
The Mechanism of Selective Recognition of Lipid Substrate by hDHHC20 Enzyme

 , ,
, ,  ,
,  and
and
Abstract
:1. Introduction
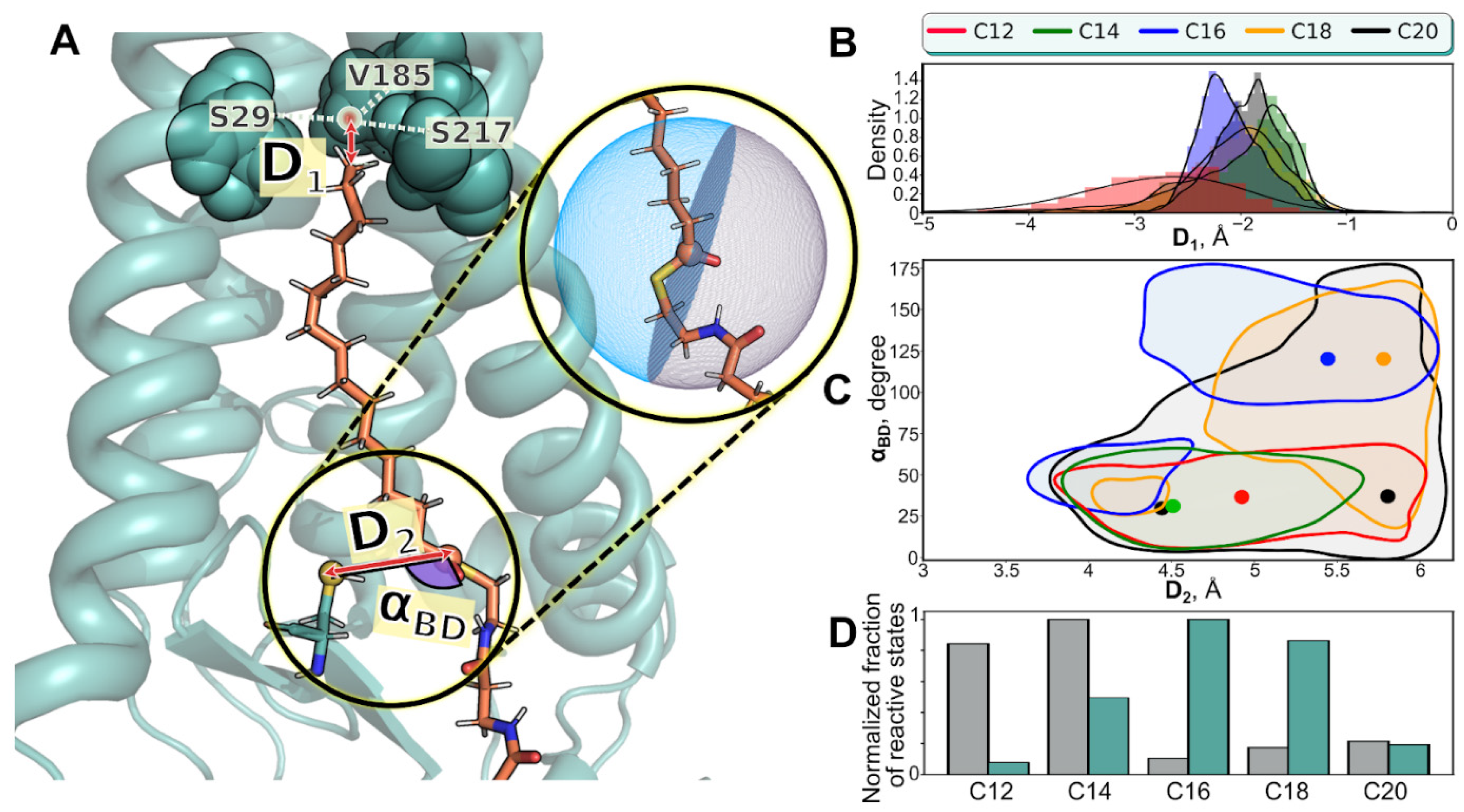
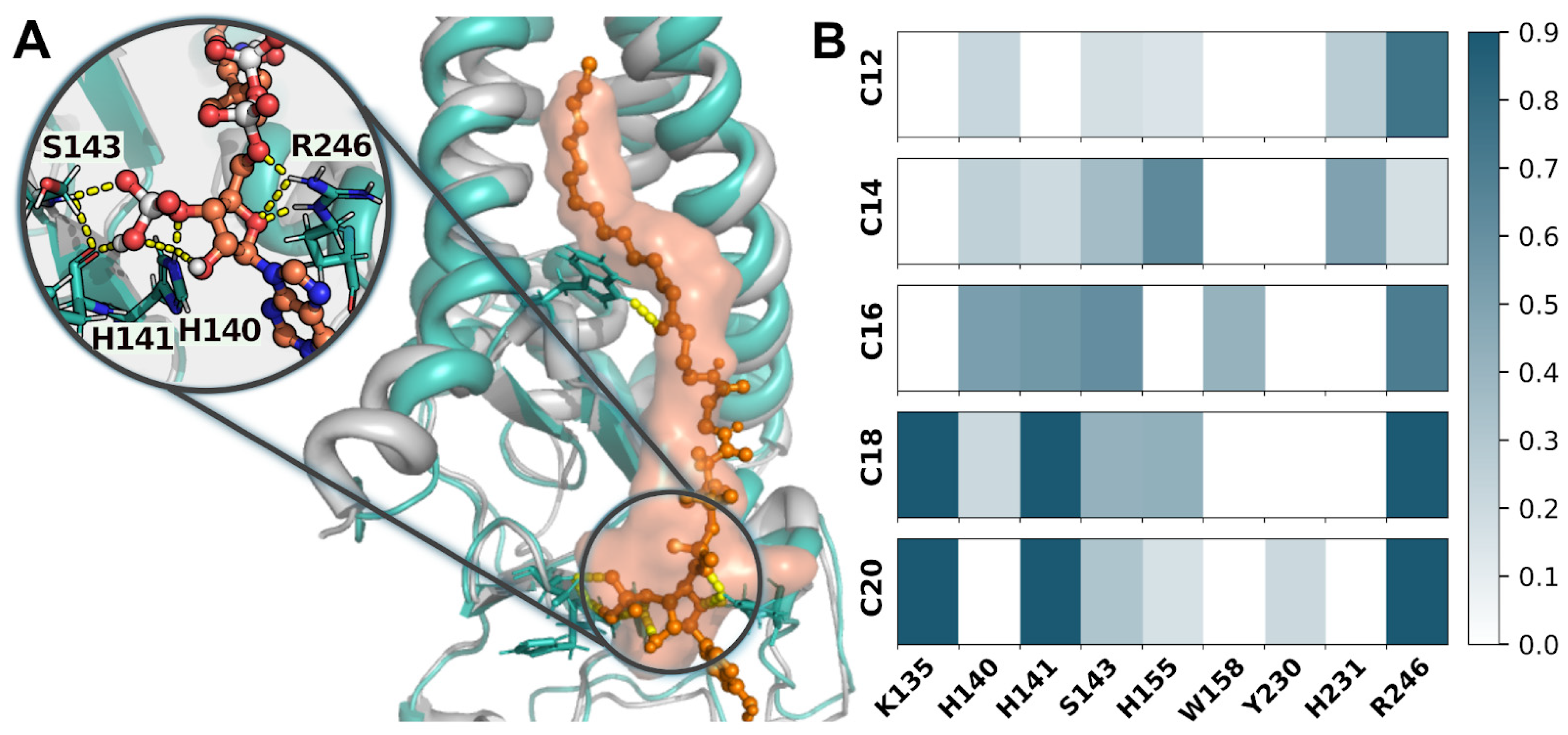
2. Results

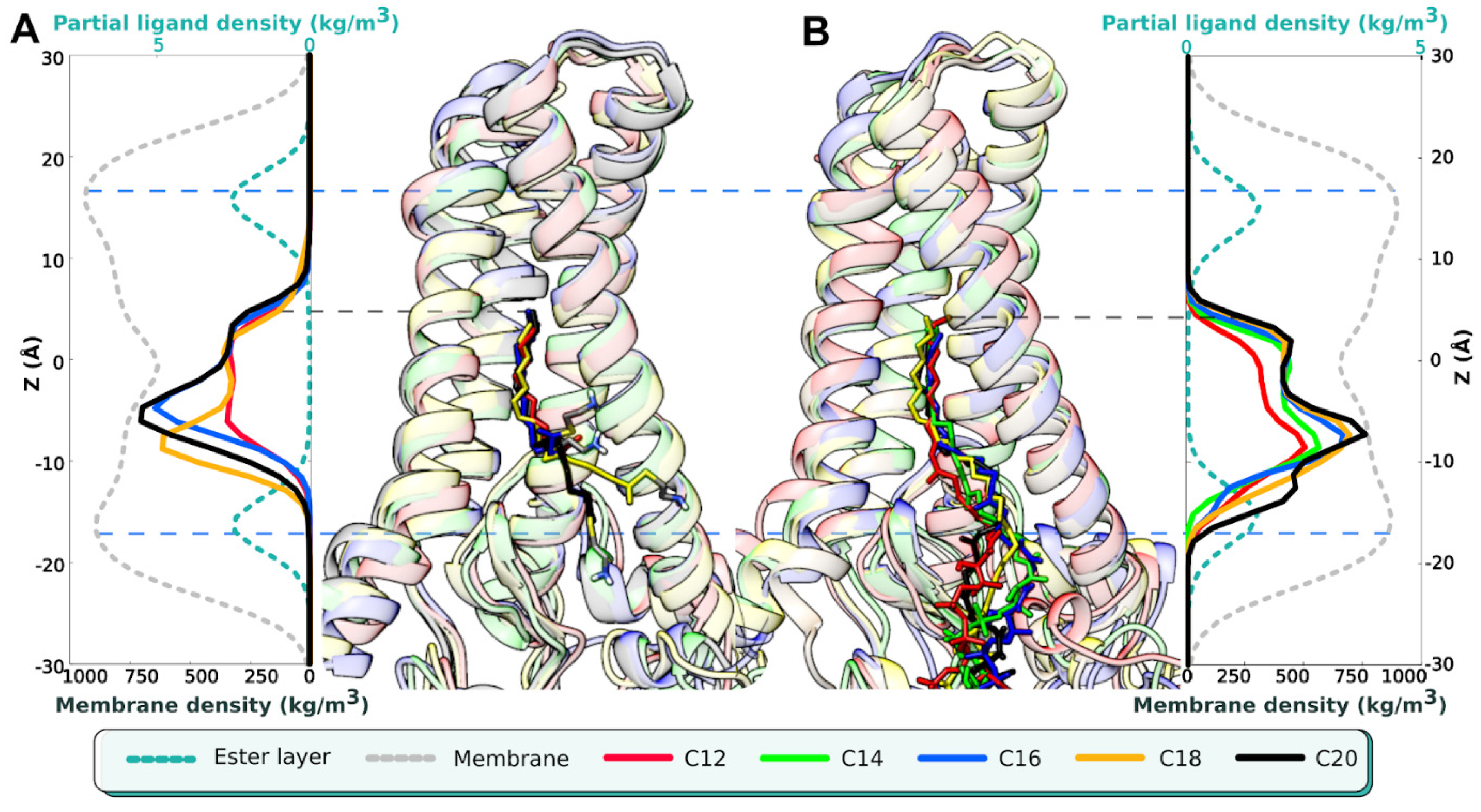
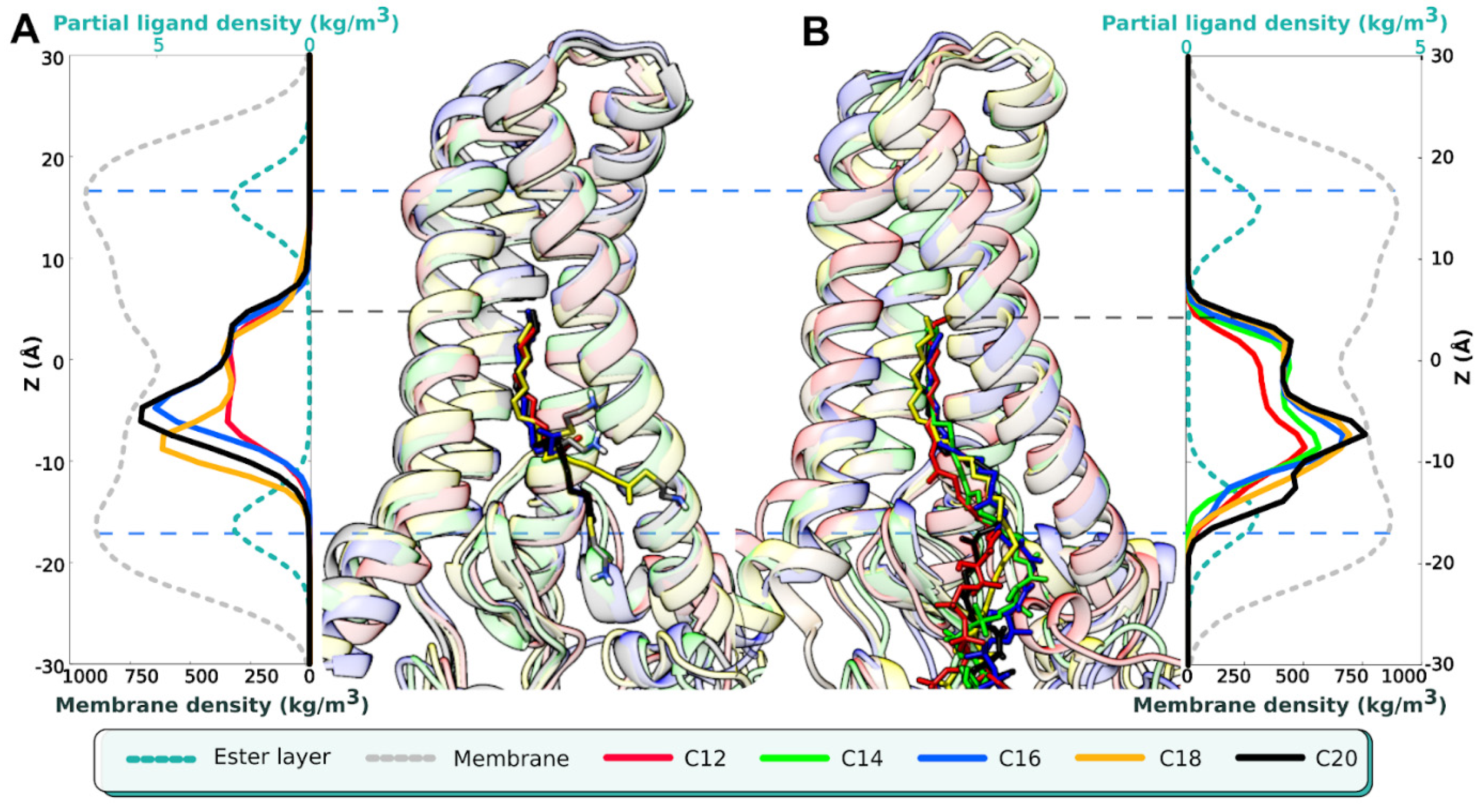{kind=link}
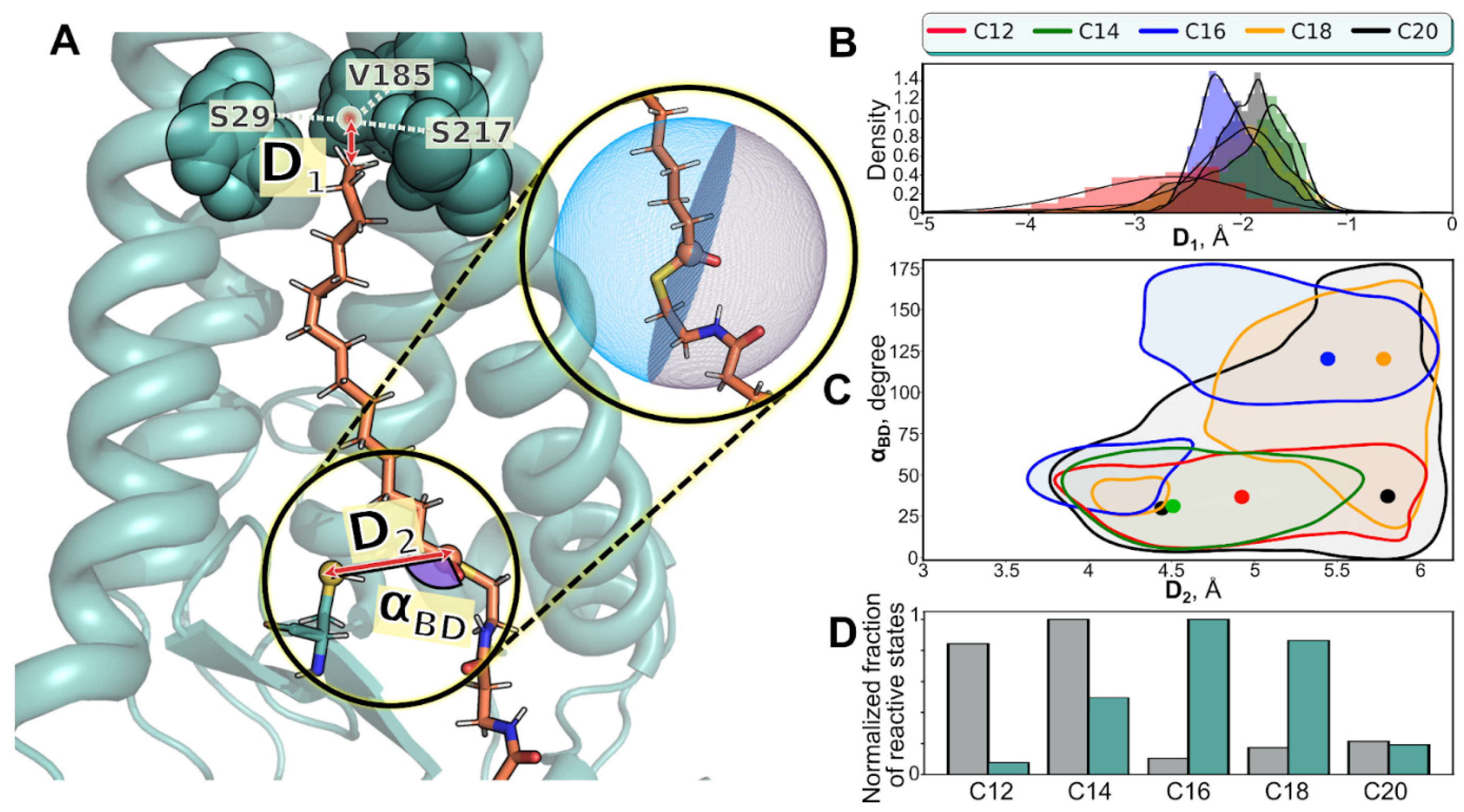{kind=link}
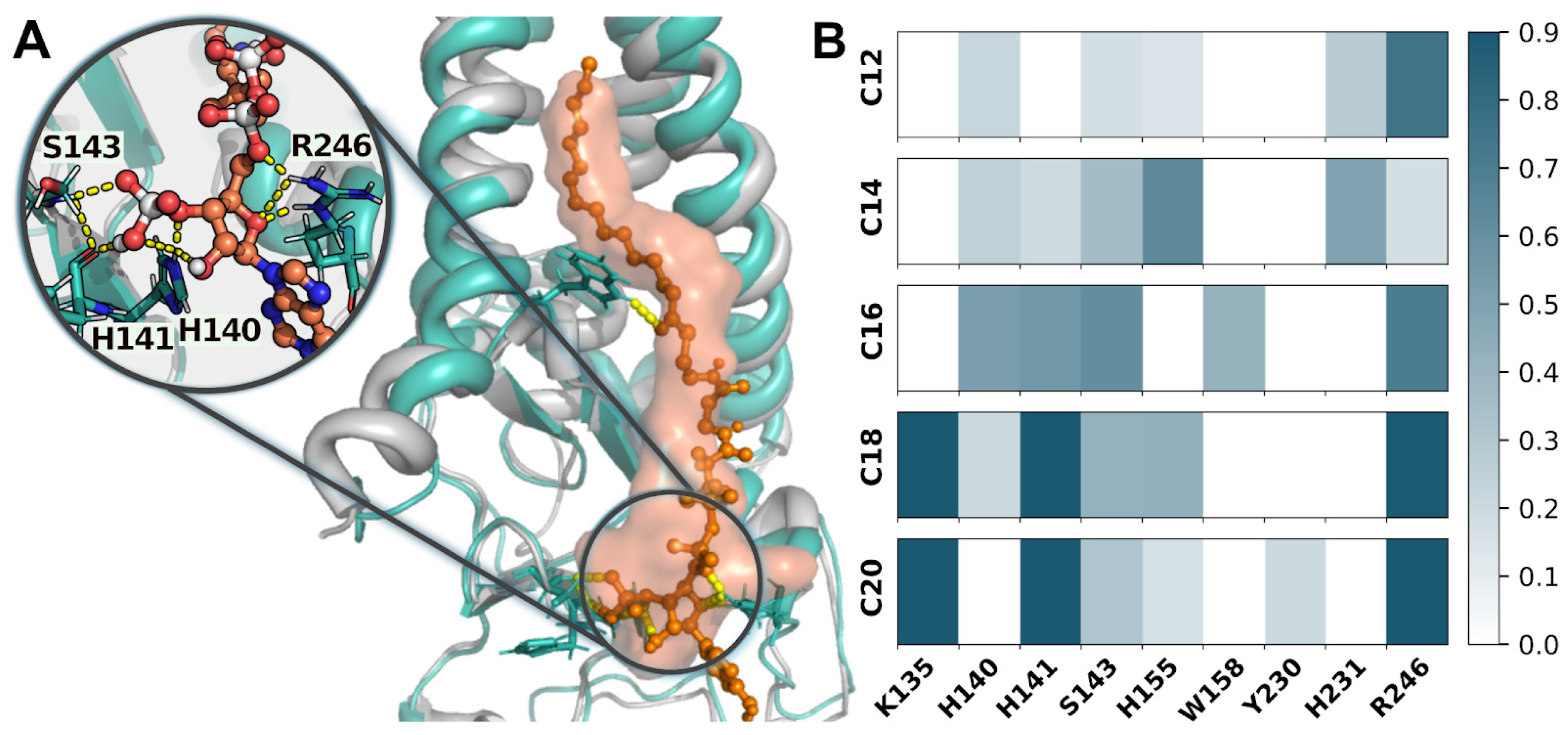{kind=link}
| Name of MD Run | System Composition | <D1>, Å | D1>, Å | <αBD>, ° |
|---|---|---|---|---|
| C12 (1) C12 (2) C12 (3) | hDHHC201/C12-CoA1/POPC238/H2O24944/Na+64/Cl−69 | −2.7 ± 0.6 −11.4 ± 3.2 −11.5 ± 1.6 | 5 ± 0.5 5.7 ± 0.3 5.2 ± 0.7 | 40 ± 20 101 ± 23 52 ± 24 |
| C14 (1) C14 (2) C14 (3) | hDHHC201/C14-CoA1/POPC238/H2O24940/Na+66/Cl−71 | −2 ± 0.6 −9.4 ± 2.8 −5.3 ± 2.3 | 4.8 ± 0.5 4.4 ± 0.5 4.9 ± 0.6 | 42 ± 27 58 ± 30 75 ±42 |
| C16 (1) C16 (2) C16 (3) | hDHHC201/C16-CoA1/POPC238/H2O24935/Na+66/Cl−71 | −2.1 ± 0.3 −1.7 ± 0.4 −1.8 ± 0.6 | 5.1 ± 0.6 5.6 ± 0.3 4.7 ± 0.5 | 112 ± 34 35 ± 23 110 ± 54 |
| C18 (1) C18 (2) C18 (3) | hDHHC201/C18-CoA1/POPC237/H2O24939/Na+66/Cl−71 | −2.1 ± 0.8 −2.4 ± 0.9 −2 ± 0.7 | 5.3 ± 0.6 5.5 ± 0.5 5.4 ± 0.4 | 107 ± 41 71 ± 38 101 ± 32 |
| C20 (1) C20 (2) C20 (3) | hDHHC201/C20-CoA1/POPC237/H2O24940/Na+66/Cl−71 | −1.9 ± 0.3 −1.6 ± 0.6 −2.3 ± 0.9 | 5.4 ± 0.5 5.5 ± 0.5 5.1 ± 0.6 | 54 ± 40 61 ± 25 66 ± 38 |
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hannoush, R.N. Synthetic Protein Lipidation. Curr. Opin. Chem. Biol. 2015, 28, 39–46. [Google Scholar] [CrossRef]
- Martin, B.R.; Wang, C.; Adibekian, A.; Tully, S.E.; Cravatt, B.F. Global Profiling of Dynamic Protein Palmitoylation. Nat. Methods 2011, 9, 84–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaskovic, S.; Blanc, M.; van der Goot, F.G. What Does S-Palmitoylation Do to Membrane Proteins? FEBS J. 2013, 280, 2766–2774. [Google Scholar] [CrossRef] [Green Version]
- Resh, M.D. Fatty Acylation of Proteins: The Long and the Short of It. Prog. Lipid Res. 2016, 63, 120–131. [Google Scholar] [CrossRef] [Green Version]
- Nůsková, H.; Serebryakova, M.V.; Ferrer-Caelles, A.; Sachsenheimer, T.; Lüchtenborg, C.; Miller, A.K.; Brügger, B.; Kordyukova, L.V.; Teleman, A.A. Stearic Acid Blunts Growth-Factor Signaling via Oleoylation of GNAI Proteins. Nat. Commun. 2021, 12, 4590. [Google Scholar] [CrossRef]
- Kordyukova, L.; Krabben, L.; Serebryakova, M.; Veit, M. S-Acylation of Proteins. Methods Mol. Biol. 2019, 1934, 265–291. [Google Scholar]
- Kordyukova, L.V.; Serebryakova, M.V.; Polyansky, A.A.; Kropotkina, E.A.; Alexeevski, A.V.; Veit, M.; Efremov, R.G.; Filippova, I.Y.; Baratova, L.A. Linker And/or Transmembrane Regions of Influenza A/Group-1, A/Group-2, and Type B Virus Hemagglutinins Are Packed Differently within Trimers. Biochim. Biophys. Acta (BBA)—Biomembr. 2011, 1808, 1843–1854. [Google Scholar] [CrossRef] [Green Version]
- Wagner, R.; Herwig, A.; Azzouz, N.; Klenk, H.D. Acylation-Mediated Membrane Anchoring of Avian Influenza Virus Hemagglutinin Is Essential for Fusion Pore Formation and Virus Infectivity. J. Virol. 2005, 79, 6449–6458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.J.; Takeda, M.; Lamb, R.A. Influenza Virus Hemagglutinin (H3 Subtype) Requires Palmitoylation of Its Cytoplasmic Tail for Assembly: M1 Proteins of Two Subtypes Differ in Their Ability to Support Assembly. J. Virol. 2005, 79, 13673–13684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kordyukova, L.V.; Serebryakova, M.V.; Khrustalev, V.V.; Veit, M. Differential S-Acylation of Enveloped Viruses. Protein Pept. Lett. 2019, 26, 588–600. [Google Scholar] [CrossRef] [PubMed]
- Rana, M.S.; Kumar, P.; Lee, C.-J.; Verardi, R.; Rajashankar, K.R.; Banerjee, A. Fatty Acyl Recognition and Transfer by an Integral Membrane S-Acyltransferase. Science 2018, 359, eaao6326. [Google Scholar] [CrossRef]
- Draper, J.M.; Smith, C.D. DHHC20: A Human Palmitoyl Acyltransferase That Causes Cellular Transformation. Mol. Membr. Biol. 2010, 27, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Gadalla, M.R.; Abrami, L.; van der Goot, F.G.; Veit, M. Hemagglutinin of Influenza A, but Not of Influenza B and C Viruses Is Acylated by ZDHHC2, 8, 15 and 20. Biochem. J. 2020, 477, 285–303. [Google Scholar] [CrossRef]
- Mesquita, F.S.; Abrami, L.; Sergeeva, O.; Turelli, P.; Qing, E.; Kunz, B.; Raclot, C.; Paz Montoya, J.; Abriata, L.A.; Gallagher, T.; et al. S-Acylation Controls SARS-CoV-2 Membrane Lipid Organization and Enhances Infectivity. Dev. Cell 2021, 56, 2790–2807.e8. [Google Scholar] [CrossRef] [PubMed]
- Puthenveetil, R.; Lun, C.M.; Murphy, R.E.; Healy, L.B.; Vilmen, G.; Christenson, E.T.; Freed, E.O.; Banerjee, A. S-Acylation of SARS-CoV-2 Spike Protein: Mechanistic Dissection, in Vitro Reconstitution and Role in Viral Infectivity. J. Biol. Chem. 2021, 297, 101112. [Google Scholar] [CrossRef]
- Wu, Z.; Zhang, Z.; Wang, X.; Zhang, J.; Ren, C.; Li, Y.; Gao, L.; Liang, X.; Wang, P.; Ma, C. Palmitoylation of SARS-CoV-2 S Protein Is Essential for Viral Infectivity. Signal Transduct. Target Ther. 2021, 6, 231. [Google Scholar] [CrossRef]
- Rana, M.S.; Lee, C.-J.; Banerjee, A. The Molecular Mechanism of DHHC Protein Acyltransferases. Biochem. Soc. Trans. 2019, 47, 157–167. [Google Scholar] [CrossRef]
- Panina, I.; Krylov, N.; Gadalla, M.R.; Aliper, E.; Kordyukova, L.; Veit, M.; Chugunov, A.; Efremov, R. Molecular Dynamics of DHHC20 Acyltransferase Suggests Principles of Lipid and Protein Substrate Selectivity. Int. J. Mol. Sci. 2022, 23, 5091. [Google Scholar] [CrossRef]
- Lee, C.-J.; Stix, R.; Rana, M.S.; Shikwana, F.; Murphy, R.E.; Ghirlando, R.; Faraldo-Gómez, J.D.; Banerjee, A. Bivalent Recognition of Fatty Acyl-CoA by a Human Integral Membrane Palmitoyltransferase. Proc. Natl. Acad. Sci. USA 2022, 119, e2022050119. [Google Scholar] [CrossRef] [PubMed]
- Burgi, H.B.; Dunitz, J.D.; Lehn, J.M.; Wipff, G. Stereochemistry of Reaction Paths at Carbonyl Centres. Tetrahedron 1974, 30, 1563–1572. [Google Scholar] [CrossRef]
- Radisky, E.S.; Koshland, D.E., Jr. A Clogged Gutter Mechanism for Protease Inhibitors. Proc. Natl. Acad. Sci. USA 2002, 99, 10316–10321. [Google Scholar] [CrossRef]
- Vilmen, G.; Banerjee, A.; Freed, E.O. Freed Rafting through the Palms: S-Acylation of SARS-CoV-2 Spike Protein Induces Lipid Reorganization. Dev. Cell 2021, 56, 2787–2789. [Google Scholar] [CrossRef] [PubMed]
- Veit, M. Palmitoylation of Virus Proteins. Biol. Cell 2012, 104, 493–515. [Google Scholar] [CrossRef]
- Sobocińska, J.; Roszczenko-Jasińska, P.; Ciesielska, A.; Kwiatkowska, K. Protein Palmitoylation and Its Role in Bacterial and Viral Infections. Front. Immunol. 2018, 8, 2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veit, M.; Serebryakova, M.V.; Kordyukova, L.V. Palmitoylation of Influenza Virus Proteins. Biochem. Soc. Trans. 2013, 41, 50–55. [Google Scholar] [CrossRef] [Green Version]
- Kordyukova, L.V.; Serebryakova, M.V.; Baratova, L.A.; Veit, M. Site-Specific Attachment of Palmitate or Stearate to Cytoplasmic versus Transmembrane Cysteines Is a Common Feature of Viral Spike Proteins. Virology 2010, 398, 49–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kordyukova, L.V.; Serebryakova, M.V.; Baratova, L.A.; Veit, M. S Acylation of the Hemagglutinin of Influenza Viruses: Mass Spectrometry Reveals Site-Specific Attachment of Stearic Acid to a Transmembrane Cysteine. J. Virol. 2008, 82, 9288–9292. [Google Scholar] [CrossRef] [Green Version]
- The PyMOL Molecular Graphics System; Schrödinger, LLC: New York, NY, USA, 2020.
- van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane Lipids: Where They Are and How They Behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef]
- Ohno, Y.; Kihara, A.; Sano, T.; Igarashi, Y. Intracellular Localization and Tissue-Specific Distribution of Human and Yeast DHHC Cysteine-Rich Domain-Containing Proteins. Biochim. Biophys. Acta 2006, 1761, 474–483. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Beglov, D.; Roux, B. Finite Representation of an Infinite Bulk System: Solvent Boundary Potential for Computer Simulations. J. Chem. Phys. 1994, 100, 9050–9063. [Google Scholar] [CrossRef]
- Yoo, J.; Aksimentiev, A. Improved Parametrization of Li+, Na+, K+, and Mg2+ Ions for All-Atom Molecular Dynamics Simulations of Nucleic Acid Systems. J. Phys. Chem. Lett. 2012, 3, 45–50. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef] [PubMed]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; Mackerell, A.D., Jr. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone φ, ψ and Side-Chain χ(1) and χ(2) Dihedral Angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D., Jr. CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [Green Version]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM General Force Field: A Force Field for Drug-like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [Green Version]
- Klauda, J.B.; Monje, V.; Kim, T.; Im, W. Improving the CHARMM Force Field for Polyunsaturated Fatty Acid Chains. J. Phys. Chem. B 2012, 116, 9424–9431. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Veit, M.; Siche, S. S-Acylation of Influenza Virus Proteins: Are Enzymes for Fatty Acid Attachment Promising Drug Targets? Vaccine 2015, 33, 7002–7007. [Google Scholar] [CrossRef]
- Gadalla, M.R.; Veit, M. Toward the Identification of ZDHHC Enzymes Required for Palmitoylation of Viral Protein as Potential Drug Targets. Expert Opin. Drug Discov. 2020, 15, 159–177. [Google Scholar] [CrossRef]
- Toelzer, C.; Gupta, K.; Yadav, S.K.N.; Borucu, U.; Davidson, A.D.; Kavanagh Williamson, M.; Shoemark, D.K.; Garzoni, F.; Staufer, O.; Milligan, R.; et al. Free Fatty Acid Binding Pocket in the Locked Structure of SARS-CoV-2 Spike Protein. Science 2020, 370, 725–730. [Google Scholar] [CrossRef]
- Lan, Q.; Chan, J.F.-W.; Xu, W.; Wang, L.; Jiao, F.; Zhang, G.; Pu, J.; Zhou, J.; Xia, S.; Lu, L.; et al. A Palmitic Acid-Conjugated, Peptide-Based Pan-CoV Fusion Inhibitor Potently Inhibits Infection of SARS-CoV-2 Omicron and Other Variants of Concern. Viruses 2022, 14, 549. [Google Scholar] [CrossRef]
- Senyilmaz-Tiebe, D.; Pfaff, D.H.; Virtue, S.; Schwarz, K.V.; Fleming, T.; Altamura, S.; Muckenthaler, M.U.; Okun, J.G.; Vidal-Puig, A.; Nawroth, P.; et al. Dietary Stearic Acid Regulates Mitochondria in Vivo in Humans. Nat. Commun. 2018, 9, 3129. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panina, I.S.; Krylov, N.A.; Chugunov, A.O.; Efremov, R.G.; Kordyukova, L.V. The Mechanism of Selective Recognition of Lipid Substrate by hDHHC20 Enzyme. Int. J. Mol. Sci. 2022, 23, 14791. https://doi.org/10.3390/ijms232314791
Panina IS, Krylov NA, Chugunov AO, Efremov RG, Kordyukova LV. The Mechanism of Selective Recognition of Lipid Substrate by hDHHC20 Enzyme. International Journal of Molecular Sciences. 2022; 23(23):14791. https://doi.org/10.3390/ijms232314791
Chicago/Turabian StylePanina, Irina S., Nikolay A. Krylov, Anton O. Chugunov, Roman G. Efremov, and Larisa V. Kordyukova. 2022. "The Mechanism of Selective Recognition of Lipid Substrate by hDHHC20 Enzyme" International Journal of Molecular Sciences 23, no. 23: 14791. https://doi.org/10.3390/ijms232314791
APA StylePanina, I. S., Krylov, N. A., Chugunov, A. O., Efremov, R. G., & Kordyukova, L. V. (2022). The Mechanism of Selective Recognition of Lipid Substrate by hDHHC20 Enzyme. International Journal of Molecular Sciences, 23(23), 14791. https://doi.org/10.3390/ijms232314791