
The Integration of Genome-Wide Association Study and Homology Analysis to Explore the Genomic Regions and Candidate Genes for Panicle-Related Traits in Foxtail Millet

Abstract
:1. Introduction
2. Results
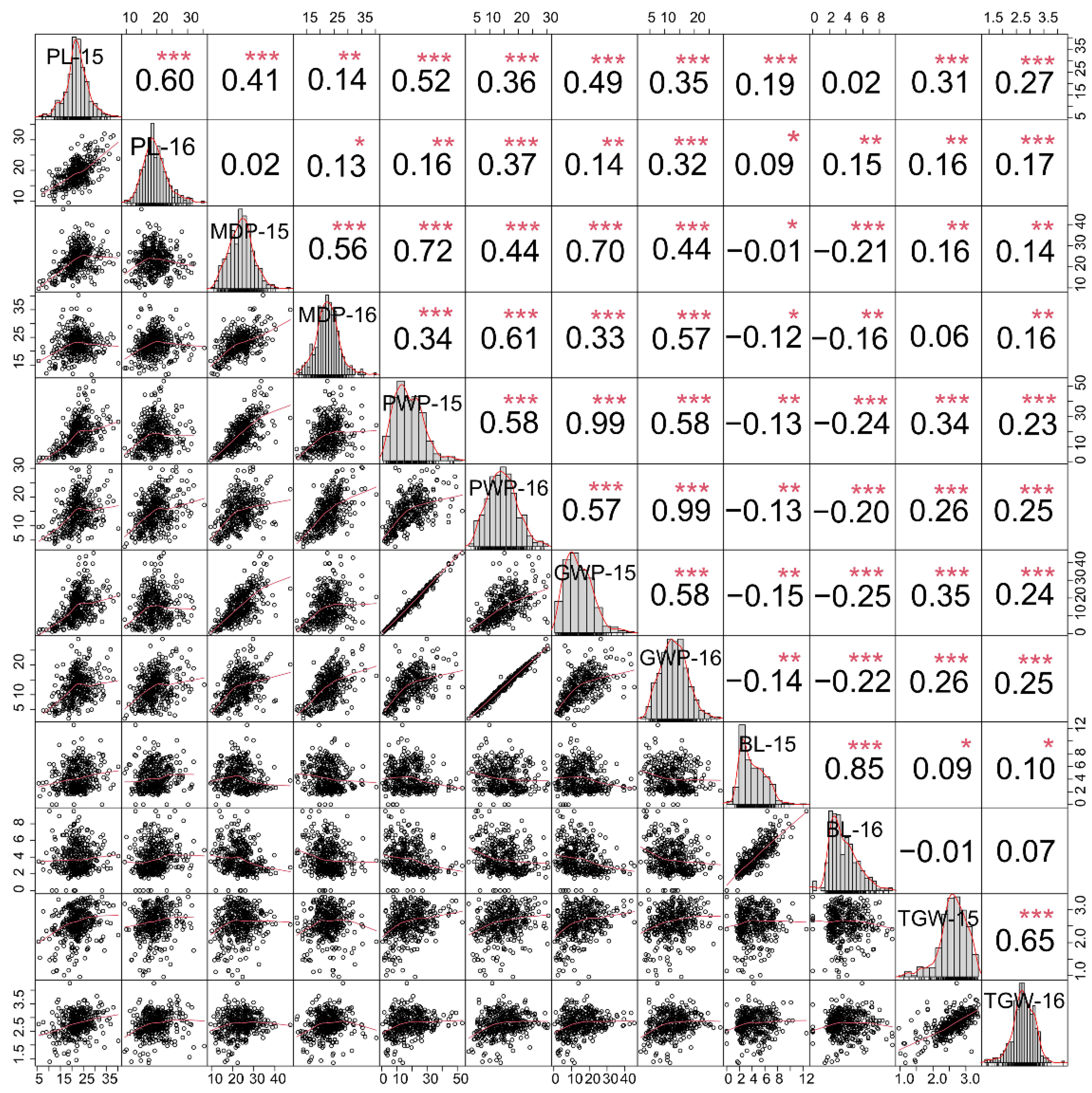
2.1. Phenotypic Characteristics of Six Panicle-Related Traits in Foxtail Millet
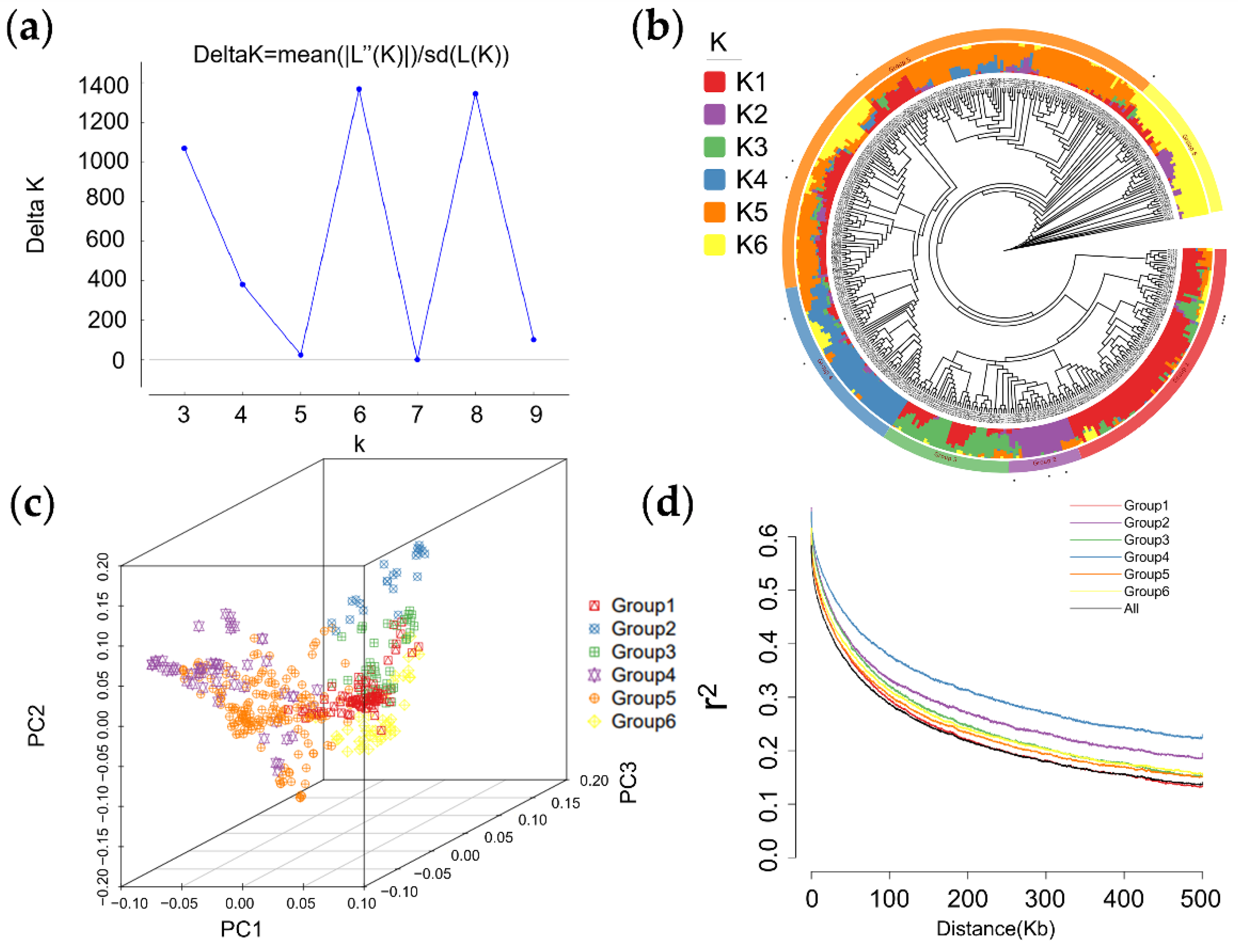
2.2. Genotyping, Population Structure, Principal Component, and Linkage Disequilibrium Analysis
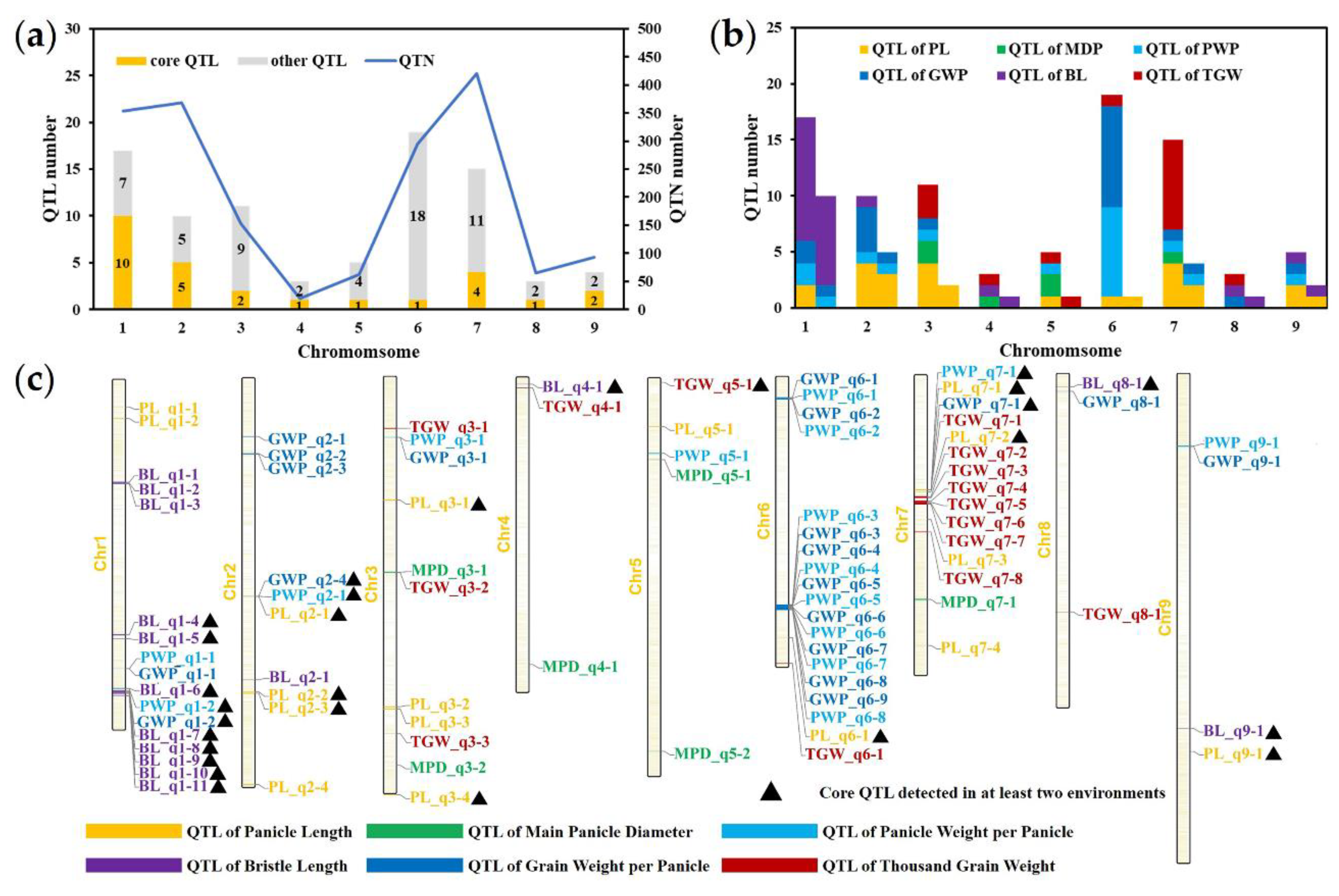
2.3. Quantitative Trait Nucleotides (QTNs) and QTL Regions for Six Panicle-Related Traits
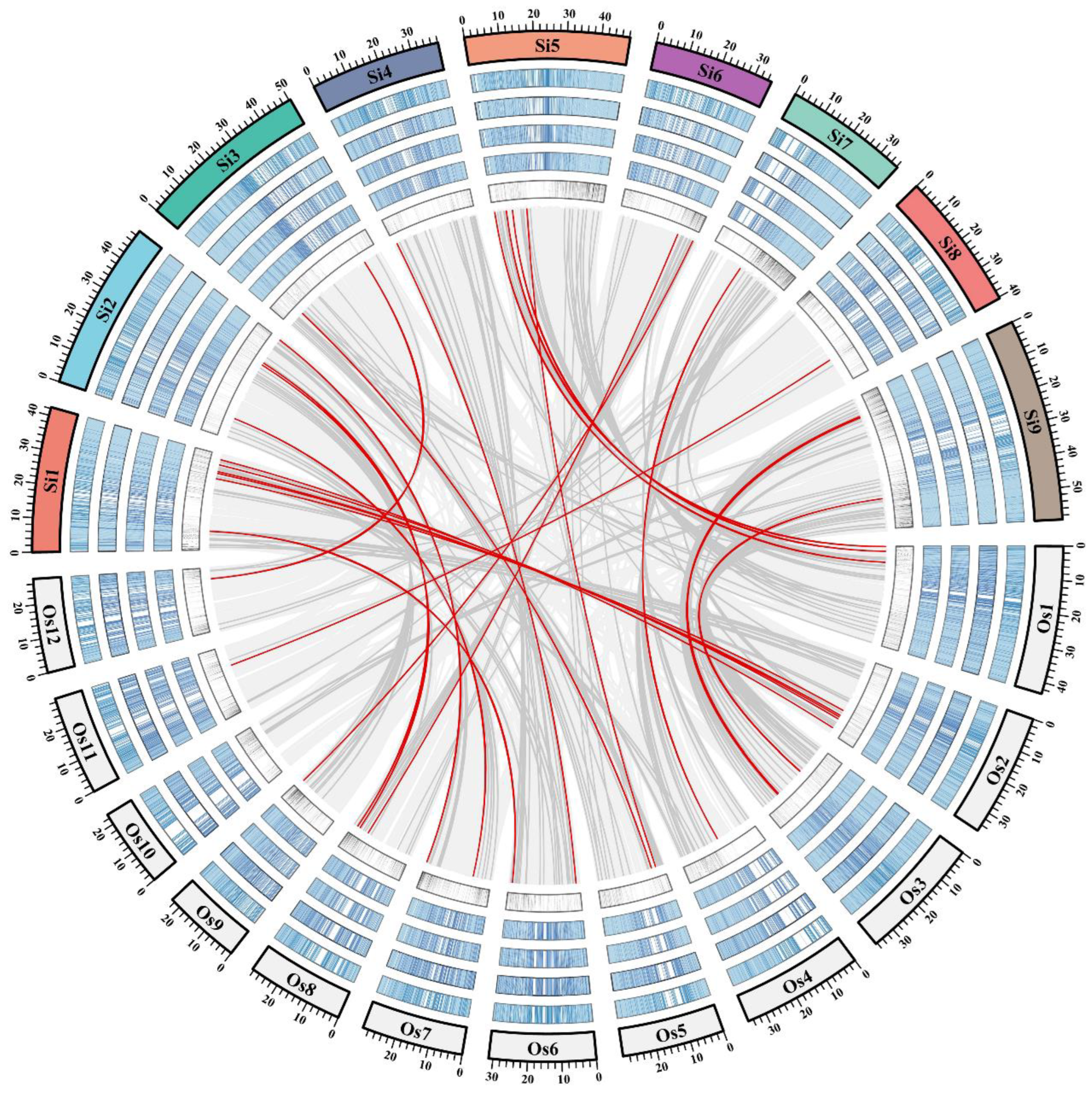
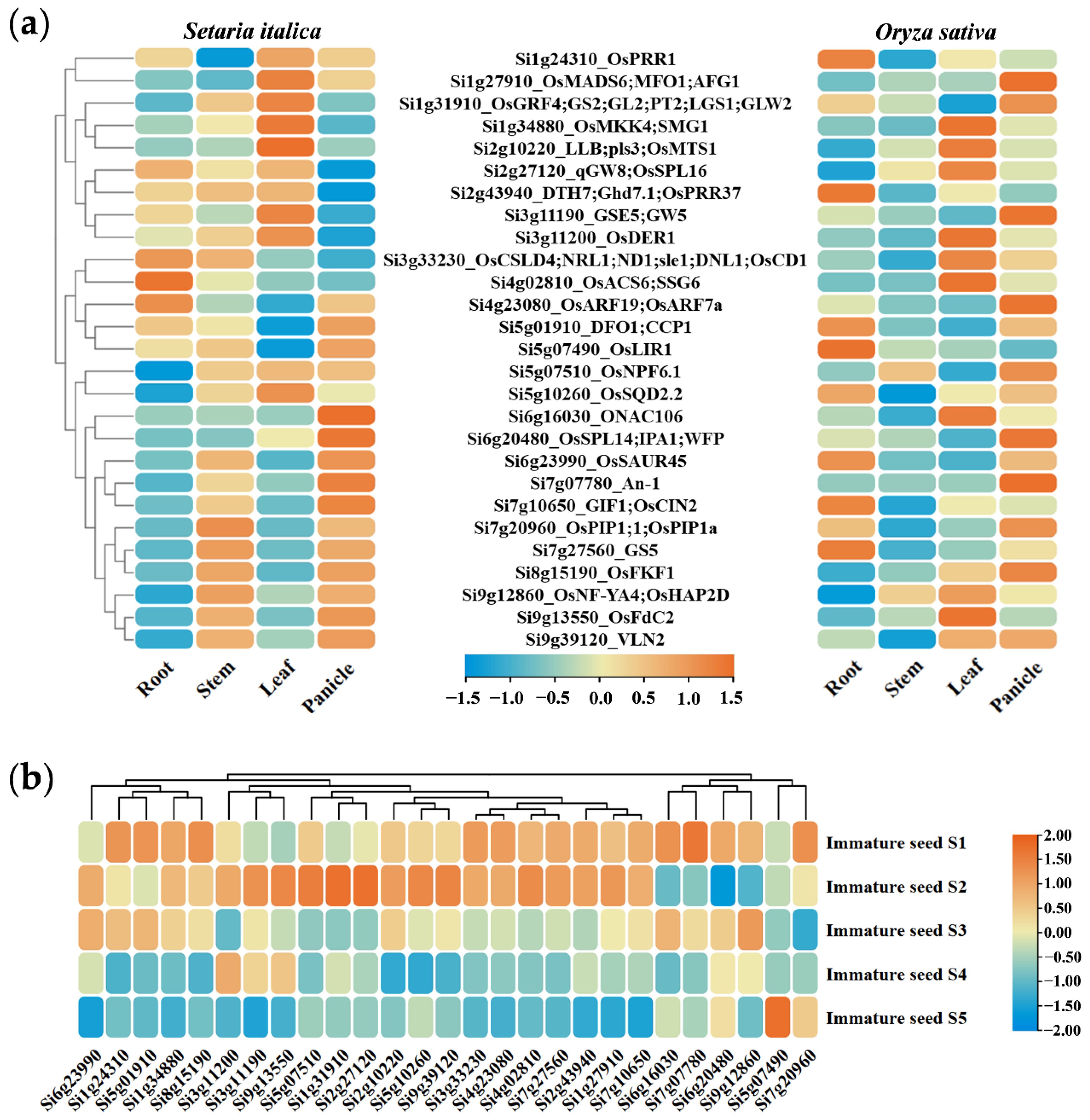
2.4. Candidate Gene Mining and Haplotype Analysis Based on Homology Comparison Strategy
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Measurement and Statistical Analysis of Panicle-Related Traits
4.3. Genotyping, Population Structure, Principal Component, and Linkage Disequilibrium Analysis
4.4. Genome-Wide Association Study
4.5. Candidate Gene Mining and Haplotype Analysis Based on Homologous Alignment
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mamidi, S.; Healey, A.; Huang, P.; Grimwood, J.; Jenkins, J.; Barry, K.; Sreedasyam, A.; Shu, S.; Lovell, J.T.; Feldman, M.; et al. A genome resource for green millet Setaria viridis enables discovery of agronomically valuable loci. Nat. Biotechnol. 2020, 38, 1203–1210. [Google Scholar] [CrossRef]
- Bettinger, R.L.; Barton, L.; Morgan, C. The origins of food production in north China: A different kind of agricultural revolution. Evol. Anthropol. Issues News Rev. 2010, 19, 9–21. [Google Scholar] [CrossRef]
- Sachdev, N.; Goomer, S.; Singh, L.R. Foxtail millet: A potential crop to meet future demand scenario for alternative sustainable protein. J. Sci. Food Agric. 2021, 101, 831–842. [Google Scholar] [CrossRef] [PubMed]
- Lata, C.; Gupta, S.; Prasad, M. Foxtail millet: A model crop for genetic and genomic studies in bioenergy grasses. Crit. Rev. Biotechnol. 2013, 33, 328–343. [Google Scholar] [CrossRef]
- Li, X.; Gao, J.; Song, J.; Guo, K.; Hou, S.; Wang, X.; He, Q.; Zhang, Y.; Zhang, Y.; Yang, Y.; et al. Multi-omics analyses of 398 foxtail millet accessions reveal genomic regions associated with domestication, metabolite traits, and anti-inflammatory effects. Mol. Plant 2022, 15, 1367–1383. [Google Scholar] [CrossRef]
- Kyozuka, J.; Tokunaga, H.; Yoshida, A. Control of grass inflorescence form by the fine-tuning of meristem phase change. Curr. Opin. Plant Biol. 2014, 17, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Suzaki, T.; Sato, M.; Ashikari, M.; Miyoshi, M.; Nagato, Y.; Hirano, H.Y. The gene FLORAL ORGAN NUMBER1 regulates floral meristem size in rice and encodes a leucine-rich repeat receptor kinase orthologous to Arabidopsis CLAVATA1. Development 2004, 131, 5649–5657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nardmann, J.; Werr, W. The shoot stem cell niche in angiosperms: Expression patterns of WUS orthologues in rice and maize imply major modifications in the course of mono- and dicot evolution. Mol. Biol. Evol. 2006, 23, 2492–2504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mjomba, F.M.; Zheng, Y.; Liu, H.; Tang, W.; Hong, Z.; Wang, F.; Wu, W. Homeobox Is Pivotal for OsWUS Controlling Tiller Development and Female Fertility in Rice. G3 2016, 6, 2013–2021. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, K.; Yasuno, N.; Sato, Y.; Yoda, M.; Yamazaki, R.; Kimizu, M.; Yoshida, H.; Nagamura, Y.; Kyozuka, J. Inflorescence meristem identity in rice is specified by overlapping functions of three AP1/FUL-like MADS box genes and PAP2, a SEPALLATA MADS box gene. Plant Cell 2012, 24, 1848–1859. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, S.; Tsuji, H.; Matsumoto, A.; Fujita, A.; Shimatani, Z.; Terada, R.; Sakamoto, T.; Kurata, T.; Shimamoto, K. FT-like proteins induce transposon silencing in the shoot apex during floral induction in rice. Proc. Natl. Acad. Sci. USA 2015, 112, E901–E910. [Google Scholar] [CrossRef] [Green Version]
- Ashikari, M.; Sakakibara, H.; Lin, S.; Yamamoto, T.; Takashi, T.; Nishimura, A.; Angeles, E.R.; Qian, Q.; Kitano, H.; Matsuoka, M. Cytokinin oxidase regulates rice grain production. Science 2005, 309, 741–745. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, K.; Ito, Y.; Sato, Y.; Kurata, N. Positive autoregulation of a KNOX gene is essential for shoot apical meristem maintenance in rice. Plant Cell 2011, 23, 4368–4381. [Google Scholar] [CrossRef] [Green Version]
- Doust, A.N.; Devos, K.M.; Gadberry, M.D.; Gale, M.D.; Kellogg, E.A. The genetic basis for inflorescence variation between foxtail and green millet (poaceae). Genetics 2005, 169, 1659–1672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, X.; Dong, K.; Wang, X.; Liu, T.; He, J.; Ren, R.; Zhang, L.; Liu, R.; Liu, X.; Li, M.; et al. A high density genetic map and QTL for agronomic and yield traits in Foxtail millet [Setaria italica (L.) P. Beauv]. BMC Genom. 2016, 17, 336. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Wang, J.; Peng, J.; Du, X.; Jiang, M.; Li, Y.; Han, F.; Du, G.; Yang, H.; Lian, S.; et al. QTL mapping for 11 agronomic traits based on a genome-wide Bin-map in a large F2 population of foxtail millet (Setaria italica (L.) P. Beauv). Mol. Breed. 2019, 39, 18. [Google Scholar] [CrossRef]
- Zhi, H.; He, Q.; Tang, S.; Yang, J.; Zhang, W.; Liu, H.; Jia, Y.; Jia, G.; Zhang, A.; Li, Y.; et al. Genetic control and phenotypic characterization of panicle architecture and grain yield-related traits in foxtail millet (Setaria italica). Theor. Appl. Genet. 2021, 134, 3023–3036. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Amo, A.; Wei, D.; Chai, Y.; Zheng, J.; Qiao, P.; Cui, C.; Lu, S.; Chen, L.; Hu, Y.G. Large-scale integration of meta-QTL and genome-wide association study discovers the genomic regions and candidate genes for yield and yield-related traits in bread wheat. Theor. Appl. Genet. 2021, 134, 3083–3109. [Google Scholar] [CrossRef]
- Saleem, K.; Shokat, S.; Waheed, M.Q.; Arshad, H.M.I.; Arif, M.A.R. A GBS-Based GWAS Analysis of Leaf and Stripe Rust Resistance in Diverse Pre-Breeding Germplasm of Bread Wheat (Triticum aestivum L.). Plants 2022, 11, 2363. [Google Scholar] [CrossRef]
- Jabbari, M.; Fakheri, B.A.; Aghnoum, R.; Mahdi Nezhad, N.; Ataei, R. GWAS analysis in spring barley (Hordeum vulgare L.) for morphological traits exposed to drought. PLoS ONE 2018, 13, e0204952. [Google Scholar] [CrossRef]
- Yano, K.; Yamamoto, E.; Aya, K.; Takeuchi, H.; Lo, P.C.; Hu, L.; Yamasaki, M.; Yoshida, S.; Kitano, H.; Hirano, K.; et al. Genome-wide association study using whole-genome sequencing rapidly identifies new genes influencing agronomic traits in rice. Nat. Genet. 2016, 48, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Rayaprolu, L.; Selvanayagam, S.; Rao, D.M.; Gupta, R.; Das, R.R.; Rathore, A.; Gandham, P.; Kiranmayee, K.N.S.U.; Deshpande, S.P.; Are, A.K. Genome-Wide Association Study for Major Biofuel Traits in Sorghum Using Minicore Collection. Protein Pept. Lett. 2021, 28, 909–928. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, V.; Gupta, S.; Gahlaut, V.; Muthamilarasan, M.; Bandyopadhyay, T.; Ramchiary, N.; Prasad, M. Genome-Wide Association Study of Major Agronomic Traits in Foxtail Millet (Setaria italica L.) Using ddRAD Sequencing. Sci. Rep. 2019, 9, 5020. [Google Scholar] [CrossRef] [Green Version]
- Jia, G.; Huang, X.; Zhi, H.; Zhao, Y.; Zhao, Q.; Li, W.; Chai, Y.; Yang, L.; Liu, K.; Lu, H.; et al. A haplotype map of genomic variations and genome-wide association studies of agronomic traits in foxtail millet (Setaria italica). Nat. Genet. 2013, 45, 957–961. [Google Scholar] [CrossRef]
- Zhang, K.; Fan, G.; Zhang, X.; Zhao, F.; Wei, W.; Du, G.; Feng, X.; Wang, X.; Wang, F.; Song, G.; et al. Identification of QTLs for 14 Agronomically Important Traits in Setaria italica Based on SNPs Generated from High-Throughput Sequencing. G3 2017, 7, 1587–1594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; He, J.; Dong, K.; Wang, X.; Wang, W.; Yang, P.; Ren, R.; Zhang, L.; Zhang, Z.; Yang, T. QTL mapping of yield component traits on bin map generated from resequencing a RIL population of foxtail millet (Setaria italica). BMC Genom. 2020, 21, 141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, Y.; Kato, T.; Yamashino, T.; Murakami, M.; Mizuno, T. Characterization of a set of phytochrome-interacting factor-like bHLH proteins in Oryza sativa. Biosci. Biotechnol. Biochem. 2007, 71, 1183–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, S.R.; Khanday, I.; Majhi, B.B.; Veluthambi, K.; Vijayraghavan, U. Auxin-responsive OsMGH3, a common downstream target of OsMADS1 and OsMADS6, controls rice floret fertility. Plant Cell Physiol. 2011, 52, 2123–2135. [Google Scholar] [CrossRef] [Green Version]
- Choi, D.; Kim, J.H.; Kende, H. Whole genome analysis of the OsGRF gene family encoding plant-specific putative transcription activators in rice (Oryza sativa L.). Plant Cell Physiol. 2004, 45, 897–904. [Google Scholar] [CrossRef] [Green Version]
- Kishi-Kaboshi, M.; Okada, K.; Kurimoto, L.; Murakami, S.; Umezawa, T.; Shibuya, N.; Yamane, H.; Miyao, A.; Takatsuji, H.; Takahashi, A.; et al. A rice fungal MAMP-responsive MAPK cascade regulates metabolic flow to antimicrobial metabolite synthesis. Plant J. 2010, 63, 599–612. [Google Scholar] [CrossRef]
- Tamiru, M.; Takagi, H.; Abe, A.; Yokota, T.; Kanzaki, H.; Okamoto, H.; Saitoh, H.; Takahashi, H.; Fujisaki, K.; Oikawa, K.; et al. A chloroplast-localized protein LESION AND LAMINA BENDING affects defence and growth responses in rice. New Phytol. 2016, 210, 1282–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Y.L.; Hong, C.Y.; Chen, K.Y. Functional characterization of the stunt lemma palea 1 mutant allele in rice. Plant Growth Regul. 2014, 73, 257–265. [Google Scholar] [CrossRef]
- Koo, B.H.; Yoo, S.C.; Park, J.W.; Kwon, C.T.; Lee, B.D.; An, G.; Zhang, Z.; Li, J.; Li, Z.; Paek, N.C. Natural variation in OsPRR37 regulates heading date and contributes to rice cultivation at a wide range of latitudes. Mol. Plant 2013, 6, 1877–1888. [Google Scholar] [CrossRef] [Green Version]
- Shomura, A.; Izawa, T.; Ebana, K.; Ebitani, T.; Kanegae, H.; Konishi, S.; Yano, M. Deletion in a gene associated with grain size increased yields during rice domestication. Nat. Genet. 2008, 40, 1023–1028. [Google Scholar] [CrossRef] [PubMed]
- Qian, D.; Chen, G.; Tian, L.; Qu, L.Q. OsDER1 Is an ER-Associated Protein Degradation Factor That Responds to ER Stress. Plant Physiol. 2018, 178, 402–412. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, T.; Eiguchi, M.; Hibara, K.; Ito, J.; Nagato, Y. Rice slender leaf 1 gene encodes cellulose synthase-like D4 and is specifically expressed in M-phase cells to regulate cell proliferation. J. Exp. Bot. 2013, 64, 2049–2061. [Google Scholar] [CrossRef] [Green Version]
- Matsushima, R.; Maekawa, M.; Kusano, M.; Tomita, K.; Kondo, H.; Nishimura, H.; Crofts, N.; Fujita, N.; Sakamoto, W. Amyloplast Membrane Protein SUBSTANDARD STARCH GRAIN6 Controls Starch Grain Size in Rice Endosperm. Plant Physiol. 2016, 170, 1445–1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, Y.; Nishimura, A.; Ito, M.; Ashikari, M.; Hirano, H.Y.; Matsuoka, M. Auxin response factor family in rice. Genes Genet. Syst. 2001, 76, 373–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, M.; Wang, Y.; Wang, Y.; Wang, C.; Ren, Y.; Lv, J.; Peng, C.; Wu, T.; Liu, K.; Zhao, S.; et al. DEFORMED FLORAL ORGAN1 (DFO1) regulates floral organ identity by epigenetically repressing the expression of OsMADS58 in rice (Oryza sativa). New Phytol. 2015, 206, 1476–1490. [Google Scholar] [CrossRef]
- Yang, C.; Hu, H.; Ren, H.; Kong, Y.; Lin, H.; Guo, J.; Wang, L.; He, Y.; Ding, X.; Grabsztunowicz, M.; et al. LIGHT-INDUCED RICE1 Regulates Light-Dependent Attachment of LEAF-TYPE FERREDOXIN-NADP+ OXIDOREDUCTASE to the Thylakoid Membrane in Rice and Arabidopsis. Plant Cell 2016, 28, 712–728. [Google Scholar] [CrossRef]
- Tang, W.; Ye, J.; Yao, X.; Zhao, P.; Xuan, W.; Tian, Y.; Zhang, Y.; Xu, S.; An, H.; Chen, G.; et al. Genome-wide associated study identifies NAC42-activated nitrate transporter conferring high nitrogen use efficiency in rice. Nat. Commun. 2019, 10, 5279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, X.; Shen, Q.; Wang, X.; Hong, Y. The Sulfoquinovosyltransferase-like Enzyme SQD2.2 is Involved in Flavonoid Glycosylation, Regulating Sugar Metabolism and Seed Setting in Rice. Sci. Rep. 2017, 7, 4685. [Google Scholar] [CrossRef]
- Sakuraba, Y.; Piao, W.; Lim, J.H.; Han, S.H.; Kim, Y.S.; An, G.; Paek, N.C. Rice ONAC106 Inhibits Leaf Senescence and Increases Salt Tolerance and Tiller Angle. Plant Cell. Physiol. 2015, 56, 2325–2339. [Google Scholar] [CrossRef]
- Miura, K.; Ikeda, M.; Matsubara, A.; Song, X.J.; Ito, M.; Asano, K.; Matsuoka, M.; Kitano, H.; Ashikari, M. OsSPL14 promotes panicle branching and higher grain productivity in rice. Nat. Genet. 2010, 42, 545–549. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.X.; Xiao, M.Z.; Liu, Y.; Fu, J.L.; He, Y.; Jiang, D.A. The small auxin-up RNA OsSAUR45 affects auxin synthesis and transport in rice. Plant Mol. Biol. 2017, 94, 97–107. [Google Scholar] [CrossRef]
- Luo, J.; Liu, H.; Zhou, T.; Gu, B.; Huang, X.; Shangguan, Y.; Zhu, J.; Li, Y.; Zhao, Y.; Wang, Y.; et al. An-1 encodes a basic helix-loop-helix protein that regulates awn development, grain size, and grain number in rice. Plant Cell 2013, 25, 3360–3376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- French, S.R.; Abu-Zaitoon, Y.; Uddin, M.M.; Bennett, K.; Nonhebel, H.M. Auxin and Cell Wall Invertase Related Signaling during Rice Grain Development. Plants 2014, 3, 95–112. [Google Scholar] [CrossRef] [Green Version]
- Sakurai, J.; Ishikawa, F.; Yamaguchi, T.; Uemura, M.; Maeshima, M. Identification of 33 rice aquaporin genes and analysis of their expression and function. Plant Cell. Physiol. 2005, 46, 1568–1577. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Liu, Y.; Li, Y.; Xu, X.; Xu, C.; Li, X.; Xiao, J.; Zhang, Q. Differential expression of GS5 regulates grain size in rice. J. Exp. Bot. 2015, 66, 2611–2623. [Google Scholar] [CrossRef] [Green Version]
- Han, S.H.; Yoo, S.C.; Lee, B.D.; An, G.; Paek, N.C. Rice FLAVIN-BINDING, KELCH REPEAT, F-BOX 1 (OsFKF1) promotes flowering independent of photoperiod. Plant Cell. Environ. 2015, 38, 2527–2540. [Google Scholar] [CrossRef]
- Lee, D.K.; Kim, H.I.; Jang, G.; Chung, P.J.; Jeong, J.S.; Kim, Y.S.; Bang, S.W.; Jung, H.; Choi, Y.D.; Kim, J.K. The NF-YA transcription factor OsNF-YA7 confers drought stress tolerance of rice in an abscisic acid independent manner. Plant Sci. 2015, 241, 199–210. [Google Scholar] [CrossRef]
- He, L.; Li, M.; Qiu, Z.; Chen, D.; Zhang, G.; Wang, X.; Chen, G.; Hu, J.; Gao, Z.; Dong, G.; et al. Primary leaf-type ferredoxin 1 participates in photosynthetic electron transport and carbon assimilation in rice. Plant J. 2020, 104, 44–58. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Xie, Y.; Zhang, J.; Ren, Y.; Zhang, X.; Wang, J.; Guo, X.; Wu, F.; Sheng, P.; Wang, J.; et al. VLN2 Regulates Plant Architecture by Affecting Microfilament Dynamics and Polar Auxin Transport in Rice. Plant Cell 2015, 27, 2829–2845. [Google Scholar]
- Chen, W.; Chen, L.; Zhang, X.; Yang, N.; Guo, J.; Wang, M.; Ji, S.; Zhao, X.; Yin, P.; Cai, L.; et al. Convergent selection of a WD40 protein that enhances grain yield in maize and rice. Science 2022, 375, eabg7985. [Google Scholar] [CrossRef]
- Hussin, S.H.; Wang, H.; Tang, S.; Zhi, H.; Tang, C.; Zhang, W.; Jia, G.; Diao, X. SiMADS34, an E-class MADS-box transcription factor, regulates inflorescence architecture and grain yield in Setaria italica. Plant Mol. Biol. 2021, 105, 419–434. [Google Scholar] [CrossRef] [PubMed]
- Ilg, A.; Yu, Q.; Schaub, P.; Beyer, P.; Al-Babili, S. Overexpression of the rice carotenoid cleavage dioxygenase 1 gene in Golden Rice endosperm suggests apocarotenoids as substrates in planta. Planta 2010, 232, 691–699. [Google Scholar] [CrossRef]
- He, L.; Cheng, L.; Wang, J.; Liu, J.; Cheng, J.; Yang, Z.; Cao, R.; Han, Y.; Li, H.; Zhang, B. Carotenoid Cleavage Dioxygenase 1 Catalyzes Lutein Degradation to Influence Carotenoid Accumulation and Color Development in Foxtail Millet Grains. J. Agric. Food Chem. 2022, 70, 9283–9294. [Google Scholar] [CrossRef]
- Giri, J.; Bhosale, R.; Huang, G.; Pandey, B.K.; Parker, H.; Zappala, S.; Yang, J.; Dievart, A.; Bureau, C.; Ljung, K.; et al. Rice auxin influx carrier OsAUX1 facilitates root hair elongation in response to low external phosphate. Nat. Commun. 2018, 9, 1408. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.; Shahriari, M.; Xiang, J.; Pasternak, T.; Igolkina, A.; Aminizade, S.; Zhi, H.; Gao, Y.; Roodbarkelari, F.; Sui, Y.; et al. The role of AUX1 during lateral root development in the domestication of the model C4 grass Setaria italica. J. Exp. Bot. 2022, 73, 2021–2034. [Google Scholar] [CrossRef] [PubMed]
- Spielmeyer, W.; Ellis, M.H.; Chandler, P.M. Semidwarf (sd-1), “green revolution” rice, contains a defective gibberellin 20-oxidase gene. Proc. Natl. Acad. Sci. USA 2002, 99, 9043–9048. [Google Scholar] [CrossRef]
- Bates, D.; Maechler, M.; Bolker, B.; Walker, S. lme4: Linear mixedeffects models using Eigen and S4. J. Stat. Softw. 2014, 67, 1–9. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Traits | Environments | Range | Mean | SD | CV | h2 | Genotype | Environment |
|---|---|---|---|---|---|---|---|---|
| Panicle length (PL, cm) | 2015 | 4.50–43.90 | 22.20 | 5.50 | 20% | 0.78 | ** | ** |
| 2016 | 8.80–40.00 | 19.30 | 4.40 | 20% | ||||
| BLUP | 10.72–31.27 | 20.81 | 3.12 | 15% | ||||
| Main panicle diameter (MPD, mm) | 2015 | 9.40–48.24 | 23.31 | 6.36 | 27% | 0.74 | ** | ** |
| 2016 | 10.19–42.82 | 22.63 | 4.69 | 21% | ||||
| BLUP | 14.80–35.47 | 22.95 | 3.03 | 13% | ||||
| Panicle weight per panicle (PWP, g) | 2015 | 0.58–57.69 | 17.86 | 9.69 | 54% | 0.73 | ** | ** |
| 2016 | 1.06–35.74 | 14.71 | 5.89 | 40% | ||||
| BLUP | 7.64–30.45 | 16.26 | 4.32 | 27% | ||||
| Grain weight per panicle (GWP, g) | 2015 | 0.50–48.88 | 15.06 | 8.51 | 57% | 0.74 | ** | ** |
| 2016 | 1.36–36.34 | 12.51 | 5.29 | 42% | ||||
| BLUP | 6.30–26.78 | 13.73 | 3.88 | 28% | ||||
| Bristle length (BL, mm) | 2015 | 0.00–14.37 | 4.23 | 2.04 | 48% | 0.91 | ** | * |
| 2016 | 0.00–10.48 | 3.86 | 1.76 | 46% | ||||
| BLUP | 0.35–10.41 | 4.03 | 1.60 | 40% | ||||
| Thousand-grain weight (TGW, g) | 2015 | 0.72–3.62 | 2.58 | 0.48 | 19% | 0.81 | ** | ** |
| 2016 | 1.08–4.36 | 2.76 | 0.44 | 16% | ||||
| BLUP | 1.55–3.31 | 2.66 | 0.31 | 11% |
| Traits | Environments | Number of Significant SNPs | Range of R2 (%) | Average of R2 (%) |
|---|---|---|---|---|
| Panicle length | 2015 | 82 | 0.04–12.81 | 4.89 |
| 2016 | 111 | 0.25–8.04 | 2.62 | |
| BLUP | 371 | 4.06–6.63 | 4.90 | |
| Main panicle diameter | 2015 | 13 | 0.04–20.21 | 4.37 |
| 2016 | 41 | 0.06–7.89 | 3.45 | |
| BLUP | 55 | 4.03–6.67 | 4.72 | |
| Panicle weight per panicle | 2015 | 231 | 0.44–25.85 | 5.69 |
| 2016 | 28 | 0.01–12.37 | 6.92 | |
| BLUP | 36 | 4.06–5.95 | 4.63 | |
| Grain weight per panicle | 2015 | 270 | 0.24–25.13 | 5.76 |
| 2016 | 48 | 0.89–12.41 | 6.02 | |
| BLUP | 37 | 4.09–6.27 | 4.62 | |
| Bristle length | 2015 | 36 | 0.03–26.78 | 16.01 |
| 2016 | 24 | 2.33–23.30 | 11.91 | |
| BLUP | 320 | 3.99–12.44 | 4.91 | |
| Thousand-grain weight | 2015 | 154 | 0.01–11.53 | 1.70 |
| 2016 | 65 | 0.03–10.08 | 3.13 | |
| BLUP | 334 | 3.89–7.35 | 4.61 | |
| Total | - | 1830 | 0.01–26.78 | 5.60 |
| No. | QTL | Chr | QTL Region (Mb) | No. of QTN | QTN | Pos | R2 (%) | Trait Environment |
|---|---|---|---|---|---|---|---|---|
| 1 | PL_q2-1 | 2 | 26.405–26.442 | 8 | 2:26442425 | 26442425 | 9.30 | PL.15, PL.blup |
| 2 | PL_q2-2 | 2 | 37.921–37.974 | 112 | 2:37940934 | 37940934 | 6.63 | PL.16, PL.blup |
| 3 | PL_q2-3 | 2 | 38.066–38.179 | 37 | 2:38117003 | 38117003 | 4.70 | PL.16, PL.blup |
| 4 | PL_q3-1 | 3 | 14.919–15.034 | 31 | 3:14994886 | 14994886 | 8.21 | PL.15, PL.blup |
| 5 | PL_q3-4 | 3 | 50.531–50.635 | 9 | 3:50544489 | 50544489 | 4.80 | PL.15, PL.blup |
| 6 | PL_q6-1 | 6 | 31.569–31.578 | 4 | 6:31578055 | 31578055 | 5.65 | PL.16, PL.blup |
| 7 | PL_q7-1 | 7 | 13.851–13.948 | 26 | 7:13860822 | 13860822 | 12.81 | PL.15, PL.blup |
| 8 | PL_q7-2 | 7 | 14.113–14.169 | 6 | 7:14120111 | 14120111 | 10.85 | PL.15, PL.16, PL.blup |
| 9 | PL_q9-1 | 9 | 45.700–45.712 | 26 | 9:45712081 | 45712081 | 5.62 | PL.15, PL.blup |
| 10 | PWP_q1-2 | 1 | 37.334–37.415 | 11 | 1:37334234 | 37334234 | 25.28 | PWP.15, PWP.blup |
| 11 | PWP_q2-1 | 2 | 26.393–26.414 | 4 | 2:26392717 | 26392717 | 4.55 | PWP.16, PWP.blup |
| 12 | PWP_q7-1 | 7 | 13.928–13.937 | 4 | 7:13927683 | 13927683 | 10.78 | PWP.15, PWP.blup |
| 13 | GWP_q1-2 | 1 | 37.334–37.415 | 12 | 1:37334234 | 37334234 | 24.66 | GWP.15, GWP.blup |
| 14 | GWP_q2-4 | 2 | 26.392–26.414 | 4 | 2:26392093 | 26392093 | 4.96 | GWP.16, GWP.blup |
| 15 | GWP_q7-1 | 7 | 13.853–13.958 | 8 | 7:13927683 | 13927683 | 10.60 | GWP.15, GWP.16, GWP.blup |
| 16 | BL_q1-4 | 1 | 30.818–30.944 | 16 | 1:30890043 | 30890043 | 12.67 | BL.15, BL.16, BL.blup |
| 17 | BL_q1-5 | 1 | 31.336–31.388 | 19 | 1:31384418 | 31384418 | 13.65 | BL.15, BL.blup |
| 18 | BL_q1-6 | 1 | 37.312–37.386 | 190 | 1:37322139 | 37322139 | 23.46 | BL.15, BL.16, BL.blup |
| 19 | BL_q1-7 | 1 | 37.626–37.728 | 39 | 1:37724771 | 37724771 | 24.65 | BL.15, BL.16, BL.blup |
| 20 | BL_q1-8 | 1 | 37.730–37.818 | 62 | 1:37751602 | 37751602 | 24.16 | BL.15, BL.16, BL.blup |
| 21 | BL_q1-9 | 1 | 37.854–37.942 | 15 | 1:37855555 | 37855555 | 12.44 | BL.15, BL.16, BL.blup |
| 22 | BL_q1-10 | 1 | 38.004–38.100 | 4 | 1:38099726 | 38099726 | 23.57 | BL.15, BL.blup |
| 23 | BL_q1-11 | 1 | 38.240–38.293 | 20 | 1:38251263 | 38251263 | 21.12 | BL.15, BL.16, BL.blup |
| 24 | BL_q4-1 | 4 | 0.833–0.893 | 6 | 4:893187 | 893187 | 7.54 | BL.16, BL.blup |
| 25 | BL_q8-1 | 8 | 1.635–1.668 | 18 | 8:1635250 | 1635250 | 8.93 | BL.16, BL.blup |
| 26 | BL_q9-1 | 9 | 42.909–42.944 | 20 | 9:42943571 | 42943571 | 14.14 | BL.15, BL.blup |
| 27 | TGW_q5-1 | 5 | 0.546–0.549 | 4 | 5:545898 | 545898 | 8.32 | TGW.16, TGW.blup |
| QTL Regions | Chr | QTL Region (Mb) | Previous Reported QTL | Position of Previous QTL (Mb) | Ref. |
|---|---|---|---|---|---|
| BL_q1-5 | 1 | 31.336–31.388 | qMPL1.1 (PL), qMPD1.1 (PD), qPWP1.1 (PWP), qGWP1.1 (GWP), qpl1 (PL), qpd1 (PD), C1.31690904 (GY) | 31.728–31.929 | [15,23,25] |
| PWP_q1-1, GWP_q1-1 | 1 | 34.970–34.987 | unnamed (BL) | 35.883–35.869 | [24] |
| PL_q2-2, PL_2-3 | 2 | 37.921–38.179 | qMPL2.1 (PL) | 39.726 | [15] |
| PL_q2-4 | 2 | 49.117–49.200 | unnamed (GY) | 49.065 | [24] |
| TGW_q3-1 | 3 | 6.258–6.342 | unnamed (PWP) | 6.768 | [24] |
| PWP_q3-1, GWP_q3-1 | 3 | 7.353–7.409 | qGWP3.1 (GWP), unnamed (TGW) | 7.412–7.806 | [24,26] |
| PL_q3-1 | 3 | 14.919–15.034 | unnamed (PL) | 14.391 | [23] |
| MPD_q3-1, TGW_q3-2 | 3 | 23.616–23.733 | unnamed (GY) | 24.672 | [24] |
| PL_q3-2 | 3 | 39.875–39.995 | qPWP3.1 (PWP), qGWP3.2 (GWP) | 39.037 | [26] |
| TGW_q3-3 | 3 | 43.160–43.182 | unnamed (PL), unnamed (TGW) | 43.132–43.267 | [23,24] |
| MPD_q3-2 | 3 | 47.020–47.023 | qPWP3.3 (PWP) | 46.338–46.477 | [26] |
| PL_q3-4 | 3 | 50.531–50.635 | unnamed (GY) | 50.073 | [23] |
| BL_q4-1 | 4 | 0.833–0.893 | unnamed (BL) | 0.871 | [24] |
| MPD_q4-1 | 4 | 34.740–34.746 | qtgw4 (TGW) | 34.306 | [25] |
| MPD_q5-2 | 5 | 45.138–45.166 | qMPD5.2 (MPD), qTGW5.1 (TGW), qpw5 (PWP), qpd5(PD) | 44.125–44.511 | [15,25] |
| PWP_q6-1, GWP_q6-1, PWP_q6-2, GWP_q6-2 | 6 | 2.535–2.794 | qPWP6.2 (PWP), unnamed (GY) | 2.631–3.434 | [23,26] |
| PWP_q6-7, GWP_q6-7, GWP_q6-9, PWP_q6-8, GWP_q6-9 | 6 | 27.924–28.247 | qPNL6 (PL) | 28.589 | [16] |
| TGW_q6-1 | 6 | 34.670–34.733 | qMPD6.1 (PD) | 34.199 | [15] |
| PL_q7-1, TGW_q7-1, PWP_q7-1, GWP_q7-1 | 7 | 13.851–13.961 | qGWP7.1 (GWP), qPWP7.2 (PWP) | 13.405–13.643 | [26] |
| PL_q7-2, TGW_q7-2, TGW_q7-3 | 7 | 14.113–14.893 | unnamed (GY) | 14.094 | [23] |
| TGW_q7-8 | 7 | 18.967–19.040 | unnamed (GY) | 19.465 | [23] |
| GWP_q8-1 | 8 | 2.181–2.191 | qGWP8.1 (GWP), qTGW8.1 (TGW) | 2.223–2.605 | [26] |
| TGW_q8-1 | 8 | 28.829–28.851 | qPD8.1 (PD) | 29.501 | [16] |
| Gene ID | Chr | Position | QTL Region | Homologs in Rice | The Function of Orthologue in Rice |
|---|---|---|---|---|---|
| Si1g24310 | 1 | 31.649-31.652 | BL_q1-5 | OsPRR1 (LOC_Os02g40510) [27] | Internode length, spike length, tiller number, grain size, and number of primary branches |
| Si1g27910 | 1 | 34.867-34.874 | GWP_q1-1 PWP_q1-1 | OsMADS6 (LOC_Os02g45770) [28] | Protein content, sterility or low fertility, gelatinization temperature, amylose content, seed setting rate, grain width, grain length, floral organ development characteristics, and total starch content |
| Si1g31910 | 1 | 37.921-37.926 | BL_q1-10 | OsGRF4; GS2; GL2; PT2; LGS1; GLW2(LOC_Os02g47280) [29] | Grain shape, ear length, grain size, grain yield, seed drop, cold tolerance, grain width, grain length, plant dry weight, 1000-grain weight, plant cell size, plant fresh weight, and nitrogen use efficiency |
| Si1g34880 | 1 | 40.251-40.252 | PL_q1-2 | OsMKK4; SMG1 (LOC_Os02g54600) [30] | Plant height, grain size, panicle type, filled grains per panicle, grain width, grain length, lignin content, cytokinin content, diterpenoid phytoalexin content, and 1000-grain weight |
| Si2g10220 | 2 | 9.256-9.262 | GWP_q2-3 | LLB; pls3; OsMTS1 (LOC_Os07g14350) [31] | Rice blast resistance, bacterial blight resistance, leaf color, chlorophyll content, grain yield, leaf inclination, leaf senescence, lesion-like lesion, hydrogen peroxide, jasmonic acid, and aboveground biomass |
| Si2g27120 | 2 | 36.896-36.902 | BL_q2-1 | qGW8; OsSPL16 (LOC_Os08g41940) [32] | Grain shape, cooking quality, grain size, mitotic cycle, and 1000-grain weight |
| Si2g43940 | 2 | 49.297-49.308 | PL_q2-4 | DTH7; Ghd7.1; OsPRR37 (LOC_Os07g49460) [33] | Plant height, heading date, photoperiod sensitivity, growth period, and grain number per panicle |
| Si3g11190 | 3 | 7.375-7.379 | PWP_q3-1 GWP_q3-1 | GSE5; GW5 (LOC_Os05g09520) [34] | Grain shape, grain size, grain width, grain length, and 1000-grain weight |
| Si3g11200 | 3 | 7.390-7.394 | PWP_q3-1 GWP_q3-1 | OsDER1 (LOC_Os05g09550) [35] | Seed development characteristics, grain width, grain length, and 1000-grain weight |
| Si3g33230 | 3 | 43.050-43.055 | TGW_q3-3 | OsCSLD4; NRL1; ND1; OsCD1 (LOC_Os12g36890) [36] | Plant height, leaf width, ear length, leaf inclination, leaf roll, grain number per ear, and sugar content |
| Si4g02810 | 4 | 1.359-1.363 | TGW_q4-1 | OsACS6; SSG6 (LOC_Os06g03990) [37] | Grain size, grain width, grain length, starch granule shape, 1000-grain weight, chloroplast development, and starch granule size |
| Si4g23080 | 4 | 34.377-34.386 | MPD_q4-1 | OsARF19; OsARF7a (LOC_Os06g48950) [38] | Internode length, leaf width, leaf length, grain yield, leaf inclination, floral organ development characteristics, auxin content, 1000-grain weight, and plant cell size |
| Si5g01910 | 5 | 0.793-0.800 | TGW_q5-1 | DFO1; CCP1 (LOC_Os01g12890) [39] | Plant height, male sterility, seed morphology and anatomy, ear length, tiller number, lemma morphology and anatomy, seed setting rate, and primary branch number |
| Si5g07490 | 5 | 5.957-5.958 | PL_q5-1 | OsLIR1 (LOC_Os01g01340) [40] | Vegetative growth potential, seed setting rate, photosynthetic capacity, and spikelet fertility |
| Si5g07510 | 5 | 5.969-5.971 | PL_q5-1 | OsNPF6.1 (LOC_Os01g01360) [41] | Plant height, panicle number, nitrogen content, nitrate transport, yield per plant, and nitrogen use efficiency |
| Si5g10260 | 5 | 8.652-8.657 | PWP_q5-1 | OsSQD2.2 (LOC_Os01g04920) [42] | Tiller number, seed setting rate, total starch content, and total soluble sugar content |
| Si6g16030 | 6 | 27.100-27.101 | PWP_q6-3 GWP_q6-3 | ONAC106 (LOC_Os08g33670) [43] | Leaf color, fertility, ear length, grain yield, salt tolerance, tiller angle, ear number, grain number per ear, oriented gravity, and 1000-grain weight |
| Si6g20480 | 6 | 31.677-31.680 | PL_q6-1 | OsSPL14; IPA1; WFP (LOC_Os08g39890) [44] | Rice blast resistance, bacterial blight resistance, plant height, ear length, tiller number, grain yield, ear branches, grain number per ear, seed setting rate, lodging rate, stem diameter, and 1000-grain weight |
| Si6g23990 | 6 | 34.326-34.327 | TGW_q6-1 | OsSAUR45 (LOC_Os09g37400) [45] | Adventitious root number, plant height, root length, leaf width, and seed setting rate |
| Si7g07780 | 7 | 15.603-15.606 | TGW_q7-7 | An-1 (LOC_Os04g28280) [46] | Awn length, grain number per ear, and grain length |
| Si7g10650 | 7 | 19.276-19.279 | TGW_q7-8 | GIF1; OsCIN2 (LOC_Os04g33740) [47] | Viscous endosperm, grain size, grain yield, amylose content, amylopectin content, and invertase activity |
| Si7g20960 | 7 | 27.099-27.101 | MPD_q7-1 | OsPIP1.1; OsPIP1a (LOC_Os02g44630) [48] | Grain yield, salt tolerance, germination rate, and germination rate |
| Si7g27560 | 7 | 32.242-32.246 | PL_q7-4 | GS5 (LOC_Os05g06660) [49] | Grain size, characteristics of seed development, grain yield, grain width, and 1000-grain weight |
| Si8g15190 | 8 | 28.271-28.275 | TGW_q8-1 | OsFKF1 (LOC_Os11g34460) [50] | Fertility, heading date, number of panicles per plant, number of grains per panicle, and 1000-grain weight |
| Si9g12860 | 9 | 8.332-8.335 | PWP_q9-1 | OsNF-YA4; OsHAP2D (LOC_Os03g48970) [51] | Number of ears, number of ears, number of grains per ear, and grain weight |
| Si9g13550 | 9 | 8.916-8.920 | GWP_q9-1 | OsFdC2 (LOC_Os03g48040) [52] | Plant height, leaf color, heading date, ear number per plant, grain number per ear, seed setting rate, photosynthetic capacity, carotenoid content, chlorophyll a content, chlorophyll b content, chlorophyll a/b ratio, and chloroplast development |
| Si9g39120 | 9 | 45.984-45.994 | PL_q9-1 | VLN2 (LOC_Os03g24220) [53] | Morphological and anatomical characters of root, plant height, leaf angle, panicle type, seed setting rate, grain width, grain thickness, and 1000-grain weight |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Yang, Y.; Hou, S.; Men, Y.; Han, Y. The Integration of Genome-Wide Association Study and Homology Analysis to Explore the Genomic Regions and Candidate Genes for Panicle-Related Traits in Foxtail Millet. Int. J. Mol. Sci. 2022, 23, 14735. https://doi.org/10.3390/ijms232314735
Liu X, Yang Y, Hou S, Men Y, Han Y. The Integration of Genome-Wide Association Study and Homology Analysis to Explore the Genomic Regions and Candidate Genes for Panicle-Related Traits in Foxtail Millet. International Journal of Molecular Sciences. 2022; 23(23):14735. https://doi.org/10.3390/ijms232314735
Chicago/Turabian StyleLiu, Xiaodong, Yang Yang, Siyou Hou, Yihan Men, and Yuanhuai Han. 2022. "The Integration of Genome-Wide Association Study and Homology Analysis to Explore the Genomic Regions and Candidate Genes for Panicle-Related Traits in Foxtail Millet" International Journal of Molecular Sciences 23, no. 23: 14735. https://doi.org/10.3390/ijms232314735
APA StyleLiu, X., Yang, Y., Hou, S., Men, Y., & Han, Y. (2022). The Integration of Genome-Wide Association Study and Homology Analysis to Explore the Genomic Regions and Candidate Genes for Panicle-Related Traits in Foxtail Millet. International Journal of Molecular Sciences, 23(23), 14735. https://doi.org/10.3390/ijms232314735