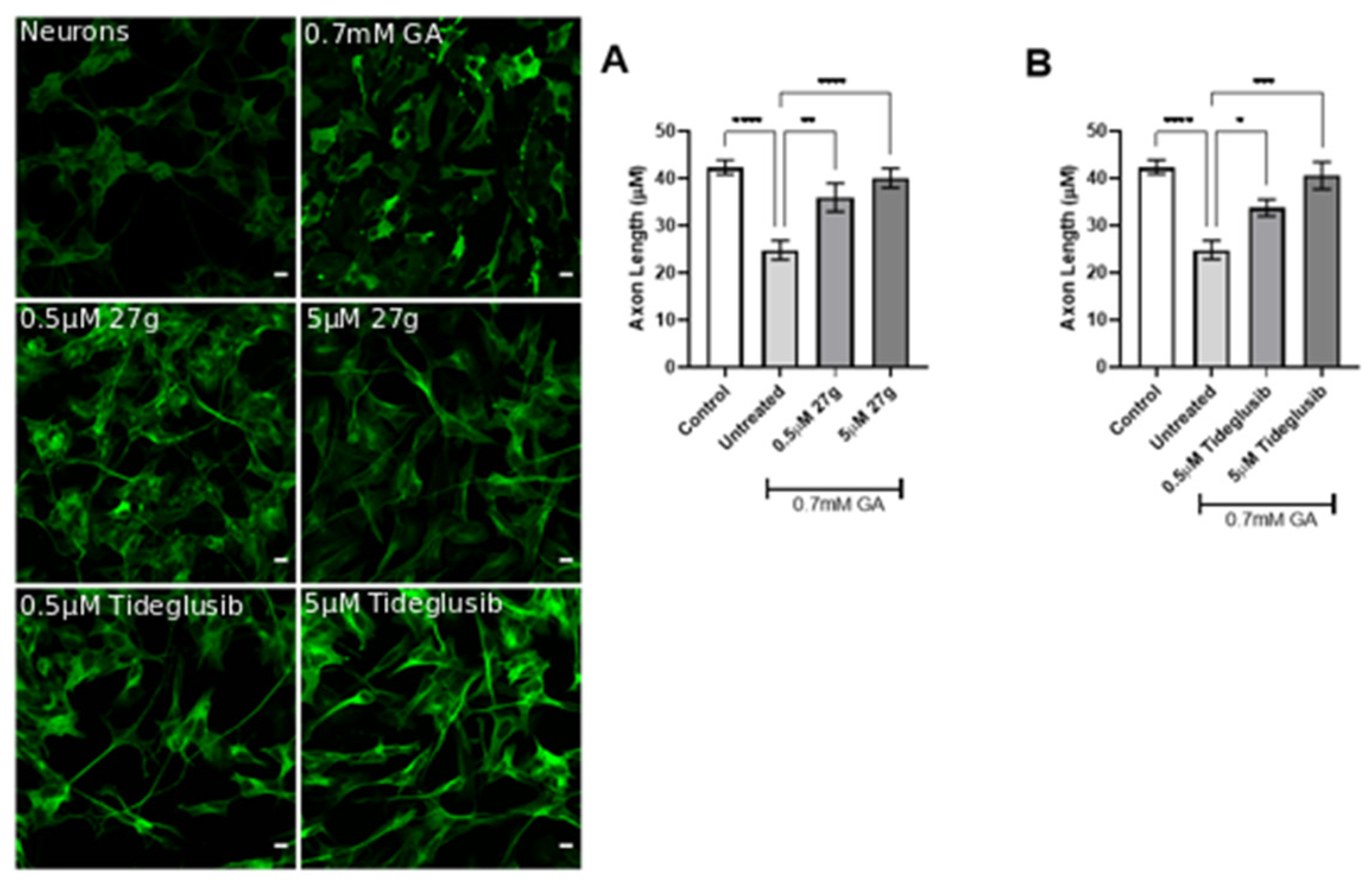
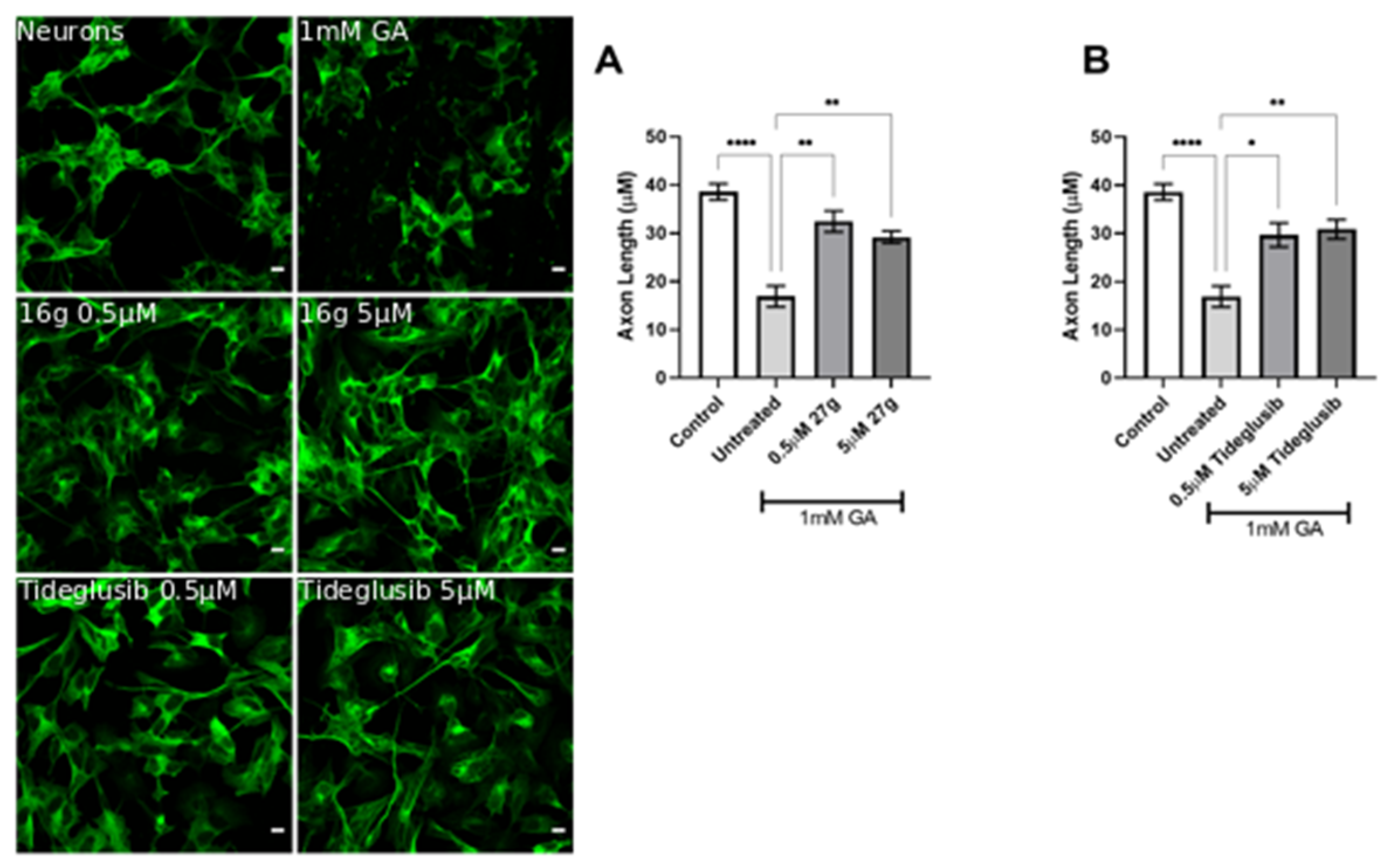
2.5. Dual AChE/GSK-3 Inhibitor 27g Prevents Tau-Induced Neurodegeneration and Protects from Neuronal Cell Death
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. SH-SY5Y Culture and Neuronal Differentiation
4.3. Glyceraldehyde Induced Tau Hyperphosphorylation
4.4. Immunostaining Cell Culture
4.5. Confocal Microscopy
4.6. pTAU Quantification
4.7. AChE Activity Assay (Ellman’s Method)
4.8. Cell Viability Assay
4.9. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alzheimer’s Association. 2019 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2019, 15, 321–387. [Google Scholar] [CrossRef]
- Merriam, A.E.; Aronson, M.K.; Gaston, P.; Wey, S.-L.; Katz, I. The Psychiatric Symptoms of Alzheimer’s Disease. J. Am. Geriatr. Soc. 1988, 36, 7–22. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.; Atluri, V.; Kaushik, A.; Yndart, A.; Nair, M. Alzheimer’s disease: Pathogenesis, diagnostics, and therapeutics. Int. J. Nanomed. 2019, 14, 5541–5554. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, J.M. A role for Akt and glycogen synthase kinase-3 as integrators of dopamine and serotonin neurotransmission in mental health. J. Psychiatry Neurosci. 2012, 37, 7–16. [Google Scholar] [CrossRef]
- Chu, J.; Lauretti, E.; Pratico, D. Caspase-3-dependent cleavage of Akt modulates tau phosphorylation via GSK3beta kinase: Implications for Alzheimer’s disease. Mol. Psychiatry 2017, 22, 1002–1008. [Google Scholar] [CrossRef]
- Hernandez, F.; Lucas, J.J.; Avila, J. GSK3 and Tau: Two Convergence Points in Alzheimer’s Disease. J. Alzheimer’s Dis. 2013, 33, S141–S144. [Google Scholar] [CrossRef]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef]
- Li, X.-H.; Lv, B.-L.; Xie, J.-Z.; Liu, J.; Zhou, X.-W.; Wang, J.-Z. AGEs induce Alzheimer-like tau pathology and memory deficit via RAGE-mediated GSK-3 activation. Neurobiol. Aging 2012, 33, 1400–1410. [Google Scholar] [CrossRef]
- Jackson, G.R.; Wiedau-Pazos, M.; Sang, T.-K.; Wagle, N.; Brown, C.A.; Massachi, S.; Geschwind, D.H. Human Wild-Type Tau Interacts with wingless Pathway Components and Produces Neurofibrillary Pathology in Drosoph. Neuron 2002, 34, 509–519. [Google Scholar] [CrossRef]
- Hernández, F.; Gómez de Barreda, E.; Fuster-Matanzo, A.; Lucas, J.J.; Avila, J. GSK3: A possible link between beta amyloid peptide and tau protein. Exp. Neurol. 2010, 223, 322–325. [Google Scholar] [CrossRef]
- Farias, G.; Cornejo, A.; Jimenez, J.; Guzman, L.; Maccioni, R.B. Mechanisms of Tau Self-Aggregation and Neurotoxicity. Curr. Alzheimer Res. 2011, 8, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Roberson, E.D.; Scearce-Levie, K.; Palop, J.J.; Yan, F.; Cheng, I.H.; Wu, T.; Gerstein, H.; Yu, G.Q.; Mucke, L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 2007, 316, 750–754. [Google Scholar] [CrossRef]
- Leroy, K.; Ando, K.; Laporte, V.; Dedecker, R.; Suain, V.; Authelet, M.; Héraud, C.; Pierrot, N.; Yilmaz, Z.; Octave, J.N.; et al. Lack of tau proteins rescues neuronal cell death and decreases amyloidogenic processing of APP in APP/PS1 mice. Am. J. Pathol. 2012, 181, 1928–1940. [Google Scholar] [CrossRef] [PubMed]
- Gasparotto, J.; Girardi, C.S.; Somensi, N.; Ribeiro, C.T.; Moreira, J.C.F.; Michels, M.; Sonai, B.; Rocha, M.; Steckert, A.V.; Barichello, T.; et al. Receptor for advanced glycation end products mediates sepsis-triggered amyloid-β accumulation, Tau phosphorylation, and cognitive impairment. J. Biol. Chem. 2018, 293, 226–244. [Google Scholar] [CrossRef] [PubMed]
- Lauretti, E.; Li, J.G.; Di Meco, A.; Praticò, D. Glucose deficit triggers tau pathology and synaptic dysfunction in a tauopathy mouse model. Transl. Psychiatry 2017, 7, e1020. [Google Scholar] [CrossRef] [PubMed]
- Bloom, G.S. Amyloid-β and Tau: The Trigger and Bullet in Alzheimer Disease Pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Götz, J.; Halliday, G.; Nisbet, R.M. Molecular Pathogenesis of the Tauopathies. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 239–261. [Google Scholar] [CrossRef] [PubMed]
- Foidl, B.M.; Humpel, C. Differential Hyperphosphorylation of Tau-S199, -T231 and -S396 in Organotypic Brain Slices of Alzheimer Mice. A Model to Study Early Tau Hyperphosphorylation Using Okadaic Acid. Front. Aging Neurosci. 2018, 10, 113. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Gannon, M.; Chen, Y.; Yan, S.; Zhang, S.; Feng, W.; Tao, J.; Sha, B.; Liu, Z.; Saito, T.; et al. β-amyloid redirects norepinephrine signaling to activate the pathogenic GSK3β/tau cascade. Sci. Transl. Med. 2020, 12, eaay6931. [Google Scholar] [CrossRef]
- Li, J.; Liu, D.; Sun, L.; Lu, Y.; Zhang, Z. Advanced glycation end products and neurodegenerative diseases: Mechanisms and perspective. J Neurol Sci 2012, 317, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Batkulwar, K.; Godbole, R.; Banarjee, R.; Kassaar, O.; Williams, R.J.; Kulkarni, M.J. Advanced Glycation End Products Modulate Amyloidogenic APP Processing and Tau Phosphorylation: A Mechanistic Link between Glycation and the Development of Alzheimer’s Disease. ACS Chem. Neurosci. 2018, 9, 988–1000. [Google Scholar] [CrossRef] [PubMed]
- Himeno, E.; Ohyagi, Y.; Ma, L.; Nakamura, N.; Miyoshi, K.; Sakae, N.; Motomura, K.; Soejima, N.; Yamasaki, R.; Hashimoto, T.; et al. Apomorphine treatment in Alzheimer mice promoting amyloid-β degradation. Ann. Neurol. 2011, 69, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.D.; Chen, X.; Schmidt, A.M.; Brett, J.; Godman, G.; Zou, Y.S.; Scott, C.W.; Caputo, C.; Frappier, T.; Smith, M.A. Glycated tau protein in Alzheimer disease: A mechanism for induction of oxidant stress. Proc. Natl. Acad. Sci. USA 1994, 91, 7787–7791. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Liu, M.; Wang, T.; Wang, L.; He, H.; Zhong, Y.; Qian, G.; An, J.; Zhu, T.; Qiu, X.; et al. Modifications of autophagy influenced the Alzheimer-like changes in SH-SY5Y cells promoted by ultrafine black carbon. Environ. Pollut. 2019, 246, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Da Pozzo, E.; La Pietra, V.; Cosimelli, B.; Da Settimo, F.; Giacomelli, C.; Marinelli, L.; Martini, C.; Novellino, E.; Taliani, S.; Greco, G. p53 Functional Inhibitors Behaving Like Pifithrin-β Counteract the Alzheimer Peptide Non-β-amyloid Component Effects in Human SH-SY5Y Cells. ACS Chem. Neurosci. 2014, 5, 390–399. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shipley, M.M. Differentiation of the SH-SY5Y Human Neuroblastoma Cell Line. J. Vis. Exp. 2016, 108, 53193. [Google Scholar] [CrossRef] [PubMed]
- Agholme, L.; Lindström, T.; Kågedal, K.; Marcusson, J.; Hallbeck, M. An In Vitro Model for Neuroscience: Differentiation of SH-SY5Y Cells into Cells with Morphological and Biochemical Characteristics of Mature Neurons. J. Alzheimer’s Dis. 2010, 20, 1069–1082. [Google Scholar] [CrossRef] [PubMed]
- Constantinescu, R.; Constantinescu, A.T.; Reichmann, H.; Janetzky, B. Neuronal differentiation and long-term culture of the human neuroblastoma line SH-SY5Y. In Neuropsychiatric Disorders an Integrative Approach; Springer: Vienna, Vienna, 2007; pp. 17–28. [Google Scholar]
- Korecka, J.A.; van Kesteren, R.E.; Blaas, E.; Spitzer, S.O.; Kamstra, J.H.; Smit, A.B.; Swaab, D.F.; Verhaagen, J.; Bossers, K. Phenotypic Characterization of Retinoic Acid Differentiated SH-SY5Y Cells by Transcriptional Profiling. PLoS ONE 2013, 8, e63862. [Google Scholar] [CrossRef] [PubMed]
- Presgraves, S.P.; Ahmed, T.; Borwege, S.; Joyce, J.N. Terminally differentiated SH-SY5Y cells provide a model system for studying neuroprotective effects of dopamine agonists. Neurotox. Res. 2003, 5, 579–598. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Simpkins, J.W. Okadaic acid induces tau phosphorylation in SH-SY5Y cells in an estrogen-preventable manner. Brain Res. 2010, 1345, 176–181. [Google Scholar] [CrossRef]
- Bretteville, A.; Marcouiller, F.; Julien, C.; El Khoury, N.B.; Petry, F.R.; Poitras, I.; Mouginot, D.; Lévesque, G.; Hébert, S.S.; Planel, E. Hypothermia-induced hyperphosphorylation: A new model to study tau kinase inhibitors. Sci. Rep. 2012, 2, 480. [Google Scholar] [CrossRef] [PubMed]
- Koriyama, Y.; Furukawa, A.; Muramatsu, M.; Takino, J.; Takeuchi, M. Glyceraldehyde caused Alzheimer’s disease-like alterations in diagnostic marker levels in SH-SY5Y human neuroblastoma cells. Sci. Rep. 2015, 5, 13313. [Google Scholar] [CrossRef]
- Wang, J.; Wang, C.; Wu, Z.; Li, X.; Xu, S.; Liu, J.; Lan, Q.; Zhu, Z.; Xu, J. Design, synthesis, biological evaluation, and docking study of 4-isochromanone hybrids bearing N-benzyl pyridinium moiety as dual binding site acetylcholinesterase inhibitors (part II). Chem. Biol. Drug Des. 2018, 91, 756–762. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wu, Z.; Cai, H.; Xu, S.; Liu, J.; Jiang, J.; Yao, H.; Wu, X.; Xu, J. Design, synthesis, biological evaluation and docking study of 4-isochromanone hybrids bearing N-benzyl pyridinium moiety as dual binding site acetylcholinesterase inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 5212–5216. [Google Scholar] [CrossRef]
- Uras, G.; Manca, A.; Zhang, P.; Markus, Z.; Mack, N.; Allen, S.; Bo, M.; Xu, S.; Xu, J.; Georgiou, M.; et al. In vivo Evaluation of a Newly Synthesized Acetylcholinesterase Inhibitor in a Transgenic Drosophila Model of Alzheimer’s Disease. Front. Neurosci. 2021, 15, 691222. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Uras, G.; Zhang, P.; Xu, S.; Yin, Y.; Liu, J.; Qin, S.; Li, X.; Allen, S.; Bai, R.; et al. Discovery of Novel Tacrine–Pyrimidone Hybrids as Potent Dual AChE/GSK-3 Inhibitors for the Treatment of Alzheimer’s Disease. J. Med. Chem. 2021, 64, 7483–7506. [Google Scholar] [CrossRef] [PubMed]
- Deane, R.; Singh, I.; Sagare, A.P.; Bell, R.D.; Ross, N.T.; LaRue, B.; Love, R.; Perry, S.; Paquette, N.; Deane, R.J.; et al. A multimodal RAGE-specific inhibitor reduces amyloid β–mediated brain disorder in a mouse model of Alzheimer disease. J. Clin. Investig. 2012, 122, 1377–1392. [Google Scholar] [CrossRef]
- Donahue, J.E.; Flaherty, S.L.; Johanson, C.E.; Duncan, J.A.; Silverberg, G.D.; Miller, M.C.; Tavares, R.; Yang, W.; Wu, Q.; Sabo, E.; et al. RAGE, LRP-1, and amyloid-beta protein in Alzheimer’s disease. Acta Neuropathol. 2006, 112, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Srikanth, V.; Maczurek, A.; Phan, T.; Steele, M.; Westcott, B.; Juskiw, D.; Münch, G. Advanced glycation endproducts and their receptor RAGE in Alzheimer’s disease. Neurobiol. Aging 2011, 32, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.D.; Chen, X.; Fu, J.; Chen, M.; Zhu, H.; Roher, A.; Slattery, T.; Zhao, L.; Nagashima, M.; Morser, J.; et al. RAGE and amyloid-β peptide neurotoxicity in Alzheimer’s disease. Nature 1996, 382, 685–691. [Google Scholar] [CrossRef]
- Cai, Z.; Liu, N.; Wang, C.; Qin, B.; Zhou, Y.; Xiao, M.; Chang, L.; Yan, L.-J.; Zhao, B. Role of RAGE in Alzheimer’s Disease. Cell. Mol. Neurobiol. 2016, 36, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Uras, G.; Onuwaje, I.; Li, W.; Yao, H.; Xu, S.; Li, X.; Li, X.; Phillips, J.; Allen, S.; et al. Novel inhibitors of AChE and Aβ aggregation with neuroprotective properties as lead compounds for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2022, 235. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Compd. | Structures |
|---|---|---|
| A | XJP-1 |  |
| Name: (E)-4-((5,6-dimethoxy-1-oxo-1,3-dihydro-2H-inden-2-ylidene)methyl)-1-(3-fluorobenzyl)pyridin-1-ium bromide | ||
| B | SAD-2 |  |
| Name: (Z)-4-((6,7-dimethoxy-4-oxoisothiochroman-3-ylidene)methyl)-1-(4-fluorobenzyl)pyridin-1-ium bromide | ||
| C | SAD-6 |  |
| Name: (Z)-3-((1-benzylpiperidin-4-yl)methylene)-6,7-dimethoxyisothiochroman-4-one | ||
| D | 24r |  |
| Name: 2-((1-(4-fluorobenzyl)piperidin-4-yl)methyl)-5,6-dimethoxy-2,3-dihydrobenzo[b]thiophene 1,1-dioxide | ||
| E | 27g |  |
| Name: 2-((7-((6-chloro-1,2,3,4-tetrahydroacridin-9-yl)amino)heptyl)amino)-6-(3-fluoropyridin-4-yl)-3-methylpyrimidin-4(3H)-one | ||
| Basic Growth Media | Differentiation Media #1 | Differentiation Media #2 | Differentiation Media #3 |
|---|---|---|---|
| EMEM | EMEM | EMEM | Neurobasal |
| 15% hiFBS | 2.5% hiFBS | 1% hiFBS | 20 mM KCl |
| 1x Pen/Strep | 1x Pen/Strep | 1x Pen/Strep | 1x Pen/Strep |
| 2 mM Glutamine | 2 mM Glutamine | 2 mM Glutamine | 2 mM Glutamax |
| 10 μM RA | 10 μM RA | 10 μM RA | |
| 50 ng/mL BDNF |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uras, G.; Li, X.; Manca, A.; Pantaleo, A.; Bo, M.; Xu, J.; Allen, S.; Zhu, Z. Development of p-Tau Differentiated Cell Model of Alzheimer’s Disease to Screen Novel Acetylcholinesterase Inhibitors. Int. J. Mol. Sci. 2022, 23, 14794. https://doi.org/10.3390/ijms232314794
Uras G, Li X, Manca A, Pantaleo A, Bo M, Xu J, Allen S, Zhu Z. Development of p-Tau Differentiated Cell Model of Alzheimer’s Disease to Screen Novel Acetylcholinesterase Inhibitors. International Journal of Molecular Sciences. 2022; 23(23):14794. https://doi.org/10.3390/ijms232314794
Chicago/Turabian StyleUras, Giuseppe, Xinuo Li, Alessia Manca, Antonella Pantaleo, Marco Bo, Jinyi Xu, Stephanie Allen, and Zheying Zhu. 2022. "Development of p-Tau Differentiated Cell Model of Alzheimer’s Disease to Screen Novel Acetylcholinesterase Inhibitors" International Journal of Molecular Sciences 23, no. 23: 14794. https://doi.org/10.3390/ijms232314794
APA StyleUras, G., Li, X., Manca, A., Pantaleo, A., Bo, M., Xu, J., Allen, S., & Zhu, Z. (2022). Development of p-Tau Differentiated Cell Model of Alzheimer’s Disease to Screen Novel Acetylcholinesterase Inhibitors. International Journal of Molecular Sciences, 23(23), 14794. https://doi.org/10.3390/ijms232314794