
DNA Damage Response in Cancer Therapy and Resistance: Challenges and Opportunities


 ,
,  and
and
Abstract
1. Introduction
2. The DNA Damage Response
2.1. Single-Strand Break Repair
2.2. Double-Strand Breaks Repair
2.3. Epigenetic Control in DNA Damage Response
3. DNA Damage Response Inhibitors in Cancer Therapy
3.1. DNA Damage Response Inhibitors in Single-Agent Approaches
3.2. DNA Damage Response Inhibitors in Combinatory Therapies
4. Biomarkers of DNA Damage Response Inhibitors
5. DNA Damage Response as a Mechanism of Cancer Therapy Resistance
6. DNA Damage Response Inhibitors to Overcome Therapy Resistance
6.1. Inhibition of PARP
6.2. Inhibition of ATM–ATR Complexes and Downstream Effectors
6.3. Inhibition of WEE1
6.4. Inhibition of DNA-Dependent Protein Kinase to Re-Sensitize Cells
7. Mechanisms of Resistance to DNA Damage Response Inhibitors
7.1. Resistance to PARP Inhibitors
7.2. Cell Cycle Regulators in DNA Damage Response Inhibitors in Resistant Cells
7.3. Activation of Alternative DNA Repair Pathways
8. Liabilities upon Treatment with DDR Inhibitors
9. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Hoeijmakers, J.H.J. DNA Damage, Aging, and Cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef]
- Ishiai, M. Regulation of the Fanconi Anemia DNA Repair Pathway by Phosphorylation and Monoubiquitination. Genes 2021, 12, 1763. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Barnieh, F.M.; Loadman, P.M.; Falconer, R.A. Progress towards a clinically-successful ATR inhibitor for cancer therapy. Curr. Res. Pharmacol. Drug Discov. 2021, 2, 100017. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Giglia-Mari, G.; Zotter, A.; Vermeulen, W. DNA damage response. Cold Spring Harb. Perspect. Biol. 2011, 3, a000745. [Google Scholar] [CrossRef]
- Tian, H.; Gao, Z.; Li, H.; Zhang, B.; Wang, G.; Zhang, Q.; Pei, D.; Zheng, J. DNA damage response—A double-edged sword in cancer prevention and cancer therapy. Cancer Lett. 2015, 358, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Pilié, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2018, 16, 81–104. [Google Scholar] [CrossRef] [PubMed]
- Baretti, M.; Le, D.T. DNA mismatch repair in cancer. Pharmacol. Ther. 2018, 189, 45–62. [Google Scholar] [CrossRef] [PubMed]
- Lau, P.J.; Kolodner, R.D. Transfer of the MSH2·MSH6 Complex from Proliferating Cell Nuclear Antigen to Mispaired Bases in DNA. J. Biol. Chem. 2003, 278, 14–17. [Google Scholar] [CrossRef] [PubMed]
- Genschel, J.; Modrich, P. Mechanism of 5′-Directed Excision in Human Mismatch Repair. Mol. Cell 2003, 12, 1077–1086. [Google Scholar] [CrossRef]
- Longley, M.J.; Pierce, A.J.; Modrich, P. DNA Polymerase δ Is Required for Human Mismatch Repair in Vitro. J. Biol. Chem. 1997, 272, 10917–10921. [Google Scholar] [CrossRef]
- Zhang, Y.; Yuan, F.; Presnell, S.R.; Tian, K.; Gao, Y.; Tomkinson, A.E.; Gu, L.; Li, G.-M. Reconstitution of 5′-Directed Human Mismatch Repair in a Purified System. Cell 2005, 122, 693–705. [Google Scholar] [CrossRef]
- Schärer, O.D. Nucleotide Excision Repair in Eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012609. [Google Scholar] [CrossRef]
- Feher, K.M.; Kolbanovskiy, A.; Durandin, A.; Shim, Y.; Min, J.-H.; Lee, Y.C.; Shafirovich, V.; Mu, H.; Broyde, S.; Geacintov, N.E. The DNA damage-sensing NER repair factor XPC-RAD23B does not recognize bulky DNA lesions with a missing nucleotide opposite the lesion. DNA Repair 2020, 96, 102985. [Google Scholar] [CrossRef]
- Tsutakawa, S.E.; Tsai, C.-L.; Yan, C.; Bralić, A.; Chazin, W.J.; Hamdan, S.M.; Schärer, O.D.; Ivanov, I.; Tainer, J.A. Envisioning how the prototypic molecular machine TFIIH functions in transcription initiation and DNA repair. DNA Repair 2020, 96, 102972. [Google Scholar] [CrossRef]
- Coin, F.; Oksenych, V.; Egly, J.-M. Distinct Roles for the XPB/p52 and XPD/p44 Subcomplexes of TFIIH in Damaged DNA Opening during Nucleotide Excision Repair. Mol. Cell 2007, 26, 245–256. [Google Scholar] [CrossRef]
- Staresincic, L.; Fagbemi, A.F.; Enzlin, J.H.; Gourdin, A.M.; Wijgers, N.; Dunand-Sauthier, I.; Giglia-Mari, G.; Clarkson, S.G.; Vermeulen, W.; Schärer, O.D. Coordination of dual incision and repair synthesis in human nucleotide excision repair. EMBO J. 2009, 28, 1111–1120. [Google Scholar] [CrossRef]
- Shivji, M.K.K.; Podust, V.N.; Huebscher, U.; Wood, R.D. Nucleotide Excision Repair DNA Synthesis by DNA Polymerase epsilon in the Presence of PCNA, RFC, and RPA. Biochemistry 1995, 34, 5011–5017. [Google Scholar] [CrossRef]
- Jacobs, A.L.; Schär, P. DNA glycosylases: In DNA repair and beyond. Chromosoma 2011, 121, 1–20. [Google Scholar] [CrossRef]
- Caldecott, K.W. Mammalian DNA base excision repair: Dancing in the moonlight. DNA Repair 2020, 93, 102921. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Fleming, A.M.; Averill, A.M.; Burrows, C.J.; Wallace, S.S. The NEIL glycosylases remove oxidized guanine lesions from telomeric and promoter quadruplex DNA structures. Nucleic Acids Res. 2015, 43, 4039–4054. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, S.; Ware, A.P.; Jayaram, P.; Gorthi, S.P.; El-Khamisy, S.F.; Satyamoorthy, K. Apurinic/Apyrimidinic Endonuclease 2 (APE2): An ancillary enzyme for contextual base excision repair mechanisms to preserve genome stability. Biochimie 2021, 190, 70–90. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.; Tapryal, N.; Islam, A.; Mitra, S.; Hazra, T. Transcription coupled base excision repair in mammalian cells: So little is known and so much to uncover. DNA Repair 2021, 107, 103204. [Google Scholar] [CrossRef]
- Caldecott, K. Protein ADP-ribosylation and the cellular response to DNA strand breaks. DNA Repair 2014, 19, 108–113. [Google Scholar] [CrossRef]
- Rouleau, M.; Patel, A.; Hendzel, M.J.; Kaufmann, S.H.; Poirier, G.G. PARP inhibition: PARP1 and beyond. Nat. Rev. Cancer 2010, 10, 293–301. [Google Scholar] [CrossRef]
- Hong, S.J.; Dawson, T.M.; Dawson, V.L. Nuclear and mitochondrial conversations in cell death: PARP-1 and AIF signaling. Trends Pharmacol. Sci. 2004, 25, 259–264. [Google Scholar] [CrossRef]
- Soldani, C.; Scovassi, A.I. Poly(ADP-ribose) polymerase-1 cleavage during apoptosis: An update. Apoptosis 2002, 7, 321–328. [Google Scholar] [CrossRef]
- Kaufmann, S.H.; Desnoyers, S.; Ottaviano, Y.; Davidson, N.E.; Poirier, G.G. Specific proteolytic cleavage of poly(ADP-ribose) polymerase: An early marker of chemotherapy-induced apoptosis. Cancer Res. 1993, 53, 3976–3985. [Google Scholar]
- Demin, A.A.; Hirota, K.; Tsuda, M.; Adamowicz, M.; Hailstone, R.; Brazina, J.; Gittens, W.; Kalasova, I.; Shao, Z.; Zha, S.; et al. XRCC1 prevents toxic PARP1 trapping during DNA base excision repair. Mol. Cell 2021, 81, 3018–3030.e5. [Google Scholar] [CrossRef]
- Padella, A.; Di Rorà, A.G.L.; Marconi, G.; Ghetti, M.; Martinelli, G.; Simonetti, G. Targeting PARP proteins in acute leukemia: DNA damage response inhibition and therapeutic strategies. J. Hematol. Oncol. 2022, 15, 10. [Google Scholar] [CrossRef] [PubMed]
- Das, B.B.; Huang, S.Y.; Murai, J.; Rehman, I.; Amé, J.C.; Sengupta, S.; Das, S.K.; Majumdar, P.; Zhang, H.; Biard, D.; et al. PARP1–TDP1 coupling for the repair of topoisomerase I–induced DNA damage. Nucleic Acids Res. 2014, 42, 4435–4449. [Google Scholar] [CrossRef] [PubMed]
- Kieffer, S.R.; Lowndes, N.F. Immediate-Early, Early, and Late Responses to DNA Double Stranded Breaks. Front. Genet. 2022, 13, 793884. [Google Scholar] [CrossRef] [PubMed]
- Caron, M.-C.; Sharma, A.K.; O’Sullivan, J.; Myler, L.R.; Ferreira, M.T.; Rodrigue, A.; Coulombe, Y.; Ethier, C.; Gagné, J.-P.; Langelier, M.-F.; et al. Poly(ADP-ribose) polymerase-1 antagonizes DNA resection at double-strand breaks. Nat. Commun. 2019, 10, 2954. [Google Scholar] [CrossRef]
- Ronato, D.; Mersaoui, S.Y.; Busatto, F.F.; Affar, E.B.; Richard, S.; Masson, J.-Y. Limiting the DNA Double-Strand Break Resectosome for Genome Protection. Trends Biochem. Sci. 2020, 45, 779–793. [Google Scholar] [CrossRef]
- Lieber, M.R. The Mechanism of Double-Strand DNA Break Repair by the Nonhomologous DNA End-Joining Pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [PubMed]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef]
- Branzei, D.; Szakal, B. DNA helicases in homologous recombination repair. Curr. Opin. Genet. Dev. 2021, 71, 27–33. [Google Scholar] [CrossRef]
- Branzei, D.; Szakal, B. DNA damage tolerance by recombination: Molecular pathways and DNA structures. DNA Repair 2016, 44, 68–75. [Google Scholar] [CrossRef]
- Koike, M. Dimerization, Translocation and Localization of Ku70 and Ku80 Proteins. J. Radiat. Res. 2002, 43, 223–236. [Google Scholar] [CrossRef]
- Bowcock, A. Molecular cloning of BRCA1: A gene for early onset familial breast and ovarian cancer. Breast Cancer Res. Treat. 1993, 28, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Panzarino, N.J.; Krais, J.J.; Cong, K.; Peng, M.; Mosqueda, M.; Nayak, S.U.; Bond, S.M.; Calvo, J.A.; Doshi, M.B.; Bere, M.; et al. Replication Gaps Underlie BRCA Deficiency and Therapy Response. Cancer Res. 2021, 81, 1388–1397. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.S. Two decades beyond BRCA1/2: Homologous recombination, hereditary cancer risk and a target for ovarian cancer therapy. Gynecol. Oncol. 2015, 137, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Krejci, L.; Altmannova, V.; Spirek, M.; Zhao, X. Homologous recombination and its regulation. Nucleic Acids Res. 2012, 40, 5795–5818. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, A.; O’Leary, C.; O’Byrne, K.J.; Burgess, J.; Richard, D.J.; Suraweera, A. Epigenetic Mechanisms in DNA Double Strand Break Repair: A Clinical Review. Front. Mol. Biosci. 2021, 8, 685440. [Google Scholar] [CrossRef]
- Sellou, H.; Lebeaupin, T.; Chapuis, C.; Smith, R.; Hegele, A.; Singh, H.R.; Kozlowski, M.; Bultmann, S.; Ladurner, A.G.; Timinszky, G.; et al. The poly(ADP-ribose)-dependent chromatin remodeler Alc1 induces local chromatin relaxation upon DNA damage. Mol. Biol. Cell 2016, 27, 3791–3799. [Google Scholar] [CrossRef]
- Karakaidos, P.; Karagiannis, D.; Rampias, T. Resolving DNA Damage: Epigenetic Regulation of DNA Repair. Molecules 2020, 25, 2496. [Google Scholar] [CrossRef]
- Lahtz, C.; Pfeifer, G.P. Epigenetic changes of DNA repair genes in cancer. J. Mol. Cell Biol. 2011, 3, 51–58. [Google Scholar] [CrossRef]
- Guan, H.; Ji, M.; Hou, P.; Liu, Z.; Wang, C.; Shan, Z.; Teng, W.; Xing, M. Hypermethylation of the DNA mismatch repair gene hMLH1 and Its association with lymph node metastasis and T1799A BRAF mutation in patients with papillary thyroid cancer. Cancer 2008, 113, 247–255. [Google Scholar] [CrossRef]
- Czerninski, R.; Krichevsky, S.; Ashhab, Y.; Gazit, D.; Patel, V.; Ben-Yehuda, D. Promoter hypermethylation of mismatch repair genes, hMLH1 and hMSH2 in oral squamous cell carcinoma. Oral Dis. 2009, 15, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Huang, H.; Mukunyadzi, P.; Suen, J.Y.; Hanna, E.; Fan, C.-Y. Promoter Hypermethylation: An Important Epigenetic Mechanism for hMLH1 Gene Inactivation in Head and Neck Squamous Cell Carcinoma. Otolaryngol. Neck Surg. 2002, 126, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-C.; Lu, Y.-P.; Tseng, R.-C.; Lin, R.-K.; Chang, J.-W.; Chen, J.-T.; Shih, C.-M.; Chen, C.-Y. Inactivation of hMLH1 and hMSH2 by promoter methylation in primary non-small cell lung tumors and matched sputum samples. J. Clin. Investig. 2003, 111, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Seedhouse, C.H.; Das-Gupta, E.P.; Russell, N.H. Methylation of the hMLH1 promoter and its association with microsatellite instability in acute myeloid leukemia. Leukemia 2003, 17, 83–88. [Google Scholar] [CrossRef]
- Fleisher, A.S.; Esteller, M.; Tamura, G.; Rashid, A.; Stine, O.C.; Yin, J.; Zou, T.-T.; Abraham, J.M.; Kong, D.; Nishizuka, S.; et al. Hypermethylation of the hMLH1 gene promoter is associated with microsatellite instability in early human gastric neoplasia. Oncogene 2001, 20, 329–335. [Google Scholar] [CrossRef]
- Gras, E.; Cortes, J.; Diez, O.; Alonso, C.; Matias-Guiu, X.; Baiget, M.; Prat, J. Loss of heterozygosity on chromosome 13q12-q14, BRCA-2 mutations and lack of BRCA-2 promoter hypermethylation in sporadic epithelial ovarian tumors. Cancer 2001, 92, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Long, X. Association of BRCA1 promoter methylation with sporadic breast cancers: Evidence from 40 studies. Sci. Rep. 2015, 5, 17869. [Google Scholar] [CrossRef]
- Yu, J.; Zhu, T.; Wang, Z.; Zhang, H.; Qian, Z.; Xu, H.; Gao, B.; Wang, W.; Gu, L.; Meng, J.; et al. A Novel Set of DNA Methylation Markers in Urine Sediments for Sensitive/Specific Detection of Bladder Cancer. Clin. Cancer Res. 2007, 13, 7296–7304. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-N.; Tseng, R.-C.; Hsu, H.-S.; Chen, J.-Y.; Tzao, C.; Ho, W.L.; Wang, Y.-C. Epigenetic Inactivation of the Chromosomal Stability Control Genes BRCA1, BRCA2, and XRCC5 in Non–Small Cell Lung Cancer. Clin. Cancer Res. 2007, 13, 832–838. [Google Scholar] [CrossRef] [PubMed]
- Bernal, C.; Vargas, M.; Ossandón, F.; Santibáñez, E.; Urrutia, J.; Luengo, V.; Zavala, L.F.; Backhouse, C.; Palma, M.; Argandoña, J.; et al. DNA methylation profile in diffuse type gastric cancer: Evidence for hypermethylation of the BRCA1 promoter region in early-onset gastric carcinogenesis. Biol. Res. 2008, 41, 303–315. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gao, D.; Herman, J.G.; Guo, M. The clinical value of aberrant epigenetic changes of DNA damage repair genes in human cancer. Oncotarget 2016, 7, 37331–37346. [Google Scholar] [CrossRef] [PubMed]
- Plantamura, I.; Cosentino, G.; Cataldo, A. MicroRNAs and DNA-Damaging Drugs in Breast Cancer: Strength in Numbers. Front. Oncol. 2018, 8, 352. [Google Scholar] [CrossRef] [PubMed]
- Petri, B.; Klinge, C.M. Regulation of breast cancer metastasis signaling by miRNAs. Cancer Metastasis Rev. 2020, 39, 837–886. [Google Scholar] [CrossRef] [PubMed]
- Fridrichova, I.; Zmetakova, I. MicroRNAs Contribute to Breast Cancer Invasiveness. Cells 2019, 8, 1361. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Gu, H.; Xiong, X.; Ao, H.; Cao, J.; Lin, W.; Yu, M.; Lin, J.; Cui, Q. MicroRNAs Involved in Carcinogenesis, Prognosis, Therapeutic Resistance, and Applications in Human Triple-Negative Breast Cancer. Cells 2019, 8, 1492. [Google Scholar] [CrossRef] [PubMed]
- Kandettu, A.; Radhakrishnan, R.; Chakrabarty, S.; Sriharikrishnaa, S.; Kabekkodu, S.P. The emerging role of miRNA clusters in breast cancer progression. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188413. [Google Scholar] [CrossRef] [PubMed]
- Kastl, L.; Brown, I.; Schofield, A.C. miRNA-34a is associated with docetaxel resistance in human breast cancer cells. Breast Cancer Res. Treat. 2011, 131, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Bao, C.; Chen, J.; Chen, D.; Lu, Y.; Lou, W.; Ding, B.; Xu, L.; Fan, W. MiR-93 suppresses tumorigenesis and enhances chemosensitivity of breast cancer via dual targeting E2F1 and CCND1. Cell Death Dis. 2020, 11, 618. [Google Scholar] [CrossRef] [PubMed]
- Mei, Z.; Su, T.; Ye, J.; Yang, C.; Zhang, S.; Xie, C. The miR-15 Family Enhances the Radiosensitivity of Breast Cancer Cells by Targeting G2 Checkpoints. Radiat. Res. 2015, 183, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Wei, R.-R.; Huang, G.-L.; Zhang, M.-Y.; Yuan, Y.-F.; Wang, H.-Y. Checkpoint kinase 1 is negatively regulated by miR-497 in hepatocellular carcinoma. Med. Oncol. 2014, 31, 844. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Qu, J.; Xu, F.; Guo, Y.; Wang, Y.; Yu, H.; Qian, B. MiR-195 suppresses non-small cell lung cancer by targeting CHEK1. Oncotarget 2015, 6, 9445–9456. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-Z.; Zhang, J.; Shao, H.-Y.; Chen, J.-P.; Zhao, H.-Y. MicroRNA-191 promotes osteosarcoma cells proliferation by targeting checkpoint kinase 2. Tumor Biol. 2015, 36, 6095–6101. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-M.; Chou, W.-C.; Hu, L.-Y.; Hsiung, C.-N.; Chu, H.-W.; Huang, Y.-L.; Hsu, H.-M.; Yu, J.-C.; Shen, C.-Y. The Effect of MicroRNA-124 Overexpression on Anti-Tumor Drug Sensitivity. PLoS ONE 2015, 10, e0128472. [Google Scholar] [CrossRef] [PubMed]
- Bisso, A.; Faleschini, M.; Zampa, F.; Capaci, V.; De Santa, J.; Santarpia, L.; Piazza, S.; Cappelletti, V.; Daidone, M.G.; Agami, R.; et al. Oncogenic miR-181a/b affect the DNA damage response in aggressive breast cancer. Cell Cycle 2013, 12, 1679–1687. [Google Scholar] [CrossRef] [PubMed]
- Moskwa, P.; Buffa, F.M.; Pan, Y.; Panchakshari, R.; Gottipati, P.; Muschel, R.J.; Beech, J.; Kulshrestha, R.; Abdelmohsen, K.; Weinstock, D.M.; et al. miR-182-Mediated Downregulation of BRCA1 Impacts DNA Repair and Sensitivity to PARP Inhibitors. Mol. Cell 2011, 41, 210–220. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Xiao, X.; Dong, L.; Wan, N.; Zhou, Z.; Deng, H.; Zhang, X. MiR-218 regulates cisplatin chemosensitivity in breast cancer by targeting BRCA1. Tumor Biol. 2014, 36, 2065–2075. [Google Scholar] [CrossRef]
- Huang, J.-W.; Wang, Y.; Dhillon, K.K.; Calses, P.; Villegas, E.; Mitchell, P.S.; Tewari, M.; Kemp, C.J.; Taniguchi, T. Systematic Screen Identifies miRNAs That Target RAD51 and RAD51D to Enhance Chemosensitivity. Mol. Cancer Res. 2013, 11, 1564–1573. [Google Scholar] [CrossRef] [PubMed]
- Espinosa-Diez, C.; Wilson, R.; Chatterjee, N.; Hudson, C.; Ruhl, R.; Hipfinger, C.; Helms, E.; Khan, O.F.; Anderson, D.G.; Anand, S. MicroRNA regulation of the MRN complex impacts DNA damage, cellular senescence, and angiogenic signaling. Cell Death Dis. 2018, 9, 632. [Google Scholar] [CrossRef]
- Chernyy, V.; Pustylnyak, V.; Kozlov, V.; Gulyaeva, L. Increased expression of miR-155 and miR-222 is associated with lymph node positive status. J. Cancer 2018, 9, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Xie, J.; Zhang, M.; Zhao, Z.; Wan, Y.; Yao, Y. miRNA-21 promotes proliferation and invasion of triple-negative breast cancer cells through targeting PTEN. Am. J. Transl. Res. 2017, 9, 953–961. [Google Scholar] [PubMed]
- Piasecka, D.; Braun, M.; Kordek, R.; Sadej, R.; Romanska, H. MicroRNAs in regulation of triple-negative breast cancer progression. J. Cancer Res. Clin. Oncol. 2018, 144, 1401–1411. [Google Scholar] [CrossRef]
- Xiao, Y.; Li, Y.; Tao, H.; Humphries, B.; Li, A.; Jiang, Y.; Yang, C.; Luo, R.; Wang, Z. Integrin α5 down-regulation by miR-205 suppresses triple negative breast cancer stemness and metastasis by inhibiting the Src/Vav2/Rac1 pathway. Cancer Lett. 2018, 433, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Damiano, V.; Brisotto, G.; Borgna, S.; di Gennaro, A.; Armellin, M.; Perin, T.; Guardascione, M.; Maestro, R.; Santarosa, M. Epigenetic silencing of miR-200c in breast cancer is associated with aggressiveness and is modulated by ZEB1. Genes Chromosom. Cancer 2016, 56, 147–158. [Google Scholar] [CrossRef] [PubMed]
- D’Ippolito, E.; Iorio, M.V. MicroRNAs and Triple Negative Breast Cancer. Int. J. Mol. Sci. 2013, 14, 22202–22220. [Google Scholar] [CrossRef] [PubMed]
- Piva, R.; Spandidos, D.A.; Gambari, R. From microRNA functions to microRNA therapeutics: Novel targets and novel drugs in breast cancer research and treatment (Review). Int. J. Oncol. 2013, 43, 985–994. [Google Scholar] [CrossRef]
- Li, Q.; Liu, J.; Jia, Y.; Li, T.; Zhang, M. miR-623 suppresses cell proliferation, migration and invasion through direct inhibition of XRCC5 in breast cancer. Aging 2020, 12, 10246–10258. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-Y.; Fu, S.-L.; Xu, S.-Q.; Zhou, X.; Liu, X.-S.; Xu, Y.-J.; Zhao, J.-P.; Wei, S. By downregulating Ku80, hsa-miR-526b suppresses non-small cell lung cancer. Oncotarget 2014, 6, 1462–1477. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer Genome Landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Huang, D.; Savage, S.R.; Calinawan, A.P.; Lin, C.; Zhang, B.; Wang, P.; Starr, T.K.; Birrer, M.J.; Paulovich, A.G. A highly annotated database of genes associated with platinum resistance in cancer. Oncogene 2021, 40, 6395–6405. [Google Scholar] [CrossRef]
- O’Connor, M.J. Targeting the DNA Damage Response in Cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef]
- Reinhardt, H.C.; Jiang, H.; Hemann, M.T.; Yaffe, M.B. Exploiting synthetic lethal interactions for targeted cancer therapy. Cell Cycle 2009, 8, 3112–3119. [Google Scholar] [CrossRef]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1949–1961. [Google Scholar] [CrossRef] [PubMed]
- Heo, Y.-A.; Duggan, S.T. Niraparib: A Review in Ovarian Cancer. Target. Oncol. 2018, 13, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Hoy, S.M. Talazoparib: First Global Approval. Drugs 2018, 78, 1939–1946. [Google Scholar] [CrossRef] [PubMed]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.-H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef]
- Boussios, S.; Karihtala, P.; Moschetta, M.; Abson, C.; Karathanasi, A.; Zakynthinakis-Kyriakou, N.; Ryan, J.E.; Sheriff, M.; Rassy, E.; Pavlidis, N. Veliparib in ovarian cancer: A new synthetically lethal therapeutic approach. Investig. New Drugs 2019, 38, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Nishio, S.; Takekuma, M.; Takeuchi, S.; Kawano, K.; Tsuda, N.; Tasaki, K.; Takahashi, N.; Abe, M.; Tanaka, A.; Nagasawa, T.; et al. Phase 1 study of veliparib with carboplatin and weekly paclitaxel in Japanese patients with newly diagnosed ovarian cancer. Cancer Sci. 2017, 108, 2213–2220. [Google Scholar] [CrossRef]
- Isakoff, S.J.; Puhalla, S.; Domchek, S.M.; Friedlander, M.; Kaufman, B.; Robson, M.; Telli, M.L.; Diéras, V.; Han, H.S.; Garber, E.J.; et al. A randomized Phase II study of veliparib with temozolomide or carboplatin/paclitaxel versus placebo with carboplatin/paclitaxel in BRCA1/2 metastatic breast cancer: Design and rationale. Futur. Oncol. 2017, 13, 307–320. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Blais, N.; Mazieres, J.; Reck, M.; Jones, C.M.; Juhasz, E.; Urban, L.; Orlov, S.; Barlesi, F.; Kio, E.; et al. Randomized, Placebo-Controlled, Phase II Study of Veliparib in Combination with Carboplatin and Paclitaxel for Advanced/Metastatic Non–Small Cell Lung Cancer. Clin. Cancer Res. 2017, 23, 1937–1944. [Google Scholar] [CrossRef]
- Klinakis, A.; Karagiannis, D.; Rampias, T. Targeting DNA repair in cancer: Current state and novel approaches. Cell Mol. Life Sci. 2020, 77, 677–703. [Google Scholar] [CrossRef]
- Tookman, L.; Krell, J.; Nkolobe, B.; Burley, L.; McNeish, I. Practical guidance for the management of side effects during rucaparib therapy in a multidisciplinary UK setting. Ther. Adv. Med. Oncol. 2020, 12, 1758835920921980. [Google Scholar] [CrossRef]
- Markman, M. Maintenance chemotherapy in the management of epithelial ovarian cancer. Cancer Metastasis Rev. 2015, 34, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Electronic Medicines Compendium (EMC). Lynparza. Datapharm. Available online: https://www.medicines.org.uk/emc/search?q=%22olaparib%22 (accessed on 3 February 2020).
- Electronic Medicines Compendium (EMC). Rubraca. Datapharm. Available online: https://www.medicines.org.uk/emc/search?q=%22Rubraca%22 (accessed on 3 February 2020).
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.-G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef] [PubMed]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Pérol, D.; González-Martín, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Mäenpää, J.; et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef] [PubMed]
- González-Martín, A.; Pothuri, B.; Vergote, I.; DePont Christensen, R.; Graybill, W.; Mirza, M.R.; McCormick, C.; Lorusso, D.; Hoskins, P.; Freyer, G.; et al. Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2391–2402. [Google Scholar] [CrossRef]
- Coleman, R.L.; Fleming, G.F.; Brady, M.F.; Swisher, E.M.; Steffensen, K.D.; Friedlander, M.; Okamoto, A.; Moore, K.N.; Efrat Ben-Baruch, N.; Werner, T.L.; et al. Veliparib with First-Line Chemotherapy and as Maintenance Therapy in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2403–2415. [Google Scholar] [CrossRef]
- Pommier, Y.; O’Connor, M.J.; de Bono, J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci. Transl. Med. 2016, 8, 362ps17. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of Poly(ADP-Ribose) Polymerase in Tumors from BRCA Mutation Carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef]
- Gelmon, K.A.; Tischkowitz, M.; Mackay, H.; Swenerton, K.; Robidoux, A.; Tonkin, K.; Hirte, H.; Huntsman, D.; Clemons, M.; Gilks, B.; et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: A phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011, 12, 852–861. [Google Scholar] [CrossRef]
- Gourley, C.; Balmaña, J.; Ledermann, J.A.; Serra, V.; Dent, R.; Loibl, S.; Pujade-Lauraine, E.; Boulton, S.J. Moving from Poly (ADP-Ribose) Polymerase Inhibition to Targeting DNA Repair and DNA Damage Response in Cancer Therapy. J. Clin. Oncol. 2019, 37, 2257–2269. [Google Scholar] [CrossRef] [PubMed]
- Pearl, L.H.; Schierz, A.C.; Ward, S.E.; Al-Lazikani, B.; Pearl, F.M. Therapeutic opportunities within the DNA damage response. Nat. Rev. Cancer 2015, 15, 166–180. [Google Scholar] [CrossRef] [PubMed]
- London, R.E. The structural basis of XRCC1-mediated DNA repair. DNA Repair 2015, 30, 90–103. [Google Scholar] [CrossRef]
- Ha, K.; Fiskus, W.; Choi, D.S.; Bhaskara, S.; Cerchietti, L.; Devaraj, S.G.T.; Shah, B.; Sharma, S.; Chang, J.C.; Melnick, A.M.; et al. Histone deacetylase inhibitor treatment induces ‘BRCAness’ and synergistic lethality with PARP inhibitor and cisplatin against human triple negative breast cancer cells. Oncotarget 2014, 5, 5637–5650. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Wilson, A.J.; Saskowski, J.; Wass, E.; Khabele, D. Suberoylanilide hydroxamic acid (SAHA) enhances olaparib activity by targeting homologous recombination DNA repair in ovarian cancer. Gynecol. Oncol. 2014, 133, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Marijon, H.; Lee, D.H.; Ding, L.-W.; Sun, H.; Gery, S.; de Gramont, A.; Koeffler, H.P. Co-targeting poly(ADP-ribose) polymerase (PARP) and histone deacetylase (HDAC) in triple-negative breast cancer: Higher synergism in BRCA mutated cells. Biomed. Pharmacother. 2018, 99, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Liu, Y.; Peng, Y.; Peng, Y.; Yu, X.; Gao, Y.; Yuan, B.; Zhu, Q.; Cao, T.; He, L.; et al. PARP inhibitor veliparib and HDAC inhibitor SAHA synergistically co-target the UHRF1/BRCA1 DNA damage repair complex in prostate cancer cells. J. Exp. Clin. Cancer Res. 2018, 37, 153. [Google Scholar] [CrossRef] [PubMed]
- Muvarak, N.E.; Chowdhury, K.; Xia, L.; Robert, C.; Choi, E.Y.; Cai, Y.; Bellani, M.; Zou, Y.; Singh, Z.N.; Duong, V.H.; et al. Enhancing the Cytotoxic Effects of PARP Inhibitors with DNA Demethylating Agents—A Potential Therapy for Cancer. Cancer Cell 2016, 30, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Thurn, K.T.; Thomas, S.; Raha, P.; Qureshi, I.; Munster, P.N. Histone Deacetylase Regulation of ATM-Mediated DNA Damage Signaling. Mol. Cancer Ther. 2013, 12, 2078–2087. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-W.; Zheng, Y.; Wang, J.-Z.; Lu, X.-X.; Wang, Z.; Chen, L.-B.; Guan, X.-X.; Tong, J.-D. Integrated analysis of DNA methylation and mRNA expression profiling reveals candidate genes associated with cisplatin resistance in non-small cell lung cancer. Epigenetics 2014, 9, 896–909. [Google Scholar] [CrossRef] [PubMed]
- Camphausen, K.; Tofilon, P.J. Inhibition of Histone Deacetylation: A Strategy for Tumor Radiosensitization. J. Clin. Oncol. 2007, 25, 4051–4056. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Carr, T.; Dimtchev, A.; Zaer, N.; Dritschilo, A.; Jung, M. Attenuated DNA Damage Repair by Trichostatin A through BRCA1 Suppression. Radiat. Res. 2007, 168, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, Y.; Mei, H.; Yin, Z.; Geng, Y.; Zhang, T.; Wu, G.; Lin, Z. The BET bromodomain inhibitor JQ1 radiosensitizes non-small cell lung cancer cells by upregulating p21. Cancer Lett. 2017, 391, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Jin, X.; Yang, J.; Yang, Y.; He, Y.; Ding, L.; Pan, Y.; Chen, S.; Jiang, J.; Huang, H. Inhibition of EZH2 by chemo- and radiotherapy agents and small molecule inhibitors induces cell death in castration-resistant prostate cancer. Oncotarget 2015, 7, 3440–3452. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Wang, Y.; Tan, L.; Fu, X. The pivotal role of DNA methylation in the radio-sensitivity of tumor radiotherapy. Cancer Med. 2018, 7, 3812–3819. [Google Scholar] [CrossRef] [PubMed]
- Ahn, D.H.; Bekaii-Saab, T. Biliary tract cancer and genomic alterations in homologous recombinant deficiency: Exploiting synthetic lethality with PARP inhibitors. Chin. Clin. Oncol. 2020, 9, 6. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; George, E.; Ragland, R.L.; Rafail, S.; Zhang, R.; Krepler, C.; Morgan, M.A.; Herlyn, M.; Brown, E.J.; Simpkins, F. Targeting the ATR/CHK1 Axis with PARP Inhibition Results in Tumor Regression in BRCA-Mutant Ovarian Cancer Models. Clin. Cancer Res. 2017, 23, 3097–3108. [Google Scholar] [CrossRef]
- Carrassa, L.; Chilà, R.; Lupi, M.; Ricci, F.; Celenza, C.; Mazzoletti, M.; Broggini, M.; Damia, G. Combined inhibition of Chk1 and Wee1: In vitro synergistic effect translates to tumor growth inhibition in vivo. Cell Cycle 2012, 11, 2507–2517. [Google Scholar] [CrossRef] [PubMed]
- Restelli, V.; Vagni, M.; Arribas, A.J.; Bertoni, F.; Damia, G.; Carrassa, L. Inhibition of CHK 1 and WEE 1 as a new therapeutic approach in diffuse large B cell lymphomas with MYC deregulation. Br. J. Haematol. 2016, 181, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Chilà, R.; Basana, A.; Lupi, M.; Guffanti, F.; Gaudio, E.; Rinaldi, A.; Cascione, L.; Restelli, V.; Tarantelli, C.; Bertoni, F.; et al. Combined inhibition of Chk1 and Wee1 as a new therapeutic strategy for mantle cell lymphoma. Oncotarget 2014, 6, 3394–3408. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.D.; Cable, P.L.; Garrus, J.E.; Sullivan, F.X.; Von Carlowitz, I.; Le Huérou, Y.; Wallace, E.; Woessner, R.D.; Gross, S. Chk1 inhibition and Wee1 inhibition combine synergistically to impede cellular proliferation. Cancer Biol. Ther. 2011, 12, 788–796. [Google Scholar] [CrossRef] [PubMed]
- Restelli, V.; Lupi, M.; Chilà, R.; Vagni, M.; Tarantelli, C.; Spriano, F.; Gaudio, E.; Bertoni, F.; Damia, G.; Carrassa, L. DNA Damage Response Inhibitor Combinations Exert Synergistic Antitumor Activity in Aggressive B-Cell Lymphomas. Mol. Cancer Ther. 2019, 18, 1255–1264. [Google Scholar] [CrossRef]
- Sanjiv, K.; Hagenkort, A.; Calderón-Montaño, J.M.; Koolmeister, T.; Reaper, P.M.; Mortusewicz, O.; Jacques, S.A.; Kuiper, R.V.; Schultz, N.; Scobie, M.; et al. Cancer-Specific Synthetic Lethality between ATR and CHK1 Kinase Activities. Cell Rep. 2016, 14, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Ha, D.-H.; Min, A.; Kim, S.; Jang, H.; Kim, S.H.; Kim, H.-J.; Ryu, H.S.; Ku, J.-L.; Lee, K.-H.; Im, S.-A. Antitumor effect of a WEE1 inhibitor and potentiation of olaparib sensitivity by DNA damage response modulation in triple-negative breast cancer. Sci. Rep. 2020, 10, 9930. [Google Scholar] [CrossRef]
- Jin, J.; Fang, H.; Yang, F.; Ji, W.; Guan, N.; Sun, Z.; Shi, Y.; Zhou, G.; Guan, X. Combined Inhibition of ATR and WEE1 as a Novel Therapeutic Strategy in Triple-Negative Breast Cancer. Neoplasia 2018, 20, 478–488. [Google Scholar] [CrossRef] [PubMed]
- Bukhari, A.; Lewis, C.W.; Pearce, J.J.; Luong, D.; Chan, G.K.; Gamper, A.M. Inhibiting Wee1 and ATR kinases produces tumor-selective synthetic lethality and suppresses metastasis. J. Clin. Investig. 2019, 129, 1329–1344. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Xu, H.; George, E.; Hallberg, D.; Kumar, S.; Jagannathan, V.; Medvedev, S.; Kinose, Y.; Devins, K.; Verma, P.; et al. Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models. Nat. Commun. 2020, 11, 3726. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Ilagan, E.; Manning, B.D. Emerging Role of mTOR in the Response to Cancer Therapeutics. Trends Cancer 2016, 2, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Vassetzky, Y.; Dokudovskaya, S. mTORC1 pathway in DNA damage response. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1293–1311. [Google Scholar] [CrossRef]
- Bandhakavi, S.; Kim, Y.-M.; Ro, S.-H.; Xie, H.; Onsongo, G.; Jun, C.-B.; Kim, D.-H.; Griffin, T.J. Quantitative Nuclear Proteomics Identifies mTOR Regulation of DNA Damage Response. Mol. Cell. Proteom. 2010, 9, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Ma, Z.; Vanderwaal, R.P.; Feng, Z.; Gonzalez-Suarez, I.; Wang, S.; Zhang, J.; Roti, J.L.R.; Gonzalo, S.; Zhang, J. The mTOR Inhibitor Rapamycin Suppresses DNA Double-Strand Break Repair. Radiat. Res. 2010, 175, 214–224. [Google Scholar] [CrossRef]
- Pópulo, H.; Lopes, J.M.; Soares, P. The mTOR Signalling Pathway in Human Cancer. Int. J. Mol. Sci. 2012, 13, 1886–1918. [Google Scholar] [CrossRef] [PubMed]
- Walsh, S.; Flanagan, L.; Quinn, C.; Evoy, D.; McDermott, E.; Pierce, A.; Duffy, M. mTOR in breast cancer: Differential expression in triple-negative and non-triple-negative tumors. Breast 2012, 21, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Montero, J.C.; Esparís-Ogando, A.; Re-Louhau, M.F.; Seoane, S.; Abad, M.; Calero, R.; Ocaña, A.; Pandiella, A. Active kinase profiling, genetic and pharmacological data define mTOR as an important common target in triple-negative breast cancer. Oncogene 2012, 33, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Guri, Y.; Hall, M.N. mTOR Signaling Confers Resistance to Targeted Cancer Drugs. Trends Cancer 2016, 2, 688–697. [Google Scholar] [CrossRef]
- Germano, G.; Lamba, S.; Rospo, G.; Barault, L.; Magrì, A.; Maione, F.; Russo, M.; Crisafulli, G.; Bartolini, A.; Lerda, G.; et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature 2017, 552, 116–120. [Google Scholar] [CrossRef]
- Zhao, P.; Li, L.; Jiang, X.; Li, Q. Mismatch repair deficiency/microsatellite instability-high as a predictor for anti-PD-1/PD-L1 immunotherapy efficacy. J. Hematol. Oncol. 2019, 12, 54. [Google Scholar] [CrossRef]
- Wang, B.; Qiao, L.; Wang, Y.; Zeng, J.; Chen, D.; Guo, H.; Zhang, Y. Mitochondrial DNA D-loop lesions with the enhancement of DNA repair contribute to gastrointestinal cancer progression. Oncol. Rep. 2018, 40, 3694–3704. [Google Scholar] [CrossRef]
- Ueta, E.; Sasabe, E.; Yang, Z.; Osaki, T.; Yamamoto, T. Enhancement of apoptotic damage of squamous cell carcinoma cells by inhibition of the mitochondrial DNA repairing system. Cancer Sci. 2008, 99, 2230–2237. [Google Scholar] [CrossRef] [PubMed]
- Shokolenko, I.N.; Alexeyev, M.F.; Robertson, F.M.; LeDoux, S.P.; Wilson, G.L. The expression of Exonuclease III from E. coli in mitochondria of breast cancer cells diminishes mitochondrial DNA repair capacity and cell survival after oxidative stress. DNA Repair 2003, 2, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Chandran, E.A.; Kennedy, I. Significant Tumor Response to the Poly (ADP-ribose) Polymerase Inhibitor Olaparib in Heavily Pretreated Patient with Ovarian Carcinosarcoma Harboring a Germline RAD51D Mutation. JCO Precis. Oncol. 2018, 2, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Gou, R.; Dong, H.; Lin, B. Application and reflection of genomic scar assays in evaluating the efficacy of platinum salts and PARP inhibitors in cancer therapy. Life Sci. 2020, 261, 118434. [Google Scholar] [CrossRef] [PubMed]
- Teyssonneau, D.; Margot, H.; Cabart, M.; Anonnay, M.; Sargos, P.; Vuong, N.-S.; Soubeyran, I.; Sevenet, N.; Roubaud, G. Prostate cancer and PARP inhibitors: Progress and challenges. J. Hematol. Oncol. 2021, 14, 51. [Google Scholar] [CrossRef]
- Cleary, J.M.; Aguirre, A.J.; Shapiro, G.I.; D’Andrea, A.D. Biomarker-Guided Development of DNA Repair Inhibitors. Mol. Cell 2020, 78, 1070–1085. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef]
- Kondrashova, O.; Topp, M.; Nesic, K.; Lieschke, E.; Ho, G.Y.; Harrell, M.I.; Zapparoli, G.V.; Hadley, A.; Holian, R.; Boehm, E.; et al. Methylation of all BRCA1 copies predicts response to the PARP inhibitor rucaparib in ovarian carcinoma. Nat. Commun. 2018, 9, 3970. [Google Scholar] [CrossRef]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M.; et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef]
- Pennington, K.P.; Walsh, T.; Harrell, M.I.; Lee, M.K.; Pennil, C.C.; Rendi, M.H.; Thornton, A.; Norquist, B.M.; Casadei, S.; Nord, A.S.; et al. Germline and Somatic Mutations in Homologous Recombination Genes Predict Platinum Response and Survival in Ovarian, Fallopian Tube, and Peritoneal Carcinomas. Clin. Cancer Res. 2014, 20, 764–775. [Google Scholar] [CrossRef]
- Arora, S.; Balasubramaniam, S.; Zhang, H.; Berman, T.; Narayan, P.; Suzman, D.; Bloomquist, E.; Tang, S.; Gong, Y.; Sridhara, R.; et al. FDA Approval Summary: Olaparib Monotherapy or in Combination with Bevacizumab for the Maintenance Treatment of Patients with Advanced Ovarian Cancer. Oncologist 2020, 26, e164–e172. [Google Scholar] [CrossRef] [PubMed]
- Minchom, A.; Aversa, C.; Lopez, J. Dancing with the DNA damage response: Next-generation anti-cancer therapeutic strategies. Ther. Adv. Med Oncol. 2018, 10, 1758835918786658. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Hampton, O.A.; Reynolds, C.P.; Kang, M.H.; Maris, J.M.; Gorlick, R.; Kolb, E.A.; Lock, R.; Carol, H.; Keir, S.T.; et al. Initial testing (stage 1) of the PARP inhibitor BMN 673 by the pediatric preclinical testing program: PALB2 mutation predicts exceptional in vivo response to BMN 673. Pediatr. Blood Cancer 2014, 62, 91–98. [Google Scholar] [CrossRef]
- Tentori, L.; Ricci-Vitiani, L.; Muzi, A.; Ciccarone, F.; Pelacchi, F.; Calabrese, R.; Runci, D.; Pallini, R.; Caiafa, P.; Graziani, G. Pharmacological inhibition of poly(ADP-ribose) polymerase-1 modulates resistance of human glioblastoma stem cells to temozolomide. BMC Cancer 2014, 14, 151. [Google Scholar] [CrossRef] [PubMed]
- Patch, A.-M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole–genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; Van Allen, E.M.; Wu, Y.-M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.-M.; Montgomery, B.; Taplin, M.-E.; Pritchard, C.C.; Attard, G.; et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 2015, 162, 454. [Google Scholar] [CrossRef]
- Miller, R.; Leary, A.; Scott, C.; Serra, V.; Lord, C.; Bowtell, D.; Chang, D.; Garsed, D.; Jonkers, J.; Ledermann, J.; et al. ESMO recommendations on predictive biomarker testing for homologous recombination deficiency and PARP inhibitor benefit in ovarian cancer. Ann. Oncol. 2020, 31, 1606–1622. [Google Scholar] [CrossRef] [PubMed]
- Abkevich, V.; Timms, K.M.; Hennessy, B.T.; Potter, J.; Carey, M.S.; Meyer, L.A.; Smith-McCune, K.; Broaddus, R.; Lu, K.H.; Chen, J.; et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br. J. Cancer 2012, 107, 1776–1782. [Google Scholar] [CrossRef] [PubMed]
- Watkins, J.A.; Irshad, S.; Grigoriadis, A.; Tutt, A.N.J. Genomic scars as biomarkers of homologous recombination deficiency and drug response in breast and ovarian cancers. Breast Cancer Res. 2014, 16, 211. [Google Scholar] [CrossRef]
- Helleday, T.; Eshtad, S.; Nik-Zainal, S. Mechanisms underlying mutational signatures in human cancers. Nat. Rev. Genet. 2014, 15, 585–598. [Google Scholar] [CrossRef]
- Popova, T.; Manié, E.; Rieunier, G.; Caux-Moncoutier, V.; Tirapo, C.; Dubois, T.; Delattre, O.; Sigal-Zafrani, B.; Bollet, M.; Longy, M.; et al. Ploidy and Large-Scale Genomic Instability Consistently Identify Basal-like Breast Carcinomas with BRCA1/2 Inactivation. Cancer Res. 2012, 72, 5454–5462. [Google Scholar] [CrossRef] [PubMed]
- Birkbak, N.J.; Wang, Z.C.; Kim, J.-Y.; Eklund, A.C.; Li, Q.; Tian, R.; Bowman-Colin, C.; Li, Y.; Greene-Colozzi, A.; Iglehart, J.D.; et al. Telomeric Allelic Imbalance Indicates Defective DNA Repair and Sensitivity to DNA-Damaging Agents. Cancer Discov. 2012, 2, 366–375. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.; Lee, E.S. Therapeutic Targeting of DNA Damage Response in Cancer. Int. J. Mol. Sci. 2022, 23, 1701. [Google Scholar] [CrossRef] [PubMed]
- Polak, P.; Kim, J.; Braunstein, L.Z.; Karlic, R.; Haradhavala, N.J.; Tiao, G.; Rosebrock, D.; Livitz, D.; Kübler, K.; Mouw, K.W.; et al. A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat. Genet. 2017, 49, 1476–1486. [Google Scholar] [CrossRef]
- Toledo, I.L.I.; Murga, M.; Zur, R.; Soria, R.; Rodriguez, A.; Martinez, S.; Oyarzabal, J.; Pastor, J.; Bischoff, J.R.; Fernandez-Capetillo, O. A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat. Struct. Mol. Biol. 2011, 18, 721–727. [Google Scholar] [CrossRef]
- Murga, M.; Campaner, S.; Lopez-Contreras, A.J.; Toledo, L.I.; Soria, R.; Montaña, M.F.; Artista, L.D.; Schleker, T.; Guerra, C.; Garcia, E.O.; et al. Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nat. Struct. Mol. Biol. 2011, 18, 1331–1335. [Google Scholar] [CrossRef] [PubMed]
- Grabocka, E.; Commisso, C.; Bar-Sagi, D. Molecular Pathways: Targeting the Dependence of Mutant RAS Cancers on the DNA Damage Response. Clin. Cancer Res. 2015, 21, 1243–1247. [Google Scholar] [CrossRef]
- Pfister, S.X.; Markkanen, E.; Jiang, Y.; Sarkar, S.; Woodcock, M.; Orlando, G.; Mavrommati, I.; Pai, C.-C.; Zalmas, L.-P.; Drobnitzky, N.; et al. Inhibiting WEE1 Selectively Kills Histone H3K36me3-Deficient Cancers by dNTP Starvation. Cancer Cell 2015, 28, 557–568. [Google Scholar] [CrossRef]
- Pilié, P.; George, A.; Yap, T. Patient selection biomarker strategies for PARP inhibitor therapy. Ann. Oncol. 2020, 31, 1603–1605. [Google Scholar] [CrossRef] [PubMed]
- Paulet, L.; Trecourt, A.; Leary, A.; Peron, J.; Descotes, F.; Devouassoux-Shisheboran, M.; Leroy, K.; You, B.; Lopez, J. Cracking the homologous recombination deficiency code: How to identify responders to PARP inhibitors. Eur. J. Cancer 2022, 166, 87–99. [Google Scholar] [CrossRef]
- Chen, Z.; Sun, T.; Yang, Z.; Zheng, Y.; Yu, R.; Wu, X.; Yan, J.; Shao, Y.W.; Shao, X.; Cao, W.; et al. Monitoring treatment efficacy and resistance in breast cancer patients via circulating tumor DNA genomic profiling. Mol. Genet. Genom. Med. 2019, 8, e1079. [Google Scholar] [CrossRef] [PubMed]
- DiBardino, D.M.; Rawson, D.W.; Saqi, A.; Heymann, J.J.; Pagan, C.A.; Bulman, W.A. Next-generation sequencing of non-small cell lung cancer using a customized, targeted sequencing panel: Emphasis on small biopsy and cytology. CytoJournal 2017, 14, 7. [Google Scholar] [CrossRef]
- Coyne, G.O.; Karlovich, C.; Wilsker, D.; Voth, A.R.; Parchment, E.R.; Chen, A.P.; Doroshow, J.H. PARP Inhibitor Applicability: Detailed Assays for Homologous Recombination Repair Pathway Components. OncoTargets Ther. 2022, 15, 165–180. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.K.; Harrell, M.I.; Oza, A.M.; Oaknin, A.; Ray-Coquard, I.; Tinker, A.V.; Helman, E.; Radke, M.R.; Say, C.; Vo, L.-T.; et al. BRCA Reversion Mutations in Circulating Tumor DNA Predict Primary and Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2019, 9, 210–219. [Google Scholar] [CrossRef]
- Bakhoum, S.F.; Landau, D.A. Chromosomal Instability as a Driver of Tumor Heterogeneity and Evolution. Cold Spring Harb. Perspect. Med. 2017, 7, a029611. [Google Scholar] [CrossRef] [PubMed]
- Jallepalli, P.V.; Lengauer, C. Chromosome segregation and cancer: Cutting through the mystery. Nat. Rev. Cancer 2001, 1, 109–117. [Google Scholar] [CrossRef]
- Lytle, N.K.; Barber, A.G.; Reya, T. Stem cell fate in cancer growth, progression and therapy resistance. Nat. Rev. Cancer 2018, 18, 669–680. [Google Scholar] [CrossRef]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2011, 31, 1869–1883. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-E.; Yin, M.; Dong, Q.; Stewart, D.J.; Merriman, K.W.; Amos, C.I.; Spitz, M.R.; Wei, Q. DNA Repair Capacity in Peripheral Lymphocytes Predicts Survival of Patients with Non–Small-Cell Lung Cancer Treated with First-Line Platinum-Based Chemotherapy. J. Clin. Oncol. 2011, 29, 4121–4128. [Google Scholar] [CrossRef] [PubMed]
- Oliver, T.G.; Mercer, K.L.; Sayles, L.C.; Burke, J.R.; Mendus, D.; Lovejoy, K.S.; Cheng, M.-H.; Subramanian, A.; Mu, D.; Powers, S.; et al. Chronic cisplatin treatment promotes enhanced damage repair and tumor progression in a mouse model of lung cancer. Genes Dev. 2010, 24, 837–852. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Kaye, S.B.; Ashworth, A. Making the best of PARP inhibitors in ovarian cancer. Nat. Rev. Clin. Oncol. 2010, 7, 508–519. [Google Scholar] [CrossRef]
- Friboulet, L.; Barrios-Gonzales, D.; Commo, F.; Olaussen, K.A.; Vagner, S.; Adam, J.; Goubar, A.; Dorvault, N.; Lazar, V.; Job, B.; et al. Molecular Characteristics of ERCC1-Negative versus ERCC1-Positive Tumors in Resected NSCLC. Clin. Cancer Res. 2011, 17, 5562–5572. [Google Scholar] [CrossRef]
- Zhang, J.; Stevens, M.F.; Bradshaw, T.D. Temozolomide: Mechanisms of Action, Repair and Resistance. Curr. Mol. Pharmacol. 2012, 5, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T.; Hamou, M.-F.; De Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT Gene Silencing and Benefit from Temozolomide in Glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Li, L.-Y.; Guan, Y.-D.; Chen, X.-S.; Yang, J.-M.; Cheng, Y. DNA Repair Pathways in Cancer Therapy and Resistance. Front. Pharmacol. 2021, 11, 629266. [Google Scholar] [CrossRef]
- Oldrini, B.; Vaquero-Siguero, N.; Mu, Q.; Kroon, P.; Zhang, Y.; Galán-Ganga, M.; Bao, Z.; Wang, Z.; Liu, H.; Sa, J.K.; et al. MGMT genomic rearrangements contribute to chemotherapy resistance in gliomas. Nat. Commun. 2020, 11, 3883. [Google Scholar] [CrossRef]
- Neboori, H.J.; Haffty, B.G.; Wu, H.; Yang, Q.; Aly, A.; Goyal, S.; Schiff, D.; Moran, M.S.; Golhar, R.; Chen, C.; et al. Low p53 Binding Protein 1 (53BP1) Expression Is Associated with Increased Local Recurrence in Breast Cancer Patients Treated with Breast-Conserving Surgery and Radiotherapy. Int. J. Radiat. Oncol. 2012, 83, e677–e683. [Google Scholar] [CrossRef]
- Ma, J.; Benitez, J.A.; Li, J.; Miki, S.; de Albuquerque, C.P.; Galatro, T.; Orellana, L.; Zanca, C.; Reed, R.; Boyer, A.; et al. Inhibition of Nuclear PTEN Tyrosine Phosphorylation Enhances Glioma Radiation Sensitivity through Attenuated DNA Repair. Cancer Cell 2019, 35, 504–518.e7. [Google Scholar] [CrossRef]
- Naidu, M.D.; Agarwal, R.; Peña, L.A.; Cunha, L.; Mezei, M.; Shen, M.; Wilson, D.M.; Liu, Y.; Sanchez, Z.; Chaudhary, P.; et al. Lucanthone and Its Derivative Hycanthone Inhibit Apurinic Endonuclease-1 (APE1) by Direct Protein Binding. PLoS ONE 2011, 6, e23679. [Google Scholar] [CrossRef]
- Li, Q.; Wei, X.; Zhou, Z.-W.; Wang, S.-N.; Jin, H.; Chen, K.-J.; Luo, J.; Westover, K.; Wang, J.-M.; Wang, D.; et al. GADD45α sensitizes cervical cancer cells to radiotherapy via increasing cytoplasmic APE1 level. Cell Death Dis. 2018, 9, 524. [Google Scholar] [CrossRef]
- Harima, Y.; Sawada, S.; Miyazaki, Y.; Kin, K.; Ishihara, H.; Imamura, M.; Sougawa, M.; Shikata, N.; Ohnishi, T. Expression of Ku80 in Cervical Cancer Correlates with Response to Radiotherapy and Survival. Am. J. Clin. Oncol. 2003, 26, e80–e85. [Google Scholar] [CrossRef]
- Hayashi, J.; Sakata, K.-I.; Someya, M.; Matsumoto, Y.; Satoh, M.; Nakata, K.; Hori, M.; Takagi, M.; Kondoh, A.; Himi, T.; et al. Analysis and results of Ku and XRCC4 expression in hypopharyngeal cancer tissues treated with chemoradiotherapy. Oncol. Lett. 2012, 4, 151–155. [Google Scholar] [CrossRef]
- Beskow, C.; Skikuniene, J.; Holgersson, Å.; Nilsson, B.; Lewensohn, R.; Kanter, L.; Viktorsson, K. Radioresistant cervical cancer shows upregulation of the NHEJ proteins DNA-PKcs, Ku70 and Ku86. Br. J. Cancer 2009, 101, 816–821. [Google Scholar] [CrossRef]
- Liu, C.; Liao, K.; Gross, N.; Wang, Z.; Li, G.; Zuo, W.; Zhong, S.; Zhang, Z.; Zhang, H.; Yang, J.; et al. Homologous recombination enhances radioresistance in hypopharyngeal cancer cell line by targeting DNA damage response. Oral Oncol. 2019, 100, 104469. [Google Scholar] [CrossRef]
- Wang, Z.; Zuo, W.; Zeng, Q.; Li, Y.; Lu, T.; Bu, Y.; Hu, G. The Homologous Recombination Repair Pathway is Associated with Resistance to Radiotherapy in Nasopharyngeal Carcinoma. Int. J. Biol. Sci. 2020, 16, 408–419. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Kettner, N.M.; Vijayaraghavan, S.; Durak, M.G.; Bui, T.; Kohansal, M.; Ha, M.J.; Liu, B.; Rao, X.; Wang, J.; Yi, M.; et al. Combined Inhibition of STAT3 and DNA Repair in Palbociclib-Resistant ER-Positive Breast Cancer. Clin. Cancer Res. 2019, 25, 3996–4013. [Google Scholar] [CrossRef]
- Pulliam, N.; Fang, F.; Ozes, A.R.; Tang, J.; Adewuyi, A.; Keer, H.; Lyons, J.; Baylin, S.B.; Matei, D.; Nakshatri, H.; et al. An Effective Epigenetic-PARP Inhibitor Combination Therapy for Breast and Ovarian Cancers Independent of BRCA Mutations. Clin. Cancer Res. 2018, 24, 3163–3175. [Google Scholar] [CrossRef]
- Lheureux, S.; Oaknin, A.; Garg, S.; Bruce, J.P.; Madariaga, A.; Dhani, N.C.; Bowering, V.; White, J.; Accardi, S.; Tan, Q.; et al. EVOLVE: A Multicenter Open-Label Single-Arm Clinical and Translational Phase II Trial of Cediranib Plus Olaparib for Ovarian Cancer after PARP Inhibition Progression. Clin. Cancer Res. 2020, 26, 4206–4215. [Google Scholar] [CrossRef]
- Parmar, K.; Kochupurakkal, B.S.; Lazaro, J.-B.; Wang, Z.C.; Palakurthi, S.; Kirschmeier, P.T.; Yang, C.; Sambel, L.A.; Färkkilä, A.; Reznichenko, E.; et al. The CHK1 Inhibitor Prexasertib Exhibits Monotherapy Activity in High-Grade Serous Ovarian Cancer Models and Sensitizes to PARP Inhibition. Clin. Cancer Res. 2019, 25, 6127–6140. [Google Scholar] [CrossRef]
- Yang, X.-D.; Kong, F.-E.; Qi, L.; Lin, J.-X.; Yan, Q.; Loong, J.H.C.; Xi, S.-Y.; Zhao, Y.; Zhang, Y.; Yuan, Y.-F.; et al. PARP inhibitor Olaparib overcomes Sorafenib resistance through reshaping the pluripotent transcriptome in hepatocellular carcinoma. Mol. Cancer 2021, 20, 20. [Google Scholar] [CrossRef]
- Patel, P.R.; Senyuk, V.; Rodriguez, N.S.; Oh, A.L.; Bonetti, E.; Mahmud, D.; Barosi, G.; Mahmud, N.; Rondelli, D. Synergistic Cytotoxic Effect of Busulfan and the PARP Inhibitor Veliparib in Myeloproliferative Neoplasms. Biol. Blood Marrow Transplant. 2019, 25, 855–860. [Google Scholar] [CrossRef]
- Sun, C.; Fang, Y.; Labrie, M.; Li, X.; Mills, G.B. Systems approach to rational combination therapy: PARP inhibitors. Biochem. Soc. Trans. 2020, 48, 1101–1108. [Google Scholar] [CrossRef]
- Higuchi, F.; Nagashima, H.; Ning, J.; Koerner, M.V.A.; Wakimoto, H.; Cahill, D.P. Restoration of Temozolomide Sensitivity by PARP Inhibitors in Mismatch Repair Deficient Glioblastoma is Independent of Base Excision Repair. Clin. Cancer Res. 2020, 26, 1690–1699. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K.; Smith, E.J.; Mladek, A.C.; Tian, S.; Decker, P.A.; Kizilbash, S.H.; Kitange, G.J.; Sarkaria, J.N. PARP Inhibitors for Sensitization of Alkylation Chemotherapy in Glioblastoma: Impact of Blood-Brain Barrier and Molecular Heterogeneity. Front. Oncol. 2019, 8, 670. [Google Scholar] [CrossRef]
- Matthews, H.K.; Bertoli, C.; de Bruin, R.A.M. Cell cycle control in cancer. Nat. Rev. Mol. Cell Biol. 2021, 23, 74–88. [Google Scholar] [CrossRef]
- Zhang, T.; Shen, Y.; Chen, Y.; Hsieh, J.-T.; Kong, Z. The ATM inhibitor KU55933 sensitizes radioresistant bladder cancer cells with DAB2IP gene defect. Int. J. Radiat. Biol. 2015, 91, 368–378. [Google Scholar] [CrossRef]
- Golding, S.E.; Rosenberg, E.; Valerie, N.; Hussaini, I.; Frigerio, M.; Cockcroft, X.F.; Wei, Y.C.; Hummersone, M.; Rigoreau, L.; Menear, K.A.; et al. Improved ATM kinase inhibitor KU-60019 radiosensitizes glioma cells, compromises insulin, AKT and ERK prosurvival signaling, and inhibits migration and invasion. Mol. Cancer Ther. 2009, 8, 2894–2902. [Google Scholar] [CrossRef]
- Karlin, J.; Allen, J.; Ahmad, S.F.; Hughes, G.; Sheridan, V.; Odedra, R.; Farrington, P.; Cadogan, E.B.; Riches, L.C.; Garcia-Trinidad, A.; et al. Orally Bioavailable and Blood–Brain Barrier-Penetrating ATM Inhibitor (AZ32) Radiosensitizes Intracranial Gliomas in Mice. Mol. Cancer Ther. 2018, 17, 1637–1647. [Google Scholar] [CrossRef]
- Fujisawa, H.; Nakajima, N.I.; Sunada, S.; Lee, Y.; Hirakawa, H.; Yajima, H.; Fujimori, A.; Uesaka, M.; Okayasu, R. VE-821, an ATR inhibitor, causes radiosensitization in human tumor cells irradiated with high LET radiation. Radiat. Oncol. 2015, 10, 175. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.-X.; Zhou, P.-K. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Transduct. Target. Ther. 2020, 5, 60. [Google Scholar] [CrossRef] [PubMed]
- Gorecki, L.; Andrs, M.; Rezacova, M.; Korabecny, J. Discovery of ATR kinase inhibitor berzosertib (VX-970, M6620): Clinical candidate for cancer therapy. Pharmacol. Ther. 2020, 210, 107518. [Google Scholar] [CrossRef] [PubMed]
- Tu, X.; Kahila, M.M.; Zhou, Q.; Yu, J.; Kalari, K.R.; Wang, L.; Harmsen, W.S.; Yuan, J.; Boughey, J.C.; Matthew, P.; et al. ATR Inhibition Is a Promising Radiosensitizing Strategy for Triple-Negative Breast Cancer. Mol. Cancer Ther. 2018, 17, 2462–2472. [Google Scholar] [CrossRef] [PubMed]
- Restelli, V.; Lupi, M.; Vagni, M.; Chilà, R.; Bertoni, F.; Damia, G.; Carrassa, L. Combining Ibrutinib with Chk1 Inhibitors Synergistically Targets Mantle Cell Lymphoma Cell Lines. Target. Oncol. 2018, 13, 235–245. [Google Scholar] [CrossRef]
- Peasland, A.; Wang, L.-Z.; Rowling, E.; Kyle, S.; Chen, T.; Hopkins, A.; Cliby, A.W.; Sarkaria, J.; Beale, G.; Edmondson, R.J.; et al. Identification and evaluation of a potent novel ATR inhibitor, NU6027, in breast and ovarian cancer cell lines. Br. J. Cancer 2011, 105, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Stefanski, C.D.; Keffler, K.; McClintock, S.; Milac, L.; Prosperi, J.R. APC loss affects DNA damage repair causing doxorubicin resistance in breast cancer cells. Neoplasia 2019, 21, 1143–1150. [Google Scholar] [CrossRef] [PubMed]
- Fokas, E.; Prevo, R.; Pollard, J.R.; Reaper, P.M.; Charlton, P.A.; Cornelissen, B.; Vallis, K.A.; Hammond, E.M.; Olcina, M.M.; Gillies McKenna, W.; et al. Targeting ATR in vivo using the novel inhibitor VE-822 results in selective sensitization of pancreatic tumors to radiation. Cell Death Dis. 2012, 3, e441. [Google Scholar] [CrossRef]
- Kwok, M.; Davies, N.; Agathanggelou, A.; Smith, E.; Oldreive, C.; Petermann, E.; Stewart, G.; Brown, J.; Lau, A.; Pratt, G.; et al. ATR inhibition induces synthetic lethality and overcomes chemoresistance in TP53- or ATM-defective chronic lymphocytic leukemia cells. Blood 2016, 127, 582–595. [Google Scholar] [CrossRef]
- Kantidze, O.L.; Velichko, A.K.; Luzhin, A.V.; Petrova, N.V.; Razin, S.V. Synthetically Lethal Interactions of ATM, ATR, and DNA-PKcs. Trends Cancer 2018, 4, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Esposito, F.; Giuffrida, R.; Raciti, G.; Puglisi, C.; Forte, S. Wee1 Kinase: A Potential Target to Overcome Tumor Resistance to Therapy. Int. J. Mol. Sci. 2021, 22, 10689. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Rohe, A.; Platzer, C.; Najjar, A.; Erdmann, F.; Sippl, W. Regulation of G2/M Transition by Inhibition of WEE1 and PKMYT1 Kinases. Molecules 2017, 22, 2045. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, M.; Ando, H.; Watanabe, N.; Kitamura, K.; Ito, K.; Okayama, H.; Miyamoto, T.; Agui, T.; Sasaki, M. Identification and characterization of human Wee1B, a new member of the Wee1 family of Cdk-inhibitory kinases. Genes Cells 2000, 5, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Heald, R.; McLoughlin, M.; McKeon, F. Human wee1 maintains mitotic timing by protecting the nucleus from cytoplasmically activated cdc2 kinase. Cell 1993, 74, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.; Broome, M.; Hunter, T. Regulation of the human WEE1Hu CDK tyrosine 15-kinase during the cell cycle. EMBO J. 1995, 14, 1878–1891. [Google Scholar] [CrossRef]
- Masaki, T.; Shiratori, Y.; Rengifo, W.; Igarashi, K.; Yamagata, M.; Kurokohchi, K.; Uchida, N.; Miyauchi, Y.; Yoshiji, H.; Watanabe, S.; et al. Cyclins and cyclin-dependent kinases: Comparative study of hepatocellular carcinoma versus cirrhosis. Hepatology 2003, 37, 534–543. [Google Scholar] [CrossRef]
- Harris, P.S.; Venkataraman, S.; Alimova, I.; Birks, D.K.; Balakrishnan, I.; Cristiano, B.; Donson, A.M.; Dubuc, A.M.; Taylor, M.D.; Foreman, N.K.; et al. Integrated genomic analysis identifies the mitotic checkpoint kinase WEE1 as a novel therapeutic target in medulloblastoma. Mol. Cancer 2014, 13, 72. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.; Hashizume, R.; Yang, X.; Kolkowitz, I.; Olow, A.; Phillips, J.; Smirnov, I.; Tom, M.W.; Prados, M.D.; James, C.D.; et al. Targeting Wee1 for the treatment of pediatric high-grade gliomas. Neuro-Oncology 2014, 16, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Slipicevic, A.; Holth, A.; Hellesylt, E.; Tropé, C.G.; Davidson, B.; Flørenes, V.A. Wee1 is a novel independent prognostic marker of poor survival in post-chemotherapy ovarian carcinoma effusions. Gynecol. Oncol. 2014, 135, 118–124. [Google Scholar] [CrossRef]
- Magnussen, G.I.; Holm, R.; Emilsen, E.; Rosnes, A.K.R.; Slipicevic, A.; Flørenes, V.A. High Expression of Wee1 Is Associated with Poor Disease-Free Survival in Malignant Melanoma: Potential for Targeted Therapy. PLoS ONE 2012, 7, e38254. [Google Scholar] [CrossRef]
- Ronco, C.; Martin, A.R.; Demange, L.; Benhida, R. ATM, ATR, CHK1, CHK2 and WEE1 inhibitors in cancer and cancer stem cells. MedChemComm 2016, 8, 295–319. [Google Scholar] [CrossRef]
- Du, X.; Li, J.; Luo, X.; Li, R.; Li, F.; Zhang, Y.; Shi, J.; He, J. Structure-activity relationships of Wee1 inhibitors: A review. Eur. J. Med. Chem. 2020, 203, 112524. [Google Scholar] [CrossRef]
- Caretti, V.; Hiddingh, L.; Lagerweij, T.; Schellen, P.; Koken, P.W.; Hulleman, E.; van Vuurden, D.G.; Vandertop, W.P.; Kaspers, G.J.; Noske, D.P.; et al. WEE1 Kinase Inhibition Enhances the Radiation Response of Diffuse Intrinsic Pontine Gliomas. Mol. Cancer Ther. 2013, 12, 141–150. [Google Scholar] [CrossRef][Green Version]
- Sarcar, B.; Kahali, S.; Prabhu, A.H.; Shumway, S.D.; Xu, Y.; Demuth, T.; Chinnaiyan, P. Targeting Radiation-Induced G(2) Checkpoint Activation with the Wee-1 Inhibitor MK-1775 in Glioblastoma Cell Lines. Mol. Cancer Ther. 2011, 10, 2405–2414. [Google Scholar] [CrossRef]
- Karnak, D.; Engelke, C.G.; Parsels, L.A.; Kausar, T.; Wei, D.; Robertson, J.R.; Marsh, K.B.; Davis, M.A.; Zhao, L.; Maybaum, J.; et al. Combined Inhibition of Wee1 and PARP1/2 for Radiosensitization in Pancreatic Cancer. Clin. Cancer Res. 2014, 20, 5085–5096. [Google Scholar] [CrossRef]
- Hirai, H.; Iwasawa, Y.; Okada, M.; Arai, T.; Nishibata, T.; Kobayashi, M.; Kimura, T.; Kaneko, N.; Ohtani, J.; Yamanaka, K.; et al. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol. Cancer Ther. 2009, 8, 2992–3000. [Google Scholar] [CrossRef] [PubMed]
- Bridges, K.A.; Hirai, H.; Buser, C.A.; Brooks, C.; Liu, H.; Buchholz, T.A.; Molkentine, J.M.; Mason, K.A.; Meyn, R.E. MK-1775, a Novel Wee1 Kinase Inhibitor, Radiosensitizes p53-Defective Human Tumor Cells. Clin. Cancer Res. 2011, 17, 5638–5648. [Google Scholar] [CrossRef] [PubMed]
- Leijen, S.; van Geel, R.M.; Sonke, G.S.; de Jong, D.; Rosenberg, E.H.; Marchetti, S.; Pluim, D.; van Werkhoven, E.; Rose, S.; Lee, M.A.; et al. Phase II Study of WEE1 Inhibitor AZD1775 Plus Carboplatin in Patients with TP53-Mutated Ovarian Cancer Refractory or Resistant to First-Line Therapy Within 3 Months. J. Clin. Oncol. 2016, 34, 4354–4361. [Google Scholar] [CrossRef]
- Osman, A.A.; Monroe, M.M.; Ortega Alves, M.V.; Patel, A.A.; Katsonis, P.; Fitzgerald, A.L.; Neskey, D.M.; Frederick, M.J.; Woo, S.H.; Caulin, C.; et al. Wee-1 Kinase Inhibition Overcomes Cisplatin Resistance Associated with High-Risk TP53 Mutations in Head and Neck Cancer through Mitotic Arrest Followed by Senescence. Mol. Cancer Ther. 2015, 14, 608–619. [Google Scholar] [CrossRef] [PubMed]
- Van Linden, A.A.; Baturin, D.; Ford, J.B.; Fosmire, S.P.; Gardner, L.; Korch, C.; Reigan, P.; Porter, C.C. Inhibition of Wee1 Sensitizes Cancer Cells to Antimetabolite Chemotherapeutics in vitro and in vivo, Independent of p53 Functionality. Mol. Cancer Ther. 2013, 12, 2675–2684. [Google Scholar] [CrossRef]
- Jette, N.; Lees-Miller, S.P. The DNA-dependent protein kinase: A multifunctional protein kinase with roles in DNA double strand break repair and mitosis. Prog. Biophys. Mol. Biol. 2015, 117, 194–205. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, H.; Liu, T.; Huang, M.; Butter, P.-P.; Li, C.; Zhang, L.; Kao, G.D.; Gong, Y.; Maity, A.; et al. Temporal DNA-PK activation drives genomic instability and therapy resistance in glioma stem cells. JCI Insight 2018, 3, e98096. [Google Scholar] [CrossRef]
- Alikarami, F.; Safa, M.; Faranoush, M.; Hayat, P.; Kazemi, A. Inhibition of DNA-PK enhances chemosensitivity of B-cell precursor acute lymphoblastic leukemia cells to doxorubicin. Biomed. Pharmacother. 2017, 94, 1077–1093. [Google Scholar] [CrossRef]
- Pospisilova, M.; Seifrtova, M.; Rezacova, M. Small molecule inhibitors of DNA-PK for tumor sensitization to anticancer therapy. J. Physiol. Pharmacol. Off. J. Pol. Physiol. Soc. 2017, 68, 337–344. [Google Scholar]
- Liang, X.-M.; Qin, Q.; Liu, B.-N.; Li, X.-Q.; Zeng, L.-L.; Wang, J.; Kong, L.-P.; Zhong, D.-S.; Sun, L.-L. Targeting DNA-PK overcomes acquired resistance to third-generation EGFR-TKI osimertinib in non-small-cell lung cancer. Acta Pharmacol. Sin. 2021, 42, 648–654. [Google Scholar] [CrossRef]
- Fang, X.; Huang, Z.; Zhai, K.; Huang, Q.; Tao, W.; Kim, L.; Wu, Q.; Almasan, A.; Yu, J.S.; Li, X.; et al. Inhibiting DNA-PK induces glioma stem cell differentiation and sensitizes glioblastoma to radiation in mice. Sci. Transl. Med. 2021, 13. [Google Scholar] [CrossRef]
- Prados-Carvajal, R.; Irving, E.; Lukashchuk, N.; Forment, J.V. Preventing and Overcoming Resistance to PARP Inhibitors: A Focus on the Clinical Landscape. Cancers 2021, 14, 44. [Google Scholar] [CrossRef]
- D’Andrea, A.D. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair 2018, 71, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.K.; Matulonis, U.A. PARP Inhibitor Resistance Mechanisms and Implications for Post-Progression Combination Therapies. Cancers 2020, 12, 2054. [Google Scholar] [CrossRef]
- Rottenberg, S.; Jaspers, J.E.; Kersbergen, A.; Van Der Burg, E.; Nygren, A.O.H.; Zander, S.A.L.; Derksen, P.W.B.; De Bruin, M.; Zevenhoven, J.; Lau, A.; et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc. Natl. Acad. Sci. USA 2008, 105, 17079–17084. [Google Scholar] [CrossRef]
- Pettitt, S.J.; Krastev, D.B.; Brandsma, I.; Dréan, A.; Song, F.; Aleksandrov, R.; Harrell, M.I.; Menon, M.; Brough, R.; Campbell, J.; et al. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat. Commun. 2018, 9, 1849. [Google Scholar] [CrossRef]
- Gogola, E.; Duarte, A.A.; de Ruiter, J.R.; Wiegant, W.W.; Schmid, J.; de Bruijn, R.; James, D.I.; Llobet, S.G.; Vis, D.J.; Annunziato, S.; et al. Selective Loss of PARG Restores PARylation and Counteracts PARP Inhibitor-Mediated Synthetic Lethality. Cancer Cell 2018, 33, 1078–1093.e12. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Ceccaldi, R.; Shapiro, G.I.; D’Andrea, A.D. Homologous Recombination Deficiency: Exploiting the Fundamental Vulnerability of Ovarian Cancer. Cancer Discov. 2015, 5, 1137–1154. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, A.R.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.-E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 2016, 535, 382–387. [Google Scholar] [CrossRef]
- Rondinelli, B.; Gogola, E.; Yücel, H.; Duarte, A.A.; Van De Ven, M.; Van Der Sluijs, R.; Konstantinopoulos, P.A.; Jonkers, J.; Ceccaldi, R.; Rottenberg, S.; et al. EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat. Cell Biol. 2017, 19, 1371–1378. [Google Scholar] [CrossRef]
- Yazinski, S.A.; Comaills, V.; Buisson, R.; Genois, M.-M.; Nguyen, H.D.; Ho, C.K.; Kwan, T.T.; Morris, R.; Lauffer, S.; Nussenzweig, A.; et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev. 2017, 31, 318–332. [Google Scholar] [CrossRef]
- Edwards, S.L.; Brough, R.; Lord, C.J.; Natrajan, R.; Vatcheva, R.; Levine, D.A.; Boyd, J.; Reis-Filho, J.S.; Ashworth, A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature 2008, 451, 1111–1115. [Google Scholar] [CrossRef] [PubMed]
- Sakai, W.; Swisher, E.M.; Karlan, B.Y.; Agarwal, M.K.; Higgins, J.; Friedman, C.; Villegas, E.; Jacquemont, C.; Farrugia, D.J.; Couch, F.J.; et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 2008, 451, 1116–1120. [Google Scholar] [CrossRef]
- Drost, R.; Bouwman, P.; Rottenberg, S.; Boon, U.; Schut, E.; Klarenbeek, S.; Klijn, C.; van der Heijden, I.; van der Gulden, H.; Wientjens, E.; et al. BRCA1 RING Function Is Essential for Tumor Suppression but Dispensable for Therapy Resistance. Cancer Cell 2011, 20, 797–809. [Google Scholar] [CrossRef]
- Noordermeer, S.M.; Adam, S.; Setiaputra, D.; Barazas, M.; Pettitt, S.J.; Ling, A.K.; Olivieri, M.; Álvarez-Quilón, A.; Moatti, N.; Zimmermann, M.; et al. The shieldin complex mediates 53BP1-dependent DNA repair. Nature 2018, 560, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Dev, H.; Chiang, T.-W.W.; Lescale, C.; De Krijger, I.; Martin, A.G.; Pilger, D.; Coates, J.; Sczaniecka-Clift, M.; Wei, W.; Ostermaier, M.; et al. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat. Cell Biol. 2018, 20, 954–965. [Google Scholar] [CrossRef]
- Clairmont, C.S.; Sarangi, P.; Ponnienselvan, K.; Galli, L.D.; Csete, I.; Moreau, L.; Adelmant, G.; Chowdhury, D.; Marto, J.A.; D’Andrea, A.D. TRIP13 regulates DNA repair pathway choice through REV7 conformational change. Nat. Cell Biol. 2020, 22, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.S.J.; Tan, D.S.P.; Lim, J.S.J.; Tan, D.S.P. Understanding Resistance Mechanisms and Expanding the Therapeutic Utility of PARP Inhibitors. Cancers 2017, 9, 109. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Liu, J.C.; Amunugama, R.; Hajdu, I.; Primack, B.; Petalcorin, M.I.R.; O’Connor, K.W.; Konstantinopoulos, P.A.; Elledge, S.J.; Boulton, S.J.; et al. Homologous-recombination-deficient tumours are dependent on Polθ-mediated repair. Nature 2015, 518, 258–262. [Google Scholar] [CrossRef]
- Henneman, L.; van Miltenburg, M.H.; Michalak, E.M.; Braumuller, T.M.; Jaspers, J.E.; Drenth, A.P.; de Korte-Grimmerink, R.; Gogola, E.; Szuhai, K.; Schlicker, A.; et al. Selective resistance to the PARP inhibitor olaparib in a mouse model for BRCA1-deficient metaplastic breast cancer. Proc. Natl. Acad. Sci. USA 2015, 112, 8409–8414. [Google Scholar] [CrossRef]
- Vaidyanathan, A.; Sawers, L.; Gannon, A.-L.; Chakravarty, P.; Scott, A.L.; Bray, E.S.; Ferguson, M.J.; Smith, G. ABCB1 (MDR1) induction defines a common resistance mechanism in paclitaxel- and olaparib-resistant ovarian cancer cells. Br. J. Cancer 2016, 115, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.-Y.N.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Saha, P.; Kornbluth, S.; Dynlacht, B.D.; Dutta, A. Cyclin-binding motifs are essential for the function of p21CIP1. Mol. Cell. Biol. 1996, 16, 4673–4682. [Google Scholar] [CrossRef] [PubMed]
- Lapenna, S.; Giordano, A. Cell cycle kinases as therapeutic targets for cancer. Nat. Rev. Drug Discov. 2009, 8, 547–566. [Google Scholar] [CrossRef] [PubMed]
- Carrassa, L.; Damia, G. Unleashing Chk1 in cancer therapy. Cell Cycle 2011, 10, 2121–2128. [Google Scholar] [CrossRef] [PubMed]
- Restelli, V.; Chilà, R.; Lupi, M.; Rinaldi, A.; Kwee, I.; Bertoni, F.; Damia, G.; Carrassa, L. Characterization of a mantle cell lymphoma cell line resistant to the Chk1 inhibitor PF-00477736. Oncotarget 2015, 6, 37229–37240. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, A.; Woo, S.; Sarodaya, N.; Rhie, B.; Tyagi, A.; Das, S.; Suresh, B.; Ko, N.; Oh, S.; Kim, K.-S.; et al. Ubiquitin-Specific Protease 29 Regulates Cdc25A-Mediated Tumorigenesis. Int. J. Mol. Sci. 2021, 22, 5766. [Google Scholar] [CrossRef]
- Ruiz, S.; Mayor-Ruiz, C.; Lafarga, V.; Murga, M.; Vega-Sendino, M.; Ortega, S.; Fernandez-Capetillo, O. A Genome-wide CRISPR Screen Identifies CDC25A as a Determinant of Sensitivity to ATR Inhibitors. Mol. Cell 2016, 62, 307–313. [Google Scholar] [CrossRef]
- Neophytou, C.M.; Trougakos, I.P.; Erin, N.; Papageorgis, P. Apoptosis Deregulation and the Development of Cancer Multi-Drug Resistance. Cancers 2021, 13, 4363. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Saini, P.; Sriraman, A.; Dobbelstein, M. Mdm2 inhibition confers protection of p53-proficient cells from the cytotoxic effects of Wee1 inhibitors. Oncotarget 2015, 6, 32339–32352. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.J.; Chao, H.X.; Simpson, A.D.; Feng, W.; Cho, M.-G.; Roberts, V.R.; Sullivan, A.R.; Shah, S.J.; Wozny, A.-S.; Fagan-Solis, K.; et al. Dual inhibition of DNA-PK and DNA polymerase theta overcomes radiation resistance induced by p53 deficiency. NAR Cancer 2020, 2, zcaa038. [Google Scholar] [CrossRef] [PubMed]
- Schnog, J.-J.B.; Samson, M.J.; Gans, R.O.B.; Duits, A.J. An urgent call to raise the bar in oncology. Br. J. Cancer 2021, 125, 1477–1485. [Google Scholar] [CrossRef]
- Martorana, F.; Da Silva, L.A.; Sessa, C.; Colombo, I. Everything Comes with a Price: The Toxicity Profile of DNA-Damage Response Targeting Agents. Cancers 2022, 14, 953. [Google Scholar] [CrossRef] [PubMed]
- LaFargue, C.J.; Molin, G.Z.D.; Sood, A.K.; Coleman, R.L. Exploring and comparing adverse events between PARP inhibitors. Lancet Oncol. 2019, 20, e15–e28. [Google Scholar] [CrossRef] [PubMed]
- Madariaga, A.; Bowering, V.; Ahrari, S.; Oza, A.M.; Lheureux, S. Manage wisely: Poly (ADP-ribose) polymerase inhibitor (PARPi) treatment and adverse events. Int. J. Gynecol. Cancer 2020, 30, 903–915. [Google Scholar] [CrossRef] [PubMed]
- Valabrega, G.; Scotto, G.; Tuninetti, V.; Pani, A.; Scaglione, F. Differences in PARP Inhibitors for the Treatment of Ovarian Cancer: Mechanisms of Action, Pharmacology, Safety, and Efficacy. Int. J. Mol. Sci. 2021, 22, 4203. [Google Scholar] [CrossRef] [PubMed]
- Csizmar, C.M.; Saliba, A.N.; Swisher, E.M.; Kaufmann, S.H. PARP Inhibitors and Myeloid Neoplasms: A Double-Edged Sword. Cancers 2021, 13, 6385. [Google Scholar] [CrossRef] [PubMed]
- Kwan, T.T.; Oza, A.M.; Tinker, A.V.; Ray-Coquard, I.; Oaknin, A.; Aghajanian, C.; Lorusso, D.; Colombo, N.; Dean, A.; Weberpals, J.; et al. Preexisting TP53-Variant Clonal Hematopoiesis and Risk of Secondary Myeloid Neoplasms in Patients with High-grade Ovarian Cancer Treated with Rucaparib. JAMA Oncol. 2021, 7, 1772–1781. [Google Scholar] [CrossRef]
- Kusne, Y.; Xie, Z.; Patnaik, M.M. Clonal hematopoiesis: Molecular and clinical implications. Leuk. Res. 2022, 113. [Google Scholar] [CrossRef]
- Bolton, K.L.; Ptashkin, R.N.; Gao, T.; Braunstein, L.; Devlin, S.M.; Kelly, D.; Patel, M.; Berthon, A.; Syed, A.; Yabe, M.; et al. Cancer therapy shapes the fitness landscape of clonal hematopoiesis. Nat. Genet. 2020, 52, 1219–1226. [Google Scholar] [CrossRef]
- Navitski, A.; Al-Rawi, D.H.; Liu, Y.; Rubinstein, M.M.; Friedman, C.F.; Rampal, R.K.; Mandelker, D.L.; Cadoo, K.; O’Cearbhaill, R.E. Baseline risk of hematologic malignancy at initiation of frontline PARP inhibitor maintenance for BRCA1/2-associated ovarian cancer. Gynecol. Oncol. Rep. 2021, 38, 100873. [Google Scholar] [CrossRef]
- Oliveira, J.L.; Greipp, P.T.; Rangan, A.; Jatoi, A.; Nguyen, P.L. Myeloid malignancies in cancer patients treated with poly(ADP-ribose) polymerase (PARP) inhibitors: A case series. Blood Cancer J. 2022, 12, 11. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef]
- Morice, P.-M.; Leary, A.; Dolladille, C.; Chrétien, B.; Poulain, L.; González-Martín, A.; Moore, K.; O’Reilly, E.M.; Ray-Coquard, I.; Alexandre, J. Myelodysplastic syndrome and acute myeloid leukaemia in patients treated with PARP inhibitors: A safety meta-analysis of randomised controlled trials and a retrospective study of the WHO pharmacovigilance database. Lancet Haematol. 2020, 8, e122–e134. [Google Scholar] [CrossRef]
- Nitecki, R.; Melamed, A.; Gockley, A.A.; Floyd, J.; Krause, K.J.; Coleman, R.L.; Matulonis, U.A.; Giordano, S.H.; Lu, K.H.; Rauh-Hain, J.A. Incidence of myelodysplastic syndrome and acute myeloid leukemia in patients receiving poly-ADP ribose polymerase inhibitors for the treatment of solid tumors: A meta-analysis of randomized trials. Gynecol. Oncol. 2021, 161, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Sun, X.; Lu, W.; Zhao, Z.; Xu, Z.; Lyu, J.; Zhao, P.; Liu, L. Poly(ADP-ribose) polymerase inhibitor-associated myelodysplastic syndrome/acute myeloid leukemia: A pharmacovigilance analysis of the FAERS database. ESMO Open 2021, 6, 100033. [Google Scholar] [CrossRef]


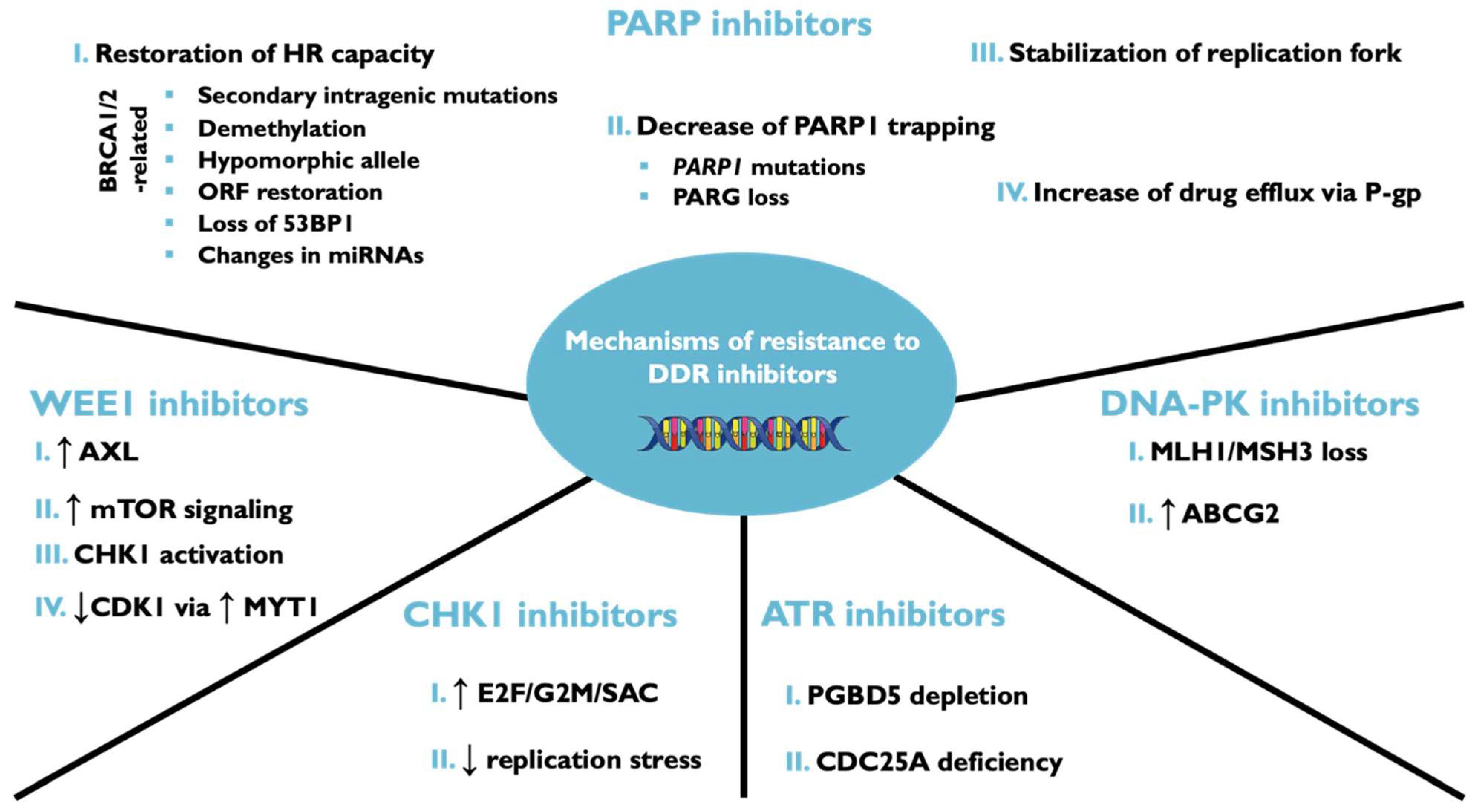{kind=link}
| Inhibitor | Class/Mechanisms of Action | Stage and Indication |
|---|---|---|
| Olaparib | PARP inhibitor | Approved: HER2-negative BC; FC; OC; PaC; PeC; PC Phase III: BC; CRC; EC; NSCLC; SCLC; SCC Phase II/III: TNBC Phase II: BlaC; CC; GC; CBM; HNC; OSC; RCC; UC; HER2-positive; Solid tumors |
| Talazoparib | PARP inhibitor | Approved: BC; HER2-negative BC Phase III: OC; PC Phase II: EC, FC; SCLC; SCC; TNBC; Solid tumors Phase I/II: AML |
| Niraparib | PARP1 inhibitor PARP2 inhibitor | Approved: FC; OC; PeC Phase III: HER2-negative BC; NSCLC; SCLC Phase II: BC; CC; CCA; CNSC; EC; GC; GBM; Glioma; HNC; Mesothelioma; NT; EsC; PaC; RCC; UC; TNBC; Solid tumors; Uveal melanoma |
| Rucaparib | PARP1 inhibitor PARP2 inhibitor | Approved: FC; OC; PeC; PC Phase II: BC; CC; EC; GC; UC; Mesothelioma; PaC; TNBC; Solid tumors Phase I/II: NSCLC |
| Pamiparib | PARP1 inhibitor PARP2 inhibitor | Approved: FC; OC; PeC (China) Phase III: Cancer Phase II: HER2-negative BC; GC Phase I/II: GBM; Glioma; Solid tumors |
| Veliparib | PARP inhibitor | Approved: advanced LSCC; OC; BC, LC Phase III: BC; HER2-negative BC; TNBC; NSCLC; OC Phase II/III: GBM Phase II: Brain metastases; CRC; GCEN; Malignant melanoma; PaC; RC; Solid tumors Phase I/II: Glioma; HNC; SCLC |
| Stenoparib | PARP1 inhibitor PARP2 inhibitorTankyrase inhibitor | Phase II: BC; OC Phase I/II: Solid tumor |
| Methoxyamine | APE1 inhibitor | Phase II. GBM; Mesothelioma; NSCLC Phase I/II: Solid tumors |
| Palbociclib | CDK4 inhibitor CDK6 inhibitor | Approved: BC Phase II/III: NSCLC Phase II: Bone metastases; Brain metastases; GIST; Mantle cell lymphoma; PaC; PC; SCC; UC Phase I/II: CRC; Malignant melanoma |
| Iniparib | Cell cycle inhibitor H2AFX protein stimulantTumor protein modulator | Phase II: Glioma |
| Ceralasertib | ATR protein inhibitor | Phase III: NSCLC Phase II: TNBC; CCA; GC; GyC; Malignant melanoma; OSC; OC; PaC; PC; SCLCN; Solid tumors Phase I/II: CLL |
| Elimusertib | ATR protein inhibitor | Phase I: HNC; Lymphoma; OC; Solid tumors |
| M 4344 | ATR protein inhibitor | Phase I: Lymphoma; Solid tumors |
| Berzosertib | ATR protein inhibitor | Phase II: FC; LS; OC; PeC; PC; SCLC; UC; Solid tumors Phase I: HNC |
| AZD 1390 | ATM protein inhibitor | Phase I: GBM; NSCLC; Soft tissue sarcoma Preclinical: BC; Meningioma |
| Prexasertib | CHEK1 inhibitor CHEK2 inhibitor | Phase II: OC; Solid tumors Phase I/II: EC; UCP reclinical: BC |
| SRA 737 | CHEK1 inhibitor | Phase II: Solid tumors Phase I/II: OC; PC |
| Peposertib | DNA-PK inhibitor | Phase I/II: RC/SCLC Phase I: GBM; Cancer; Solid tumors |
| AZD 7648 | DNA-PK inhibitor | Phase I: Soft tissue sarcoma |
| Adavosertib | WEE1 inhibitor | Phase II: TNBC; EC; FC; NSCLC; PeC; PC; RCC; SCLC; UtC; Solid tumors Phase I: Hematological disorders; HNC; UtC Preclinical: DLBCL; GC |
| Volasertib | PLK1 inhibitor | Phase III: AML Phase II: MDS; NSCLC; OC Phase I: Rhabdomyosarcoma; Solid tumors |
| Onvansertib | PLK1 inhibitor | Phase II: AML; PaC; PC; SCLC Phase I/II: CRC; TNBC Preclinical: CMML; Medulloblastoma; OC |
| Targeting Protein | Inhibitor | Clinical Status | Disease State | NCT Number |
|---|---|---|---|---|
| ATR PARP | AZD6738 Olaparib | Phase 2, Recruiting | Neoadjuvant Chemotherapy-resistant residual triple-negative breast cancer | NCT03740893 |
| PARP 1/2 and Tankyrase 1/2 | 2X-121 | Phase 2, Active not recruiting | Metastatic breast cancer, PARPi-resistant cancer | NCT03562832 |
| PARP | Talazoparib | Phase 2, Recruiting | Multiple cancers, patients with aberrations in DNA damage response genes | NCT04550494 |
| PD-1 PARP | Pembrolizumab Olaparib | Phase 2, Not recruiting | Nasopharyngeal carcinoma, resistant to platinum agents | NCT04825990 |
| Testosterone PARP | Olaparib | Phase 2, Active, not recruiting | Castration-resistant prostate cancer | NCT03516812 |
| ATR PARP | BAY1895344 Niraparib | Phase 1, Recruiting | Advanced solid tumors, ovarian cancer | NCT04267939 |
| PARP | Talazoparib Temozolomide | Phase 1,2 Recruiting | Metastatic castration-resistant prostate cancer and no mutations in DNA damage repair | NCT04019327 |
| PARP | Rucaparib | Phase 2 Recruiting | Prostate cancer metastatic, resistant to androgen deprivation therapy and who carry a DNA repair gene mutation | NCT03413995 |
| PARP | Olaparib Temozolomide IMRT | Phase 1, 2 Recruiting | Unresectable high-grade gliomas | NCT03212742 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jurkovicova, D.; Neophytou, C.M.; Gašparović, A.Č.; Gonçalves, A.C. DNA Damage Response in Cancer Therapy and Resistance: Challenges and Opportunities. Int. J. Mol. Sci. 2022, 23, 14672. https://doi.org/10.3390/ijms232314672
Jurkovicova D, Neophytou CM, Gašparović AČ, Gonçalves AC. DNA Damage Response in Cancer Therapy and Resistance: Challenges and Opportunities. International Journal of Molecular Sciences. 2022; 23(23):14672. https://doi.org/10.3390/ijms232314672
Chicago/Turabian StyleJurkovicova, Dana, Christiana M. Neophytou, Ana Čipak Gašparović, and Ana Cristina Gonçalves. 2022. "DNA Damage Response in Cancer Therapy and Resistance: Challenges and Opportunities" International Journal of Molecular Sciences 23, no. 23: 14672. https://doi.org/10.3390/ijms232314672
APA StyleJurkovicova, D., Neophytou, C. M., Gašparović, A. Č., & Gonçalves, A. C. (2022). DNA Damage Response in Cancer Therapy and Resistance: Challenges and Opportunities. International Journal of Molecular Sciences, 23(23), 14672. https://doi.org/10.3390/ijms232314672