
Modulation of the Inflammatory Response in Polycystic Ovary Syndrome (PCOS)—Searching for Epigenetic Factors


Abstract
:1. Introduction
Methodology of Literature Searching
2. Oxidative Stress and Inflammatory Signaling in PCOS
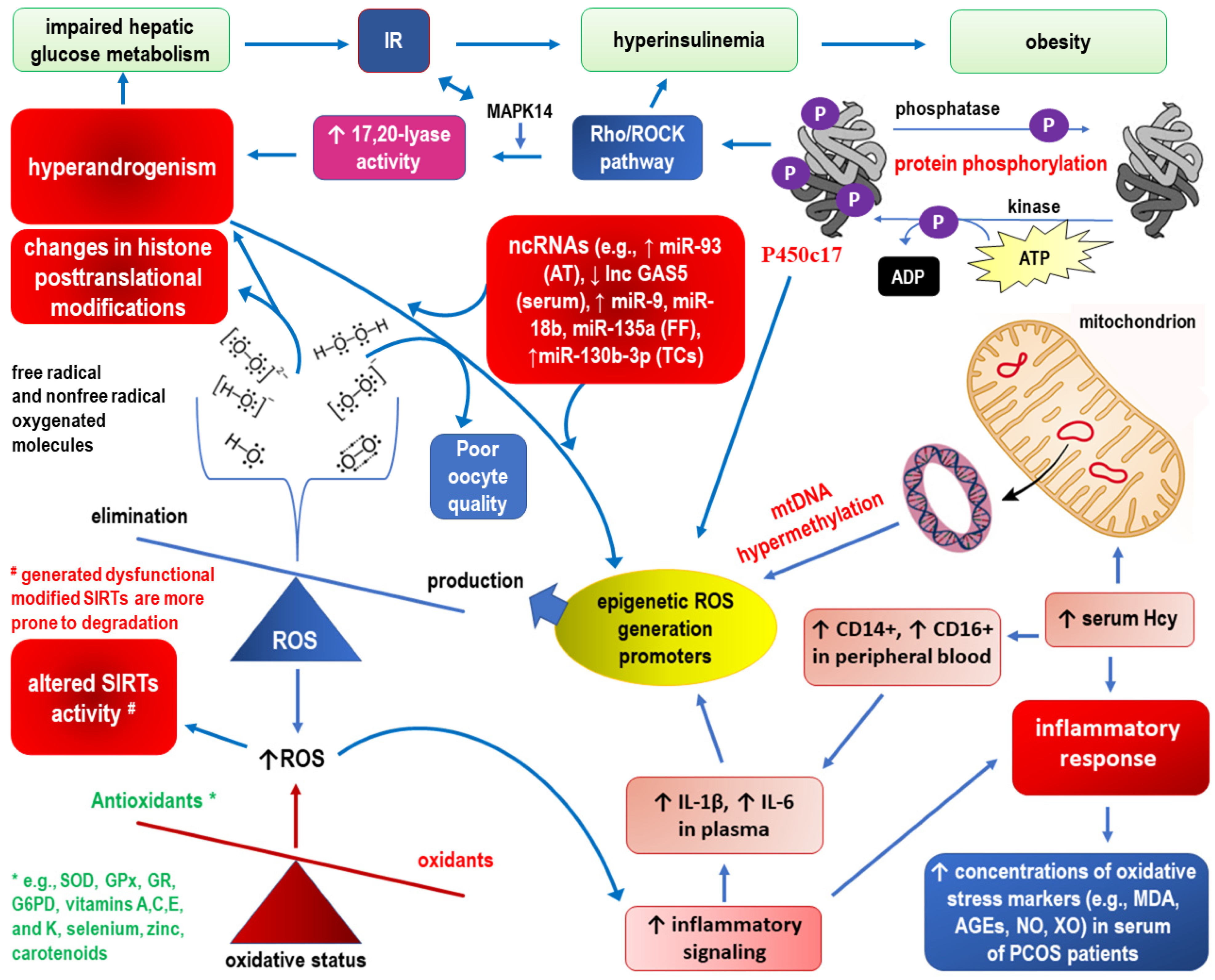
2.1. Epigenetic Landscape Related to ROS Formation in PCOS (See Also Figure 3, Which Corresponds to the Text of this Section)
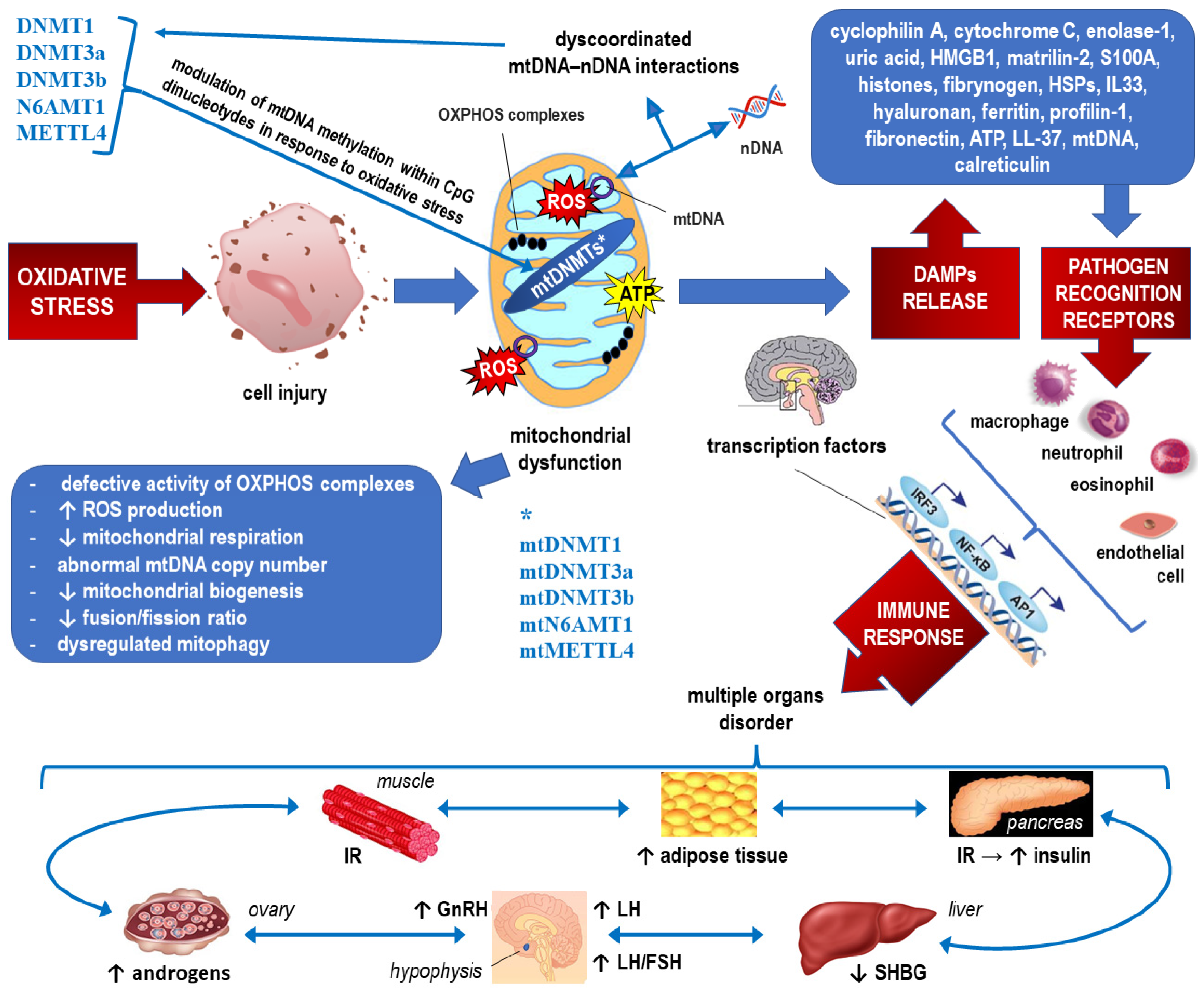
2.2. Epigenetics of Mitochondrial Dysfunction in PCOS (See Also Figure 4, which Corresponds to the Text of this Section)
3. Epigenetic Factors Influencing the Cytokine Profile and Inflammatory Markers in PCOS
4. Epigenetic Regulation of Insulin Resistance and Inflammatory Signaling in PCOS
5. Epigenetic Modulation of Androgenic Activity in Response to Inflammatory Signaling in PCOS
6. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 5-mC | 5-methylcytosine |
| 6-mA | 6-methlyl-adenine |
| ADMA | asymmetric dimethylarginine |
| AGEs | advanced glycation end products |
| AMHR | anti-Müllerian hormone receptor |
| AP-1 | activator protein 1 |
| AR | androgen receptor |
| ATP | adenosine triphosphate |
| BHMT | betaine homocysteine methyltransferase |
| BMI | body mass index |
| BMP4 | bone morphogenetic protein 4 |
| C3 | complement element 3 |
| CpG | cytosine-phosphate-guanine dinucleotides |
| CRHBP | corticotropin releasing hormone binding protein |
| CRP | C-reactive protein |
| CYP11A1 | cholesterol monooxygenase (cytochrome P450 family 11 subfamily A member 1) |
| CYP17A1 | cytochrome P450 17α-hydroxylase/17,20-lyase |
| CYP19A1 | enzyme aromatase (cytochrome P450 family 19 subfamily A member 1) |
| DAMPs | damage-associated molecular patterns |
| DENND1A | DENN domain containing 1A |
| DENND1A.V2 | DENN domain containing 1A variant 2 |
| DHEA | dehydroepiandrosterone |
| DNA | deoxyribonucleic acid |
| DNMTs | DNA methyltrasferases (e.g., DNMT, DNMT3a, and DNMT3) |
| E | estrogen |
| ER | endoplasmic reticulum |
| ET-1 | endothelin 1 |
| FOXO3A | transcription factor forkhead box protein O3 |
| FSH | follicle-stimulating hormone |
| GCs | granulosa cells |
| GNMT | glycine N-methyltransferase |
| Hcy | homocysteine |
| HDACs | histone deacetylases |
| HMGB-1 | high mobility group box-1 |
| hs-CRP | high-sensitivity C-reactive protein |
| HSD3B2 | hydroxy-delta-5-steroid dehydrogenase, 3 beta- and steroid delta-isomerase 2 |
| ICAM-1 | Intercellular Adhesion Molecule 1 (CD54) |
| IFN-γ | interferon-gamma |
| IKKβ | I-kappa-B-kinase beta |
| IL | interleukin (e.g.,: IL-6, IL-10, IL-12, IL-18, IL-34) |
| iNOS | inducible nitric oxide synthase |
| IR | insulin resistance |
| IRS | insulin receptor substrate |
| JNK1 | c-Jun N-terminal kinase 1 |
| LH | luteinizing hormone |
| LIF | leukemia inhibitory factor |
| lncRNA | long noncoding RNA |
| MALAT1 | long-chain ncRNA metastasis-related lung adenocarcinoma transcript 1 |
| MAPK14 | mitogen-activated protein kinase 14 (p38α) |
| MCP-1 | monocyte chemoattractant protein-1 |
| MDA | malondialdehyde |
| MDM2 | murine double minute 2 oncogene |
| METTL4 | methyltransferase like 4 |
| MIP-1α | macrophage inflammatory protein-1α |
| miRNA | microRNAs |
| MMP-9 | matrix metalloproteinase-9 |
| MNC | mononuclear cells |
| mtDNA | mitochondrial DNA |
| mtDNMTs | mitochondrial DNA methyltrasferases |
| mTOR | serine-threonine protein kinase mammalian target of rapamycin (mTOR), |
| N6AMT1 | N-6 adenine-specific DNA methyltrasferase 1 |
| NAD+ | nicotinamide adenine dinucleotide |
| NAFLD | nonalcoholic fatty liver disease |
| ncRNAs | noncoding RNAs |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NLRP3 | NOD-, LRR- and pyrin domain-containing protein 3 |
| NO | nitric oxide |
| NRF-1 | nuclear respiratory factor 1 |
| OXPHOS | oxidative phosphorylation |
| PAI-I | plasminogen activator inhibitor-I |
| PCOS | polycystic ovary syndrome |
| PGC-1α | peroxisome proliferator activated receptor gamma coactivator-1 alpha |
| PI3K/AKT | phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT) signaling pathway |
| PLAC2 | placenta-specific protein 2 |
| PPAR | peroxisome proliferator-activated receptor |
| Prx4 | peroxiredoxin 4 |
| RAGE | cellular membrane receptor for advanced glycation end products |
| Rho/ROCK pathway | Rho/Rho-associated coiled-coil containing protein kinase pathway |
| RNA | ribonucleic acid |
| ROS | reactive oxygen species |
| SIRTs | sirtuins (e.g., SIRT3–sirtuin 3) |
| SIX5 | transcription factor SIX homeobox protein 5 (SIX5) |
| SLE | systemic lupus erythematosus |
| SMAD4 | SMAD family member 4 (mothers against decapentaplegic homolog 4) |
| SOCS | suppressors of cytokine signaling |
| sRAGE | soluble form of receptor for advanced glycation end products |
| STAT | signal transducer and activator of transcription |
| T | testosterone |
| TFAM | mitochondrial transcription factor A p53–protein p53 |
| TGFβ | transforming growth factor β |
| TGFβR1, TGFβR2 | transforming growth factor beta receptor 1 and 2, respectively |
| TLR4 | Toll-like receptor 4 |
| TLR8 | Toll-like receptor 8 |
| TNF-α | tumor necrosis factor alpha |
| TOS | total oxidant status |
| VCAM-1 | vascular cell adhesion protein 1 (CD106) |
| VEGFs | vascular endothelial growth factors |
| WBC | white blood cell count |
| XO | xanthine oxidase |
| YY1 | transcription factor yin-yang 1 |
References
- Azziz, R. Introduction. Fertil. Steril. 2016, 106, 4–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Wu, Q.; Hao, Y.; Jiao, M.; Wang, X.; Jiang, S.; Han, L. Measuring the global disease burden of polycystic ovary syndrome in 194 countries: Global Burden of Disease Study 2017. Hum. Reprod. 2021, 36, 1108–1119. [Google Scholar] [CrossRef]
- Pundir, C.S.; Deswal, R.; Narwal, V.; Dang, A. The Prevalence of Polycystic Ovary Syndrome: A Brief Systematic Review. J. Hum. Reprod. Sci. 2020, 13, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Joham, A.E.; Piltonen, T.; Lujan, M.E.; Kiconco, S.; Tay, C.T. Challenges in diagnosis and understanding of natural history of polycystic ovary syndrome. Clin. Endocrinol. 2022, 97, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Louwers, Y.V.; Laven, J.S. Characteristics of polycystic ovary syndrome throughout life. Ther. Adv. Reprod. Health 2020, 14. [Google Scholar] [CrossRef]
- The Rotterdam ESHRE/ASRM-sponsored PCOS consensus workshop group. Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome (PCOS). Hum. Reprod. 2004, 19, 41–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenfield, R.L.; Ehrmann, D.A. The Pathogenesis of Polycystic Ovary Syndrome (PCOS): The Hypothesis of PCOS as Functional Ovarian Hyperandrogenism Revisited. Endocr. Rev. 2016, 37, 467–520. [Google Scholar] [CrossRef]
- Pirotta, S.; Joham, A.; Grieger, J.A.; Tay, C.T.; Bahri-Khomami, M.; Lujan, M.; Lim, S.S.; Moran, L.J. Obesity and the Risk of Infertility, Gestational Diabetes, and Type 2 Diabetes in Polycystic Ovary Syndrome. Semin. Reprod. Med. 2020, 38, 342–351. [Google Scholar] [CrossRef]
- Vassilatou, E. Nonalcoholic fatty liver disease and polycystic ovary syndrome. World J. Gastroenterol. 2014, 20, 8351–8363. [Google Scholar] [CrossRef]
- Doycheva, I.; Ehrmann, D.A. Nonalcoholic fatty liver disease and obstructive sleep apnea in women with polycystic ovary syndrome. Fertil. Steril. 2022, 117, 897–911. [Google Scholar] [CrossRef]
- Wang, D.; He, B. Current Perspectives on Nonalcoholic Fatty Liver Disease in Women with Polycystic Ovary Syndrome. Diabetes Metab. Syndr. Obes. Targets Ther. 2022, 15, 1281–1291. [Google Scholar] [CrossRef] [PubMed]
- Belani, M.; Deo, A.; Shah, P.; Banker, M.; Singal, P.; Gupta, S. Differential insulin and steroidogenic signaling in insulin resistant and non-insulin resistant human luteinized granulosa cells—A study in PCOS patients. J. Steroid Biochem. Mol. Biol. 2018, 178, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Artimani, T.; Karimi, J.; Mehdizadeh, M.; Yavangi, M.; Khanlarzadeh, E.; Ghorbani, M.; Asadi, S.; Kheiripour, N. Evaluation of pro-oxidant-antioxidant balance (PAB) and its association with inflammatory cytokines in polycystic ovary syndrome (PCOS). Gynecol. Endocrinol. 2017, 34, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Rudnicka, E.; Suchta, K.; Grymowicz, M.; Calik-Ksepka, A.; Smolarczyk, K.; Duszewska, A.M.; Smolarczyk, R.; Meczekalski, B. Chronic Low Grade Inflammation in Pathogenesis of PCOS. Int. J. Mol. Sci. 2021, 22, 3789. [Google Scholar] [CrossRef] [PubMed]
- Aboeldalyl, S.; James, C.; Seyam, E.; Ibrahim, E.; Shawki, H.; Amer, S. The Role of Chronic Inflammation in Polycystic Ovarian Syndrome—A Systematic Review and Meta-Analysis. Int. J. Mol. Sci. 2021, 22, 2734. [Google Scholar] [CrossRef]
- Khichar, A.; Gupta, S.; Mishra, S.; Meena, M. Assessment of Inflammatory Markers in Women with PCOS and their Correlation with Insulin Resistance. Clin. Lab. 2021, 67, 210310. [Google Scholar] [CrossRef]
- Sharkesh, E.Z.; Keshavarz, S.A.; Nazari, L.; Abbasi, B. The dietary inflammatory index is directly associated with polycystic ovary syndrome: A case-control study. Clin. Endocrinol. 2021, 96, 698–706. [Google Scholar] [CrossRef]
- Tatone, C.; Di Emidio, G.; Placidi, M.; Rossi, G.; Ruggieri, S.; Taccaliti, C.; D’Alfonso, A.; Amicarelli, F.; Guido, M. AGEs-related dysfunctions in PCOS: Evidence from animal and clinical research. J. Endocrinol. 2021, 251, R1–R9. [Google Scholar] [CrossRef]
- Luevano-Contreras, C.; Chapman-Novakofski, K. Dietary Advanced Glycation End Products and Aging. Nutrients 2010, 2, 1247–1265. [Google Scholar] [CrossRef] [Green Version]
- Sharma, C.; Kaur, A.; Thind, S.S.; Singh, B.; Raina, S. Advanced glycation End-products (AGEs): An emerging concern for processed food industries. J. Food Sci. Technol. 2015, 52, 7561–7576. [Google Scholar] [CrossRef]
- Gill, V.; Kumar, V.; Singh, K.; Kumar, A.; Kim, J.-J. Advanced Glycation End Products (AGEs) May Be a Striking Link between Modern Diet and Health. Biomolecules 2019, 9, 888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diamanti-Kandarakis, E.; Palimeri, S.; Palioura, E. Current perspectives on the health risks associated with the consumption of advanced glycation end products: Recommendations for dietary management. Diabetes Metab. Syndr. Obes. Targets Ther. 2015, 8, 415–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutkowska, A.; Diamanti-Kandarakis, E. Do Advanced Glycation End Products (AGEs) Contribute to the Comorbidities of Polycystic Ovary Syndrome (PCOS)? Curr. Pharm. Des. 2016, 22, 5558–5571. [Google Scholar] [CrossRef] [PubMed]
- Merhi, Z.; Kandaraki, E.A.; Diamanti-Kandarakis, E. Implications and Future Perspectives of AGEs in PCOS Pathophysiology. Trends Endocrinol. Metab. 2019, 30, 150–162. [Google Scholar] [CrossRef]
- Azhary, J.M.K.; Harada, M.; Kunitomi, C.; Kusamoto, A.; Takahashi, N.; Nose, E.; Oi, N.; Wada-Hiraike, O.; Urata, Y.; Hirata, T.; et al. Androgens Increase Accumulation of Advanced Glycation End Products in Granulosa Cells by Activating ER Stress in PCOS. Endocrinology 2020, 161. [Google Scholar] [CrossRef]
- Garg, D.; Merhi, Z. Relationship between Advanced Glycation End Products and Steroidogenesis in PCOS. Reprod. Biol. Endocrinol. 2016, 14, 71. [Google Scholar] [CrossRef] [Green Version]
- Shorakae, S.; Teede, H.; de Courten, B.; Lambert, G.; Boyle, J.; Moran, L.J. The Emerging Role of Chronic Low-Grade Inflammation in the Pathophysiology of Polycystic Ovary Syndrome. Semin. Reprod. Med. 2015, 33, 257–269. [Google Scholar] [CrossRef]
- Gomez, J.M.D.; VanHise, K.; Stachenfeld, N.; Chan, J.L.; Merz, N.B.; Shufelt, C. Subclinical cardiovascular disease and polycystic ovary syndrome. Fertil. Steril. 2022, 117, 912–923. [Google Scholar] [CrossRef]
- Salamun, V.; Rizzo, M.; Lovrecic, L.; Hocevar, K.; Burnik, T.P.; Janez, A.; Jensterle, M.; Bokal, E.V.; Peterlin, B.; Maver, A. The Endometrial Transcriptome of Metabolic and Inflammatory Pathways during the Window of Implantation Is Deranged in Infertile Obese Polycystic Ovarian Syndrome Women. Metab. Syndr. Relat. Disord. 2022, 20, 384–394. [Google Scholar] [CrossRef]
- Jiang, N.-X.; Li, X.-L. The Disorders of Endometrial Receptivity in PCOS and Its Mechanisms. Reprod. Sci. 2021, 29, 2465–2476. [Google Scholar] [CrossRef]
- Arffman, R.K.; Saraswat, M.; Joenväärä, S.; Khatun, M.; Agarwal, R.; Tohmola, T.; Sundström-Poromaa, I.; Renkonen, R.; Piltonen, T.T. Thromboinflammatory changes in plasma proteome of pregnant women with PCOS detected by quantitative label-free proteomics. Sci. Rep. 2019, 9, 17578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Teng, F.; Wu, Q.; Wu, Y.; Hu, L. Relationship between proinflammatory cytokines and clomiphene resistance in patients with polycystic ovary syndrome. Ann. Palliat. Med. 2021, 10, 11884–11890. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Qi, H.; Baker, P.N.; Zhen, Q.; Zeng, Q.; Shi, R.; Tong, C.; Ge, Q. Altered Circulating Inflammatory Cytokines Are Associated with Anovulatory Polycystic Ovary Syndrome (PCOS) Women Resistant to Clomiphene Citrate Treatment. Med. Sci. Monit. 2017, 23, 1083–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, J.; O’Brien, C.; Hawrelak, J.; Gersh, F.L. Polycystic Ovary Syndrome: An Evolutionary Adaptation to Lifestyle and the Environment. Int. J. Environ. Res. Public Health 2022, 19, 1336. [Google Scholar] [CrossRef] [PubMed]
- Bruni, V.; Capozzi, A.; Lello, S. The Role of Genetics, Epigenetics and Lifestyle in Polycystic Ovary Syndrome Development: The State of the Art. Reprod. Sci. 2021, 29, 668–679. [Google Scholar] [CrossRef]
- Eiras, M.C.; Pinheiro, D.P.; Romcy, K.A.M.; Ferriani, R.A.; Dos Reis, R.M.; Furtado, C.L.M. Polycystic Ovary Syndrome: The Epigenetics Behind the Disease. Reprod. Sci. 2021, 29, 680–694. [Google Scholar] [CrossRef]
- Scarfò, G.; Daniele, S.; Fusi, J.; Gesi, M.; Martini, C.; Franzoni, F.; Cela, V.; Artini, P.G. Metabolic and Molecular Mechanisms of Diet and Physical Exercise in the Management of Polycystic Ovarian Syndrome. Biomedicines 2022, 10, 1305. [Google Scholar] [CrossRef]
- Ilie, I.R.; Georgescu, C.E. Polycystic Ovary Syndrome-Epigenetic Mechanisms and Aberrant MicroRNA. Adv. Clin. Chem. 2015, 71, 25–45. [Google Scholar] [CrossRef]
- Smirnov, V.V.; Beeraka, N.M.; Butko, D.Y.; Nikolenko, V.N.; Bondarev, S.A.; Achkasov, E.E.; Sinelnikov, M.Y.; Vikram, P.R.H. Updates on Molecular Targets and Epigenetic-Based Therapies for PCOS. Reprod. Sci. 2022, 1–15. [Google Scholar] [CrossRef]
- Wright, J. Epigenetics: Reversible tags. Nature 2013, 498, S10–S11. [Google Scholar] [CrossRef]
- Conway, S.J.; Woster, P.M.; Greenlee, W.J.; Georg, G.; Wang, S. Epigenetics: Novel Therapeutics Targeting Epigenetics. J. Med. Chem. 2016, 59, 1247–1248. [Google Scholar] [CrossRef] [PubMed]
- Bajrami, E.; Spiroski, M. Genomic Imprinting. Open Access Maced. J. Med. Sci. 2016, 4, 181–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Q.; Chen, Y.; Zhou, X. The roles of microRNAs in epigenetic regulation. Curr. Opin. Chem. Biol. 2019, 51, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Martínez, E.R.; Gómez-Viais, Y.I.; García-Gómez, E.; Reyes-Mayoral, C.; Reyes-Muñoz, E.; Camacho-Arroyo, I.; Cerbón, M.A. DNA Methylation in the Pathogenesis of Polycystic Ovary Syndrome. Reproduction 2019, 158, R27–R40. [Google Scholar] [CrossRef] [PubMed]
- Rawat, K.; Sandhu, A.; Gautam, V.; Saha, P.K.; Saha, L. Role of genomic DNA methylation in PCOS pathogenesis: A systematic review and meta-analysis involving case-controlled clinical studies. Mol. Hum. Reprod. 2022, 28, gaac024. [Google Scholar] [CrossRef]
- Zhong, X.; Jin, F.; Huang, C.; Du, M.; Gao, M.; Wei, X. DNA methylation of AMHRII and INSR gene is associated with the pathogenesis of Polycystic Ovary Syndrome (PCOS). Technol. Health Care 2021, 29, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [Green Version]
- Halliwell, B.; Gutteridge, J.M.; Cross, C.E. Free radicals, antioxidants, and human disease: Where are we now? J. Lab. Clin. Med. 1992, 1992 119, 598–620. [Google Scholar]
- Riley, P.A. Free Radicals in Biology: Oxidative Stress and the Effects of Ionizing Radiation. Int. J. Radiat. Biol. 1994, 65, 27–33. [Google Scholar] [CrossRef]
- Gutteridge, J.M. Biological origin of free radicals, and mechanisms of antioxidant protection. Chem. Interact. 1994, 91, 133–140. [Google Scholar] [CrossRef]
- Papalou, O.; Victor, V.; Diamanti-Kandarakis, E. Oxidative Stress in Polycystic Ovary Syndrome. Curr. Pharm. Des. 2016, 22, 2709–2722. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, M. Oxidative stress and polycystic ovary syndrome: A brief review. Int. J. Prev. Med. 2019, 10, 86. [Google Scholar] [CrossRef] [PubMed]
- Mazloomi, S.; Sheikh, N.; Farimani, M.S.; Pilehvari, S. Association of Prx4, Total Oxidant Status, and Inflammatory Factors with Insulin Resistance in Polycystic Ovary Syndrome. Int. J. Endocrinol. 2021, 2021, 9949753. [Google Scholar] [CrossRef] [PubMed]
- Perła-Kaján, J.; Jakubowski, H. Dysregulation of Epigenetic Mechanisms of Gene Expression in the Pathologies of Hyperhomocysteinemia. Int. J. Mol. Sci. 2019, 20, 3140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esse, R.; Barroso, M.; De Almeida, I.T.; Castro, R. The Contribution of Homocysteine Metabolism Disruption to Endothelial Dysfunction: State-of-the-Art. Int. J. Mol. Sci. 2019, 20, 867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.; Wang, X.; Kong, W. Hyperhomocysteinaemia and vascular injury: Advances in mechanisms and drug targets. J. Cereb. Blood Flow Metab. 2017, 175, 1173–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Niu, H.; Zhang, J. Homocysteine induces mitochondrial dysfunction and oxidative stress in myocardial ischemia/reperfusion injury through stimulating ROS production and the ERK1/2 signaling pathway. Exp. Ther. Med. 2020, 20, 938–944. [Google Scholar] [CrossRef] [PubMed]
- Guzelmeric, K.; Alkan, N.; Pirimoglu, M.; Unal, O.; Turan, C. Chronic inflammation and elevated homocysteine levels are associated with increased body mass index in women with polycystic ovary syndrome. Gynecol. Endocrinol. 2007, 23, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Qi, X.; Zhao, Y.; Li, R.; Zhang, C.; Chang, H.-M.; Pang, Y.; Qiao, J. Elevated CD14++CD16+ Monocytes in Hyperhomocysteinemia-Associated Insulin Resistance in Polycystic Ovary Syndrome. Reprod. Sci. 2018, 25, 1629–1636. [Google Scholar] [CrossRef] [PubMed]
- Kondapaneni, V.; Gutlapalli, S.D.; Poudel, S.; Zeb, M.; Toulassi, I.A.; Cancarevic, I. Significance of Homocysteine Levels in the Management of Polycystic Ovarian Syndrome: A Literature Review. Cureus 2020, 12, e11110. [Google Scholar] [CrossRef] [PubMed]
- Sinha, P.; Bhushan, R. Correlation of serum homocysteine levels and hyperinsulinaemia with body mass index in polycystic ovarian syndrome. J. Hum. Reprod. Sci. 2022, 15, 34. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Li, J.; Chenfei, W.; Jia, Y.; Niu, Y.-J.; Wang, C.; Zhao, R. Abnormally activated one-carbon metabolic pathway is associated with mtDNA hypermethylation and mitochondrial malfunction in the oocytes of polycystic gilt ovaries. Sci. Rep. 2016, 6, 19436. [Google Scholar] [CrossRef] [Green Version]
- Jia, L.; Zeng, Y.; Hu, Y.; Liu, J.; Yin, C.; Niu, Y.; Wang, C.; Li, J.; Jia, Y.; Hong, J.; et al. Homocysteine impairs porcine oocyte quality via deregulation of one-carbon metabolism and hypermethylation of mitochondrial DNA. Biol. Reprod. 2018, 100, 907–916. [Google Scholar] [CrossRef]
- Razi, Y.; Eftekhar, M.; Fesahat, F.; Firouzabadi, R.D.; Razi, N.; Sabour, M.; Razi, M.H. Concentrations of homocysteine in follicular fluid and embryo quality and oocyte maturity in infertile women: A prospective cohort. J. Obstet. Gynaecol. 2020, 41, 588–593. [Google Scholar] [CrossRef]
- Kietzmann, T.; Petry, A.; Shvetsova, A.; Gerhold, J.; Görlach, A. The epigenetic landscape related to reactive oxygen species formation in the cardiovascular system. Br. J. Pharmacol. 2017, 174, 1533–1554. [Google Scholar] [CrossRef] [Green Version]
- Osibogun, O.; Ogunmoroti, O.; Michos, E.D. Polycystic ovary syndrome and cardiometabolic risk: Opportunities for cardiovascular disease prevention. Trends Cardiovasc. Med. 2019, 30, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-H.; Heneidi, S.; Lee, J.-M.; Layman, L.C.; Stepp, D.W.; Gamboa, G.M.; Chen, B.-S.; Chazenbalk, G.; Azziz, R. miRNA-93 Inhibits GLUT4 and Is Overexpressed in Adipose Tissue of Polycystic Ovary Syndrome Patients and Women with Insulin Resistance. Diabetes 2013, 62, 2278–2286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.; Xing, W.; Li, Y.; Xie, Y.; Tang, X.; Zhang, Q. Downregulation of serum long noncoding RNA GAS5 may contribute to insulin resistance in PCOS patients. Gynecol. Endocrinol. 2018, 34, 784–788. [Google Scholar] [CrossRef]
- Roth, L.W.; McCallie, B.; Alvero, R.; Schoolcraft, W.B.; Minjarez, D.; Katz-Jaffe, M.G. Altered microRNA and gene expression in the follicular fluid of women with polycystic ovary syndrome. J. Assist. Reprod. Genet. 2014, 31, 355–362. [Google Scholar] [CrossRef]
- McAllister, J.M.; Han, A.X.; Modi, B.; E Teves, M.; Mavodza, G.R.; Anderson, Z.L.; Shen, T.; Christenson, L.K.; Archer, K.J.; Strauss, J.F. miRNA Profiling Reveals miRNA-130b-3p Mediates DENND1A Variant 2 Expression and Androgen Biosynthesis. Endocrinology 2019, 160, 1964–1981. [Google Scholar] [CrossRef]
- Tu, M.; Wu, Y.; Mu, L.; Zhang, D. Long non-coding RNAs: Novel players in the pathogenesis of polycystic ovary syndrome. Ann. Transl. Med. 2021, 9, 173. [Google Scholar] [CrossRef] [PubMed]
- Tee, M.K.; Dong, Q.; Miller, W.L. Pathways Leading to Phosphorylation of P450c17 and to the Posttranslational Regulation of Androgen Biosynthesis. Endocrinology 2008, 149, 2667–2677. [Google Scholar] [CrossRef] [Green Version]
- De Medeiros, S.F.; Barbosa, J.S.; Yamamoto, M.M.W. Comparison of steroidogenic pathways among normoandrogenic and hyperandrogenic polycystic ovary syndrome patients and normal cycling women. J. Obstet. Gynaecol. Res. 2014, 41, 254–263. [Google Scholar] [CrossRef]
- Matoba, K.; Kawanami, D.; Ishizawa, S.; Kanazawa, Y.; Yokota, T.; Utsunomiya, K. Rho-kinase mediates TNF-α-induced MCP-1 expression via p38 MAPK signaling pathway in mesangial cells. Biochem. Biophys. Res. Commun. 2010, 402, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Han, B.; Fan, M.; Wang, N.; Wang, H.; Zhu, H.; Cheng, T.; Zhao, S.; Song, H.; Qiao, J. Oxidative stress increases the 17,20-lyase-catalyzing activity of adrenal P450c17 through p38α in the development of hyperandrogenism. Mol. Cell. Endocrinol. 2019, 484, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Diamanti-Kandarakis, E.; Pappalou, O.; Kandaraki, E.A. The Role of Androgen Excess on Insulin Sensitivity in Women. Front. Horm. Res. Basel 2019, 53, 50–64. [Google Scholar] [CrossRef]
- Vélez, L.M.; Motta, A.B. Association Between Polycystic Ovary Syndrome and Metabolic Syndrome. Curr. Med. Chem. 2014, 21, 3999–4012. [Google Scholar] [CrossRef]
- García-Giménez, J.-L.; Garcés, C.; Romá-Mateo, C.; Pallardó, F.V. Oxidative stress-mediated alterations in histone post-translational modifications. Free Radic. Biol. Med. 2021, 170, 6–18. [Google Scholar] [CrossRef]
- Niu, Y.; DesMarais, T.L.; Tong, Z.; Yao, Y.; Costa, M. Oxidative stress alters global histone modification and DNA methylation. Free Radic. Biol. Med. 2015, 82, 22–28. [Google Scholar] [CrossRef] [Green Version]
- Charążka, B.; Siejka, A. Correlations between serum sirtuin levels and cardiovascular risk factors in women with polycystic ovary syndrome. Adv. Med. Sci. 2022, 67, 123–128. [Google Scholar] [CrossRef]
- Santos, L.; Escande, C.; Denicola, A. Potential Modulation of Sirtuins by Oxidative Stress. Oxidative Med. Cell. Longev. 2016, 2016, 9831825. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Ren, J.; Wang, F.; Pan, M.; Cui, L.; Li, M.; Qu, F. Mitochondrial and glucose metabolic dysfunctions in granulosa cells induce impaired oocytes of polycystic ovary syndrome through Sirtuin 3. Free Radic. Biol. Med. 2022, 187, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Kim, M.H.; Lee, H.J.; Huh, J.-W.; Lee, S.-R.; Lee, H.-S.; Lee, D.-S. Peroxiredoxin 4 inhibits insulin-induced adipogenesis through regulation of ER stress in 3T3-L1 cells. Mol. Cell. Biochem. 2020, 468, 97–109. [Google Scholar] [CrossRef]
- Mehmeti, I.; Lortz, S.; Elsner, M.; Lenzen, S. Peroxiredoxin 4 Improves Insulin Biosynthesis and Glucose-induced Insulin Secretion in Insulin-secreting INS-1E Cells. J. Biol. Chem. 2014, 289, 26904–26913. [Google Scholar] [CrossRef] [Green Version]
- Gateva, A.T.; Velikova, T.V.; Kamenov, Z.A. Peroxiredoxin 4 levels in patients with PCOS and/or obesity. J. Gynecol. Obstet. Hum. Reprod. 2019, 48, 739–743. [Google Scholar] [CrossRef] [PubMed]
- Kreuz, S.; Fischle, W. Oxidative stress signaling to chromatin in health and disease. Epigenomics 2016, 8, 843–862. [Google Scholar] [CrossRef] [Green Version]
- Dabravolski, S.; Nikiforov, N.; Eid, A.; Nedosugova, L.; Starodubova, A.; Popkova, T.; Bezsonov, E.; Orekhov, A. Mitochondrial Dysfunction and Chronic Inflammation in Polycystic Ovary Syndrome. Int. J. Mol. Sci. 2021, 22, 3923. [Google Scholar] [CrossRef]
- Chatterjee, D.; Das, P.; Chakrabarti, O. Mitochondrial Epigenetics Regulating Inflammation in Cancer and Aging. Front. Cell Dev. Biol. 2022, 10. [Google Scholar] [CrossRef]
- Grazioli, S.; Pugin, J. Mitochondrial Damage-Associated Molecular Patterns: From Inflammatory Signaling to Human Diseases. Front. Immunol. 2018, 9, 832. [Google Scholar] [CrossRef] [Green Version]
- Zeng, X.; Huang, Q.; Long, S.L.; Zhong, Q.; Mo, Z. Mitochondrial Dysfunction in Polycystic Ovary Syndrome. DNA Cell Biol. 2020, 39, 1401–1409. [Google Scholar] [CrossRef]
- Zhang, J.; Bao, Y.; Zhou, X.; Zheng, L. Polycystic ovary syndrome and mitochondrial dysfunction. Reprod. Biol. Endocrinol. 2019, 17, 67. [Google Scholar] [CrossRef] [PubMed]
- Yasukawa, T.; Kang, D. An overview of mammalian mitochondrial DNA replication mechanisms. J. Biochem. 2018, 164, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Pasala, M.S.; Prakash, A. Mitochondrial DNA: Epigenetics and environment. Environ. Mol. Mutagen. 2019, 60, 668–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinnery, P.F.; Elliott, H.R.; Hudson, G.; Samuels, D.C.; Relton, C.L. Epigenetics, epidemiology and mitochondrial DNA diseases. Int. J. Epidemiol. 2012, 41, 177–187. [Google Scholar] [CrossRef] [Green Version]
- Lopes, A.F.C. Mitochondrial metabolism and DNA methylation: A review of the interaction between two genomes. Clin. Epigenetics 2020, 12, 182. [Google Scholar] [CrossRef]
- Sato, M.; Sato, K. Maternal inheritance of mitochondrial DNA. Autophagy 2012, 8, 424–425. [Google Scholar] [CrossRef] [Green Version]
- Luo, S.; Valencia, C.A.; Zhang, J.; Lee, N.-C.; Slone, J.; Gui, B.; Wang, X.; Li, Z.; Dell, S.; Brown, J.; et al. Biparental Inheritance of Mitochondrial DNA in Humans. Proc. Natl. Acad. Sci. USA 2018, 115, 13039–13044. [Google Scholar] [CrossRef] [Green Version]
- Holt, I.J. Zen and the art of mitochondrial DNA maintenance. Trends Genet. 2010, 26, 103–109. [Google Scholar] [CrossRef]
- Rebelo, A.P.; Williams, S.L.; Moraes, C.T. In vivo methylation of mtDNA reveals the dynamics of protein–mtDNA interactions. Nucleic Acids Res. 2009, 37, 6701–6715. [Google Scholar] [CrossRef] [Green Version]
- Hao, Z.; Wu, T.; Cui, X.; Zhu, P.; Tan, C.; Dou, X.; Hsu, K.-W.; Lin, Y.-T.; Peng, P.-H.; Zhang, L.-S.; et al. N6-Deoxyadenosine Methylation in Mammalian Mitochondrial DNA. Mol. Cell 2020, 78, 382–395.e8. [Google Scholar] [CrossRef]
- Shock, L.S.; Thakkar, P.V.; Peterson, E.J.; Moran, R.G.; Taylor, S.M. DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proc. Natl. Acad. Sci. USA 2011, 108, 3630–3635. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.-L.; Zhu, S.; He, M.; Chen, D.; Zhang, Q.; Chen, Y.; Yu, G.; Liu, J.; Xie, S.-Q.; Luo, F.; et al. N6-Methyladenine DNA Modification in the Human Genome. Mol. Cell 2018, 71, 306–318.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandhu, J.K.; Waqar, A.; Jain, A.; Joseph, C.; Srivastava, K.; Ochuba, O.; Alkayyali, T.; Ruo, S.W.; Poudel, S. Oxidative Stress in Polycystic Ovarian Syndrome and the Effect of Antioxidant N-Acetylcysteine on Ovulation and Pregnancy Rate. Cureus 2021, 13, e17887. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Han, J.; Wang, X.; Liu, Y.; Zhang, Z. Roles of HIF-1α/BNIP3 mediated mitophagy in mitochondrial dysfunction of letrozole-induced PCOS rats. Histochem. J. 2022, 53, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.; Ren, L.; Zhang, C.; Miao, K.; Tan, K.; Yang, Q.; Hu, Y.; Xi, G.; Luo, G.; Yang, M.; et al. Mitochondrial genome undergoes de novo DNA methylation that protects mtDNA against oxidative damage during the peri-implantation window. Proc. Natl. Acad. Sci. USA 2022, 119, e2201168119. [Google Scholar] [CrossRef] [PubMed]
- Malamouli, M.; Levinger, I.; McAinch, A.J.; Trewin, A.J.; Rodgers, R.J.; Moreno-Asso, A. The mitochondrial profile in women with polycystic ovary syndrome: Impact of exercise. J. Mol. Endocrinol. 2022, 68, R11–R23. [Google Scholar] [CrossRef]
- Reddy, T.V.; Govatati, S.; Deenadayal, M.; Shivaji, S.; Bhanoori, M. Polymorphisms in the TFAM and PGC1-α genes and their association with polycystic ovary syndrome among South Indian women. Gene 2018, 641, 129–136. [Google Scholar] [CrossRef]
- Huang, J.; Wu, S.; Wang, P.; Wang, G. Non-coding RNA Regulated Cross-Talk between Mitochondria and Other Cellular Compartments. Front. Cell Dev. Biol. 2021, 9, 688523. [Google Scholar] [CrossRef]
- Jin, L.; Yang, Q.; Zhou, C.; Liu, L.; Wang, H.; Hou, M.; Wu, Y.; Shi, F.; Sheng, J.; Huang, H. Profiles for long non-coding RNAs in ovarian granulosa cells from women with PCOS with or without hyperandrogenism. Reprod. Biomed. Online 2018, 37, 613–623. [Google Scholar] [CrossRef]
- Cunningham, J.T.; Rodgers, J.T.; Arlow, D.H.; Vazquez, F.; Mootha, V.K.; Puigserver, P. mTOR controls mitochondrial oxidative function through a YY1–PGC-1α transcriptional complex. Nature 2007, 450, 736–740. [Google Scholar] [CrossRef]
- Liu, J.; Wu, D.-C.; Qu, L.-H.; Liao, H.-Q.; Li, M.-X. The role of mTOR in ovarian Neoplasms, polycystic ovary syndrome, and ovarian aging. Clin. Anat. 2018, 31, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.R.; Han, J. Mitochondrial Nucleoid: Shield and Switch of the Mitochondrial Genome. Oxidative Med. Cell. Longev. 2017, 2017, 8060949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, S.C.; Uchiyama, L.F.; Nunnari, J. ER-mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science 2016, 353, aaf5549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzone, R.; Zwergel, C.; Artico, M.; Taurone, S.; Ralli, M.; Greco, A.; Mai, A. The emerging role of epigenetics in human autoimmune disorders. Clin. Epigenetics 2019, 11, 34. [Google Scholar] [CrossRef] [Green Version]
- Hiam, D.; Simar, D.; Laker, R.; Altıntaş, A.; Gibson-Helm, M.; Fletcher, E.; Moreno-Asso, A.; Trewin, A.J.; Barres, R.; Stepto, N.K. Epigenetic Reprogramming of Immune Cells in Women with PCOS Impact Genes Controlling Reproductive Function. J. Clin. Endocrinol. Metab. 2019, 104, 6155–6170. [Google Scholar] [CrossRef]
- Duleba, A.J.; Dokras, A. Is PCOS an inflammatory process? Fertil. Steril. 2012, 97, 7–12. [Google Scholar] [CrossRef] [Green Version]
- Cai, H.; Jin, S.; Lin, J.; Yu, L.; Qian, P.; Chen, W. IL -34 was high in serum of women with polycystic ovary syndrome and may function as potential diagnostic biomarker and therapeutic target. J. Obstet. Gynaecol. Res. 2022, 48, 973–979. [Google Scholar] [CrossRef]
- Christakou, C.; Economou, F.; Livadas, S.; Piperi, C.; Adamopoulos, C.; Marinakis, E.; Diamanti-Kandarakis, E. Strong and positive association of Endothelin-1 with AGEs in PCOS: A causal relationship or a bystander? Hormones 2011, 10, 292–297. [Google Scholar] [CrossRef] [Green Version]
- Dambala, K.; Paschou, S.A.; Michopoulos, A.; Siasos, G.; Goulis, D.G.; Vavilis, D.; Tarlatzis, B.C. Biomarkers of Endothelial Dysfunction in Women with Polycystic Ovary Syndrome. Angiology 2019, 70, 797–801. [Google Scholar] [CrossRef]
- Diamanti-Kandarakis, E.; Alexandraki, K.; Piperi, C.; Protogerou, A.; Katsikis, I.; Paterakis, T.; Lekakis, J.; Panidis, D. Inflammatory and endothelial markers in women with polycystic ovary syndrome. Eur. J. Clin. Investig. 2006, 36, 691–697. [Google Scholar] [CrossRef]
- Garg, D.; Grazi, R.; Lambert-Messerlian, G.M.; Merhi, Z. Correlation between follicular fluid levels of sRAGE and vitamin D in women with PCOS. J. Assist. Reprod. Genet. 2017, 34, 1507–1513. [Google Scholar] [CrossRef] [PubMed]
- Borchiellini, M.; Ummarino, S.; Di Ruscio, A. The Bright and Dark Side of DNA Methylation: A Matter of Balance. Cells 2019, 8, 1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, M.; Wu, Y.; Wang, F.; Huang, Y.; Qian, Y.; Li, J.; Lv, P.; Ying, Y.; Liu, J.; Liu, Y.; et al. Effect of lncRNA MALAT1 on the Granulosa Cell Proliferation and Pregnancy Outcome in Patients with PCOS. Front. Endocrinol. 2022, 13, 825431. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Tang, H.-Y.; Tan, L.; Zhao, D.-M. MALAT1 is involved in the pathophysiological process of PCOS by modulating TGFβ signaling in granulosa cells. Mol. Cell. Endocrinol. 2019, 499, 110589. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhao, Y.; Aguilar, A.; Bernard, D.; Yang, C.-Y. Targeting the MDM2–p53 Protein–Protein Interaction for New Cancer Therapy: Progress and Challenges. Cold Spring Harb. Perspect. Med. 2017, 7, a026245. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Xiang, Y.; Song, Y.; Zhang, D.; Tan, L. MALAT1 downregulation is associated with polycystic ovary syndrome via binding with MDM2 and repressing P53 degradation. Mol. Cell. Endocrinol. 2021, 543, 111528. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, Y.; Cui, X.; He, Q.; Li, H. Down-regulation of MALAT1 aggravates polycystic ovary syndrome by regulating MiR-302d-3p-mediated leukemia inhibitory factor activity. Life Sci. 2021, 277, 119076. [Google Scholar] [CrossRef]
- Komatsu, K.; Koya, T.; Wang, J.; Yamashita, M.; Kikkawa, F.; Iwase, A. Analysis of the Effect of Leukemia Inhibitory Factor on Follicular Growth in Cultured Murine Ovarian Tissue. Biol. Reprod. 2015, 93, 18. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Wang, Y.; Wang, J.; Chen, G.; Wang, H. Long non-coding RNA placenta-specific protein 2 regulates micorRNA-19a/tumor necrosis factor α to participate in polycystic ovary syndrome. Bioengineered 2021, 13, 856–862. [Google Scholar] [CrossRef]
- Thathapudi, S.; Kodati, V.; Erukkambattu, J.; Katragadda, A.; Addepally, U.; Hasan, Q. Tumor Necrosis Factor-Alpha and Polycystic Ovarian Syndrome: A Clinical, Biochemical, and Molecular Genetic Study. Genet. Test. Mol. Biomark. 2014, 18, 605–609. [Google Scholar] [CrossRef] [Green Version]
- Xu, N.; Liu, M.; Wang, Z.; Yang, S.; Zhang, W.; He, S.; Hu, C.; Zhu, H.; Quan, L.; Bai, J. TNF-α is a novel target of miR-19a. Int. J. Oncol. 2011, 38, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Huang, J.; Chen, Y.; Yang, Y.; Li, R.; Li, Y.; Chen, X.; Yang, D. Identification of several circulating microRNAs from a genome-wide circulating microRNA expression profile as potential biomarkers for impaired glucose metabolism in polycystic ovarian syndrome. Endocrine 2016, 53, 280–290. [Google Scholar] [CrossRef]
- Statello, L.; Guo, C.-J.; Chen, L.-L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef]
- Yu, Y.; Li, G.; He, X.; Lin, Y.; Chen, Z.; Lin, X.; Xu, H. MicroRNA-21 regulate the cell apoptosis and cell proliferation of polycystic ovary syndrome (PCOS) granulosa cells through target toll like receptor TLR8. Bioengineered 2021, 12, 5789–5796. [Google Scholar] [CrossRef] [PubMed]
- De Luca, C.; Olefsky, J.M. Inflammation and insulin resistance. FEBS Lett. 2008, 582, 97–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shepherd, P.R.; Withers, D.; Siddle, K. Phosphoinositide 3-kinase: The key switch mechanism in insulin signalling. Biochem. J. 1998, 333, 471–490. [Google Scholar] [CrossRef] [Green Version]
- Cantley, L.C. The Phosphoinositide 3-Kinase Pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef]
- Avruch, J. MAP kinase pathways: The first twenty years. Biochim. Biophys. Acta 2007, 1773, 1150–1160. [Google Scholar] [CrossRef] [Green Version]
- Mardilovich, K.; Pankratz, S.L.; Shaw, L.M. Expression and function of the insulin receptor substrate proteins in cancer. Cell Commun. Signal. 2009, 7, 14. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhu, X.; Bi, X.; Huang, J.; Zhou, L. The Insulin Receptor: An Important Target for the Development of Novel Medicines and Pesticides. Int. J. Mol. Sci. 2022, 23, 7793. [Google Scholar] [CrossRef]
- Sobah, M.L.; Liongue, C.; Ward, A.C. SOCS Proteins in Immunity, Inflammatory Diseases, and Immune-Related Cancer. Front. Med. 2021, 8, 727987. [Google Scholar] [CrossRef] [PubMed]
- Lule, K.O.; Akarsu, E.; Sayiner, Z.A.; Lule, N.O.; Balci, S.O.; Demirel, C.; Bozdag, Z.; Korkmaz, M.; Yilmaz, I. The effects of metformin, pioglitazone, exenatide and exercise on fatty liver in obese diabetic rats: The role of IRS-1 and SOCS-3 molecules. Inflammopharmacology 2022, 30, 243–250. [Google Scholar] [CrossRef]
- Li, X.; Zhang, H.; An, G.; Liu, M.; Han, S.; Jin, Q.; Song, Y.; Lin, Y.; Dong, B.; Wang, S.; et al. S-Nitrosylation of Akt by organic nitrate delays revascularization and the recovery of cardiac function in mice following myocardial infarction. J. Cell. Mol. Med. 2020, 25, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Guo, S. Insulin signaling, resistance, and metabolic syndrome: Insights from mouse models into disease mechanisms. J. Endocrinol. 2013, 220, T1–T23. [Google Scholar] [CrossRef] [Green Version]
- Calcaterra, V.; Verduci, E.; Cena, H.; Magenes, V.; Todisco, C.; Tenuta, E.; Gregorio, C.; De Giuseppe, R.; Bosetti, A.; Di Profio, E.; et al. Polycystic Ovary Syndrome in Insulin-Resistant Adolescents with Obesity: The Role of Nutrition Therapy and Food Supplements as a Strategy to Protect Fertility. Nutrients 2021, 13, 1848. [Google Scholar] [CrossRef]
- Szczuko, M.; Kikut, J.; Szczuko, U.; Szydłowska, I.; Nawrocka-Rutkowska, J.; Ziętek, M.; Verbanac, D.; Saso, L. Nutrition Strategy and Life Style in Polycystic Ovary Syndrome—Narrative Review. Nutrients 2021, 13, 2452. [Google Scholar] [CrossRef]
- Menichini, D.; Ughetti, C.; Monari, F.; Di Vinci, P.L.; Neri, I.; Facchinetti, F. Nutraceuticals and polycystic ovary syndrome: A systematic review of the literature. Gynecol. Endocrinol. 2022, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Pang, Y. Systemic and ovarian inflammation in women with polycystic ovary syndrome. J. Reprod. Immunol. 2022, 151, 103628. [Google Scholar] [CrossRef]
- Velez, L.M.; Seldin, M.; Motta, A.B. Inflammation and reproductive function in women with polycystic ovary syndrome. Biol. Reprod. 2021, 104, 1205–1217. [Google Scholar] [CrossRef]
- Barrea, L.; Muscogiuri, G.; Pugliese, G.; de Alteriis, G.; Colao, A.; Savastano, S. Metabolically Healthy Obesity (MHO) vs. Metabolically Unhealthy Obesity (MUO) Phenotypes in PCOS: Association with Endocrine-Metabolic Profile, Adherence to the Mediterranean Diet, and Body Composition. Nutrients 2021, 13, 3925. [Google Scholar] [CrossRef]
- Samy, N.; Hashim, M.; Sayed, M.; Said, M. Clinical significance of inflammatory markers in polycystic ovary syndrome: Their relationship to insulin resistance and Body Mass Index. Dis. Markers 2009, 26, 163–170. [Google Scholar] [CrossRef] [PubMed]
- White, M.F.; Kahn, C.R. Insulin action at a molecular level—100 years of progress. Mol. Metab. 2021, 52, 101304. [Google Scholar] [CrossRef] [PubMed]
- Haeusler, R.A.; McGraw, T.E.; Accili, D. Biochemical and cellular properties of insulin receptor signalling. Nat. Rev. Mol. Cell Biol. 2017, 19, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Barajas-Olmos, F.; Centeno-Cruz, F.; Zerrweck, C.; Imaz-Rosshandler, I.; Martínez-Hernández, A.; Córdova, E.J.; Rangel-Escareño, C.; Gálvez, F.; Castillo, A.; Maydón, H.; et al. Altered DNA methylation in liver and adipose tissues derived from individuals with obesity and type 2 diabetes. BMC Med. Genet. 2018, 19, 28. [Google Scholar] [CrossRef]
- Maude, H.; Sanchez-Cabanillas, C.; Cebola, I. Epigenetics of Hepatic Insulin Resistance. Front. Endocrinol. 2021, 12, 681356. [Google Scholar] [CrossRef]
- Sang, Q.; Zhang, S.; Zou, S.; Wang, H.; Feng, R.; Li, Q.; Jin, L.; He, L.; Xing, Q.; Wang, L. Quantitative analysis of follistatin (FST) promoter methylation in peripheral blood of patients with polycystic ovary syndrome. Reprod. Biomed. Online 2012, 26, 157–163. [Google Scholar] [CrossRef] [Green Version]
- Sang, Q.; Li, X.; Wang, H.; Wang, H.; Zhang, S.; Feng, R.; Xu, Y.; Li, Q.; Zhao, X.; Xing, Q.; et al. Quantitative Methylation Level of the EPHX1 Promoter in Peripheral Blood DNA Is Associated with Polycystic Ovary Syndrome. PLoS ONE 2014, 9, e88013. [Google Scholar] [CrossRef]
- Shen, H.-R.; Qiu, L.-H.; Zhang, Z.-Q.; Qin, Y.-Y.; Cao, C.; Di, W. Genome-Wide Methylated DNA Immunoprecipitation Analysis of Patients with Polycystic Ovary Syndrome. PLoS ONE 2013, 8, e64801. [Google Scholar] [CrossRef]
- Ting, W.; Yanyan, Q.; Jian, H.; Keqin, H.; Duan, M. The Relationship Between Insulin Resistance and CpG Island Methylation of LMNA Gene in Polycystic Ovary Syndrome. Cell Biophys. 2013, 67, 1041–1047. [Google Scholar] [CrossRef]
- Wang, P.; Zhao, H.; Li, T.; Zhang, W.; Wu, K.; Li, M.; Bian, Y.; Liu, H.; Ning, Y.; Li, G.; et al. Hypomethylation of the LH/Choriogonadotropin Receptor Promoter Region Is a Potential Mechanism Underlying Susceptibility to Polycystic Ovary Syndrome. Endocrinology 2014, 155, 1445–1452. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Zhu, D.; Duan, H.; Ren, A.; Glintborg, D.; Andersen, M.; Skov, V.; Thomassen, M.; Kruse, T.; Tan, Q. Differential DNA methylation patterns of polycystic ovarian syndrome in whole blood of Chinese women. Oncotarget 2016, 8, 20656–20666. [Google Scholar] [CrossRef] [PubMed]
- Sagvekar, P.; Mangoli, V.; Desai, S.; Patil, A.; Mukherjee, S. LINE1 CpG-DNA Hypomethylation in Granulosa Cells and Blood Leukocytes Is Associated with PCOS and Related Traits. J. Clin. Endocrinol. Metab. 2017, 102, 1396–1405. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Zhao, Y.; Ren, Y.; Li, M.; Li, T.; Li, R.; Yu, Y.; Qiao, J. Epigenetic regulation of an adverse metabolic phenotype in polycystic ovary syndrome: The impact of the leukocyte methylation of PPARGC1A promoter. Fertil. Steril. 2016, 107, 467–474.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arpón, A.; Milagro, F.I.; Ramos-Lopez, O.; Mansego, M.L.; Santos, J.L.; Riezu-Boj, J.-I.; Martínez, J.A. Epigenome-wide association study in peripheral white blood cells involving insulin resistance. Sci. Rep. 2019, 9, 2445. [Google Scholar] [CrossRef] [Green Version]
- Day, S.; Coletta, R.L.; Kim, J.Y.; Garcia, L.A.; Campbell, L.E.; Benjamin, T.R.; Roust, L.R.; De Filippis, E.A.; Mandarino, L.J.; Coletta, D.K. Potential epigenetic biomarkers of obesity-related insulin resistance in human whole-blood. Epigenetics 2017, 12, 254–263. [Google Scholar] [CrossRef]
- Liu, J.; Carnero-Montoro, E.; van Dongen, J.; Lent, S.; Nedeljkovic, I.; Ligthart, S.; Tsai, P.-C.; Martin, T.C.; Mandaviya, P.R.; Jansen, R.; et al. An integrative cross-omics analysis of DNA methylation sites of glucose and insulin homeostasis. Nat. Commun. 2019, 10, 2581. [Google Scholar] [CrossRef] [Green Version]
- Chambers, J.C.; Loh, M.; Lehne, B.; Drong, A.; Kriebel, J.; Motta, V.; Wahl, S.; Elliott, H.R.; Rota, F.; Scott, W.R.; et al. Epigenome-wide association of DNA methylation markers in peripheral blood from Indian Asians and Europeans with incident type 2 diabetes: A nested case-control study. Lancet Diabetes Endocrinol. 2015, 3, 526–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dayeh, T.; Tuomi, T.; Almgren, P.; Perfilyev, A.; Jansson, P.-A.; de Mello, V.D.; Pihlajamäki, J.; Vaag, A.; Groop, L.; Nilsson, E.; et al. DNA methylation of loci within ABCG1 and PHOSPHO1 in blood DNA is associated with future type 2 diabetes risk. Epigenetics 2016, 11, 482–488. [Google Scholar] [CrossRef] [Green Version]
- Wahl, S.; Drong, A.; Lehne, B.; Loh, M.; Scott, W.R.; Kunze, S.; Tsai, P.-C.; Ried, J.S.; Zhang, W.; Yang, Y.; et al. Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity. Nature 2016, 541, 81–86. [Google Scholar] [CrossRef] [Green Version]
- Cardona, A.; Day, F.R.; Perry, J.R.B.; Loh, M.; Chu, A.Y.; Lehne, B.; Paul, D.S.; Lotta, L.A.; Stewart, I.D.; Kerrison, N.D.; et al. Epigenome-Wide Association Study of Incident Type 2 Diabetes in a British Population: EPIC-Norfolk Study. Diabetes 2019, 68, 2315–2326. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Goldberg, J.; Bremner, J.D.; Vaccarino, V. Global DNA Methylation Is Associated with Insulin Resistance. Diabetes 2012, 61, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Nano, J.; Ding, J.; Zheng, Y.; Hennein, R.; Liu, C.; Speliotes, E.K.; Huan, T.; Song, C.; Mendelson, M.M.; et al. A Peripheral Blood DNA Methylation Signature of Hepatic Fat Reveals a Potential Causal Pathway for Nonalcoholic Fatty Liver Disease. Diabetes 2019, 68, 1073–1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nano, J.; Ghanbari, M.; Wang, W.; de Vries, P.; Dhana, K.; Muka, T.; Uitterlinden, A.G.; van Meurs, J.B.; Hofman, A.; Franco, O.; et al. Epigenome-Wide Association Study Identifies Methylation Sites Associated with Liver Enzymes and Hepatic Steatosis. Gastroenterology 2017, 153, 1096–1106.e2. [Google Scholar] [CrossRef] [PubMed]
- Lambertini, L.; Saul, S.R.; Copperman, A.B.; Hammerstad, S.S.; Yi, Z.; Zhang, W.; Tomer, Y.; Kase, N. Intrauterine Reprogramming of the Polycystic Ovary Syndrome: Evidence from a Pilot Study of Cord Blood Global Methylation Analysis. Front. Endocrinol. 2017, 8, 352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.-Y.; Sun, C.-X.; Liu, Y.-K.; Li, Y.; Wang, L.; Zhang, W. Promoter Methylation of CYP19A1 Gene in Chinese Polycystic Ovary Syndrome Patients. Gynecol. Obstet. Investig. 2013, 76, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.-Y.; Sun, C.-X.; Liu, Y.-K.; Li, Y.; Wang, L.; Zhang, W. Genome-wide screen of ovary-specific DNA methylation in polycystic ovary syndrome. Fertil. Steril. 2015, 104, 145–153.e6. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-X.; Wei, J.-Z.; Jiao, J.; Jiang, S.-Y.; Yu, D.-H.; Li, D. Genome-wide DNA methylation and gene expression patterns provide insight into polycystic ovary syndrome development. Oncotarget 2014, 5, 6603–6610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, F.; Wang, F.-F.; Yin, R.; Ding, G.-L.; El-Prince, M.; Gao, Q.; Shi, B.-W.; Pan, H.-H.; Huang, Y.-T.; Jin, M.; et al. A molecular mechanism underlying ovarian dysfunction of polycystic ovary syndrome: Hyperandrogenism induces epigenetic alterations in the granulosa cells. Klin. Wochenschr. 2012, 90, 911–923. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Bao, X.; Peng, Z.; Wang, L.; Du, L.; Niu, W.; Sun, Y. Comprehensive analysis of genome-wide DNA methylation across human polycystic ovary syndrome ovary granulosa cell. Oncotarget 2016, 7, 27899–27909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, L.-L.; Xie, J.-K.; Cui, J.-Q.; Wei, D.; Yin, B.-L.; Zhang, Y.-N.; Chen, Y.-H.; Han, X.; Wang, Q.; Zhang, C.-L. Promoter methylation of yes-associated protein (YAP1) gene in polycystic ovary syndrome. Medicine 2017, 96, e5768. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, E.A.; Matte, A.; Perfilyev, A.; de Mello, V.; Käkelä, P.; Pihlajamäki, J.; Ling, C. Epigenetic Alterations in Human Liver From Subjects With Type 2 Diabetes in Parallel With Reduced Folate Levels. J. Clin. Endocrinol. Metab. 2015, 100, E1491–E1501. [Google Scholar] [CrossRef] [PubMed]
- Kirchner, H.; Sinha, I.; Gao, H.; Ruby, M.A.; Schönke, M.; Lindvall, J.M.; Barrès, R.; Krook, A.; Näslund, E.; Dahlman-Wright, K.; et al. Altered DNA methylation of glycolytic and lipogenic genes in liver from obese and type 2 diabetic patients. Mol. Metab. 2016, 5, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Abderrahmani, A.; Yengo, L.; Caiazzo, R.; Canouil, M.; Cauchi, S.; Raverdy, V.; Plaisance, V.; Pawlowski, V.; Lobbens, S.; Maillet, J.; et al. Increased Hepatic PDGF-AA Signaling Mediates Liver Insulin Resistance in Obesity-Associated Type 2 Diabetes. Diabetes 2018, 67, 1310–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotta, K.; Kitamoto, T.; Kitamoto, A.; Ogawa, Y.; Honda, Y.; Kessoku, T.; Yoneda, M.; Imajo, K.; Tomeno, W.; Saito, S.; et al. Identification of the genomic region under epigenetic regulation during non-alcoholic fatty liver disease progression. Hepatol. Res. 2017, 48, E320–E334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, S.; Yang, H.; Moylan, C.A.; Pang, H.; Dellinger, A.; Abdelmalek, M.; Garrett, M.E.; Ashley-Koch, A.; Suzuki, A.; Tillmann, H.L.; et al. Relationship Between Methylome and Transcriptome in Patients with Nonalcoholic Fatty Liver Disease. Gastroenterology 2013, 145, 1076–1087. [Google Scholar] [CrossRef] [Green Version]
- De Mello, V.D.; Matte, A.; Perfilyev, A.; Männistö, V.; Rönn, T.; Nilsson, E.; Käkelä, P.; Ling, C.; Pihlajamäki, J. Human liver epigenetic alterations in non-alcoholic steatohepatitis are related to insulin action. Epigenetics 2017, 12, 287–295. [Google Scholar] [CrossRef] [Green Version]
- Bysani, M.; Perfilyev, A.; de Mello, V.D.; Rönn, T.; Nilsson, E.; Pihlajamäki, J.; Ling, C. Epigenetic alterations in blood mirror age-associated DNA methylation and gene expression changes in human liver. Epigenomics 2017, 9, 105–122. [Google Scholar] [CrossRef] [Green Version]
- Kokosar, M.; Benrick, A.; Perfilyev, A.; Fornes, R.; A Nilsson, E.; Maliqueo, M.; Behre, C.J.; Sazonova, A.; Ohlsson, C.; Ling, C.; et al. Epigenetic and Transcriptional Alterations in Human Adipose Tissue of Polycystic Ovary Syndrome. Sci. Rep. 2016, 6, 22883. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, E.; Benrick, A.; Kokosar, M.; Krook, A.; Lindgren, E.; Källman, T.; Martis, M.M.; Højlund, K.; Ling, C.; Stener-Victorin, E. Transcriptional and Epigenetic Changes Influencing Skeletal Muscle Metabolism in Women with Polycystic Ovary Syndrome. J. Clin. Endocrinol. Metab. 2018, 103, 4465–4477. [Google Scholar] [CrossRef] [Green Version]
- Yao, Z.-G.; Liu, Y.; Zhang, L.; Huang, L.; Ma, C.-M.; Xu, Y.-F.; Zhu, H.; Qin, C. Co-location of HDAC2 and Insulin Signaling Components in the Adult Mouse Hippocampus. Cell. Mol. Neurobiol. 2012, 32, 1337–1342. [Google Scholar] [CrossRef]
- Albosta, M.; Bakke, J. Intermittent fasting: Is there a role in the treatment of diabetes? A review of the literature and guide for primary care physicians. Clin. Diabetes Endocrinol. 2021, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Sun, S.; Wei, X.; Zhang, M.; Chen, Y.; Mao, X.; Chen, G.; Liu, C. Short-term moderate caloric restriction in a high-fat diet alleviates obesity via AMPK/SIRT1 signaling in white adipocytes and liver. Food Nutr. Res. 2022, 66. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; Chen, S.; Han, Y.; Zhang, D.; Tan, Y.; He, Y.; Liu, M. Ameliorating Inflammation in Insulin-resistant Rat Adipose Tissue with Abdominal Massage Regulates SIRT1/NF-κB Signaling. Cell Biochem. Biophys. 2022, 80, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.Y.; Christensen, S.M.; Duong, N.; Tran, Q.-A.; Xiong, H.M.; Huang, J.; James, S.; Vallabh, D.; Talbott, G.; Rose, M.; et al. Sirt3 Pharmacologically Promotes Insulin Sensitivity through PI3/AKT/mTOR and Their Downstream Pathway in Adipocytes. Int. J. Mol. Sci. 2022, 23, 3740. [Google Scholar] [CrossRef]
- Cirillo, F.; Catellani, C.; Lazzeroni, P.; Sartori, C.; Nicoli, A.; Amarri, S.; La Sala, G.B.; Street, M.E. MiRNAs Regulating Insulin Sensitivity Are Dysregulated in Polycystic Ovary Syndrome (PCOS) Ovaries and Are Associated with Markers of Inflammation and Insulin Sensitivity. Front. Endocrinol. 2019, 10, 879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vijayakumar, E.C.; Bhatt, L.; Prabhavalkar, K.S. High Mobility Group Box-1 (HMGB1): A Potential Target in Therapeutics. Curr. Drug Targets 2019, 20, 1474–1485. [Google Scholar] [CrossRef]
- Guo, J.; Zhang, R.; Zhao, Y.; Wang, J. MiRNA-29c-3p Promotes Intestinal Inflammation via Targeting Leukemia Inhibitory Factor in Ulcerative Colitis. J. Inflamm. Res. 2021, 14, 2031–2043. [Google Scholar] [CrossRef]
- Wang, R.; Li, Q.; He, Y.; Yang, Y.; Ma, Q.; Li, C. miR-29c-3p inhibits microglial NLRP3 inflammasome activation by targeting NFAT5 in Parkinson’s disease. Genes Cells 2020, 25, 364–374. [Google Scholar] [CrossRef]
- Li, Z.; Yi, N.; Chen, R.; Meng, Y.; Wang, Y.; Liu, H.; Cao, W.; Hu, Y.; Gu, Y.; Tong, C.; et al. miR-29b-3p protects cardiomyocytes against endotoxin-induced apoptosis and inflammatory response through targeting FOXO3A. Cell. Signal. 2020, 74, 109716. [Google Scholar] [CrossRef]
- Chen, H.; Fu, Y.; Guo, Z.; Zhou, X. MicroRNA-29c-3p participates in insulin function to modulate polycystic ovary syndrome via targeting Forkhead box O 3. Bioengineered 2022, 13, 4361–4371. [Google Scholar] [CrossRef]
- Qin, Y.; Wang, Y.; Zhao, H.; Yang, Z.; Kang, Y. Aberrant miRNA-mRNA regulatory network in polycystic ovary syndrome is associated with markers of insulin sensitivity and inflammation. Ann. Transl. Med. 2021, 9, 1405. [Google Scholar] [CrossRef] [PubMed]
- Sathishkumar, C.; Prabu, P.; Mohan, V.; Balasubramanyam, M. Linking a role of lncRNAs (long non-coding RNAs) with insulin resistance, accelerated senescence, and inflammation in patients with type 2 diabetes. Hum. Genom. 2018, 12, 41. [Google Scholar] [CrossRef] [PubMed]
- Reyes-García, J.; Montaño, L.M.; Carbajal-García, A.; Wang, Y.-X. Sex Hormones and Lung Inflammation. Adv. Exp. Med. Biol. 2021, 1304, 259–321. [Google Scholar] [CrossRef] [PubMed]
- Trigunaite, A.; Dimo, J.; Jørgensen, T.N. Suppressive effects of androgens on the immune system. Cell. Immunol. 2015, 294, 87–94. [Google Scholar] [CrossRef]
- Klein, S.L.; Flanagan, K.L. Sex differences in immune responses. Nat. Rev. Immunol. 2016, 16, 626–638. [Google Scholar] [CrossRef] [PubMed]
- Pace, S.; Werz, O. Impact of Androgens on Inflammation-Related Lipid Mediator Biosynthesis in Innate Immune Cells. Front. Immunol. 2020, 11, 1356. [Google Scholar] [CrossRef]
- Bupp, M.R.G.; Jorgensen, T.N. Androgen-Induced Immunosuppression. Front. Immunol. 2018, 9, 794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angum, F.; Khan, T.; Kaler, J.; Siddiqui, L.; Hussain, A. The Prevalence of Autoimmune Disorders in Women: A Narrative Review. Cureus 2020, 12, e8094. [Google Scholar] [CrossRef] [PubMed]
- Fox, C.W.; Zhang, L.; Sohni, A.; Doblado, M.; Wilkinson, M.F.; Chang, R.J.; Duleba, A.J. Inflammatory Stimuli Trigger Increased Androgen Production and Shifts in Gene Expression in Theca-Interstitial Cells. Endocrinology 2019, 160, 2946–2958. [Google Scholar] [CrossRef] [Green Version]
- González, F.; Sia, C.L.; Bearson, D.M.; Blair, H.E. Hyperandrogenism Induces a Proinflammatory TNFα Response to Glucose Ingestion in a Receptor-Dependent Fashion. J. Clin. Endocrinol. Metab. 2014, 99, E848–E854. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Ruan, Y.C.; Yang, Y.-J.; Wang, K.; Liang, S.-S.; Han, Y.-B.; Teng, X.-M.; Yang, J.-Z. Follicular hyperandrogenism downregulates aromatase in luteinized granulosa cells in polycystic ovary syndrome women. Reproduction 2015, 150, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Qiao, J. Association of Insulin Resistance and Elevated Androgen Levels with Polycystic Ovarian Syndrome (PCOS): A Review of Literature. J. Health Eng. 2022, 2022, 9240569. [Google Scholar] [CrossRef]
- Combs, J.C.; Hill, M.J.; DeCherney, A.H. Polycystic Ovarian Syndrome Genetics and Epigenetics. Clin. Obstet. Gynecol. 2020, 64, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Shamsi, M.; Ghazavi, A.; Saeedifar, A.M.; Mosayebi, G.; Pour, S.K.; Ganji, A. The immune system’s role in PCOS. Mol. Biol. Rep. 2022, 49, 10689–10702. [Google Scholar] [CrossRef] [PubMed]
- Panghiyangani, R.; Soeharso, P.; Andrijono; Suryandari, D.A.; Wiweko, B.; Kurniati, M.; Pujianto, D.A. CYP19A1 gene expression in patients with polycystic ovarian syndrome. J. Hum. Reprod. Sci. 2020, 13, 100–103. [Google Scholar] [CrossRef]
- Catteau-Jonard, S.; Jamin, S.; Leclerc, A.; Gonzalès, J.; Dewailly, D.; Di Clemente, N. Anti-Mullerian Hormone, Its Receptor, FSH Receptor, and Androgen Receptor Genes Are Overexpressed by Granulosa Cells from Stimulated Follicles in Women with Polycystic Ovary Syndrome. J. Clin. Endocrinol. Metab. 2008, 93, 4456–4461. [Google Scholar] [CrossRef]
- Liu, X.; Qiao, P.; Jiang, A.; Jiang, J.; Han, H.; Wang, L.; Ren, C. Paracrine Regulation of Steroidogenesis in Theca Cells by Granulosa Cells Derived from Mouse Preantral Follicles. BioMed Res. Int. 2015, 2015, 9240569. [Google Scholar] [CrossRef] [Green Version]
- Bao, D.; Li, M.; Zhou, D.; Zhuang, C.; Ge, Z.; Wei, Q.; Zhang, L. miR-130b-3p is high-expressed in polycystic ovarian syndrome and promotes granulosa cell proliferation by targeting SMAD4. J. Steroid Biochem. Mol. Biol. 2021, 209, 105844. [Google Scholar] [CrossRef]
- Liu, Y.; Du, S.-Y.; Ding, M.; Dou, X.; Zhang, F.-F.; Wu, Z.-Y.; Qian, S.-W.; Zhang, W.; Tang, Q.-Q.; Xu, C.-J. The BMP4-Smad signaling pathway regulates hyperandrogenism development in a female mouse model. J. Biol. Chem. 2017, 292, 11740–11750. [Google Scholar] [CrossRef] [Green Version]
- Maucher, D.; Schmidt, B.; Schumann, J. Loss of Endothelial Barrier Function in the Inflammatory Setting: Indication for a Cytokine-Mediated Post-Transcriptional Mechanism by Virtue of Upregulation of miRNAs miR-29a-3p, miR-29b-3p, and miR-155-5p. Cells 2021, 10, 2843. [Google Scholar] [CrossRef]
- Lin, H.-Y.; Yang, Y.-L.; Wang, P.-W.; Wang, F.-S.; Huang, Y.-H. The Emerging Role of MicroRNAs in NAFLD: Highlight of MicroRNA-29a in Modulating Oxidative Stress, Inflammation, and Beyond. Cells 2020, 9, 1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, C.-H.; Chang, C.-M.; Li, H.-Y.; Shen, H.-Y.; Lieu, F.-K.; Wang, P.S.-G. Dysregulated immunological and metabolic functions discovered by a polygenic integrative analysis for PCOS. Reprod. Biomed. Online 2020, 40, 160–167. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.; Zhang, Y.; Li, X.; Cui, P.; Li, J.; Brännström, M.; Shao, L.R.; Billig, H. Alterations of endometrial epithelial–mesenchymal transition and MAPK signalling components in women with PCOS are partially modulated by metformin in vitro. Mol. Hum. Reprod. 2020, 26, 312–326. [Google Scholar] [CrossRef]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204–7218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, J.; Li, X.; Liu, P.; Li, K.; Sha, L.; Yang, X.; Zhu, L.; Wang, Z.; Dong, Y.; Zhang, L.; et al. Inulin and metformin ameliorate polycystic ovary syndrome via anti-inflammation and modulating gut microbiota in mice. Endocr. J. 2019, 66, 859–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duleba, A.J. Medical management of metabolic dysfunction in PCOS. Steroids 2012, 77, 306–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fruzzetti, F.; Capozzi, A.; Canu, A.; Lello, S. Treatment with d-chiro-inositol and alpha lipoic acid in the management of polycystic ovary syndrome. Gynecol. Endocrinol. 2019, 35, 506–510. [Google Scholar] [CrossRef]
- Mancini, A.; Bruno, C.; Vergani, E.; D′abate, C.; Giacchi, E.; Silvestrini, A. Oxidative Stress and Low-Grade Inflammation in Polycystic Ovary Syndrome: Controversies and New Insights. Int. J. Mol. Sci. 2021, 22, 1667. [Google Scholar] [CrossRef]
- Szukiewicz, D.; Uilenbroek, J.T. Polycystic ovary syndrome--searching for an animal model. J. Med. 1998, 29, 259–275. [Google Scholar]
- Corrie, L.; Gulati, M.; Singh, S.K.; Kapoor, B.; Khursheed, R.; Awasthi, A.; Vishwas, S.; Chellappan, D.K.; Gupta, G.; Jha, N.K.; et al. Recent updates on animal models for understanding the etiopathogenesis of polycystic ovarian syndrome. Life Sci. 2021, 280, 119753. [Google Scholar] [CrossRef]
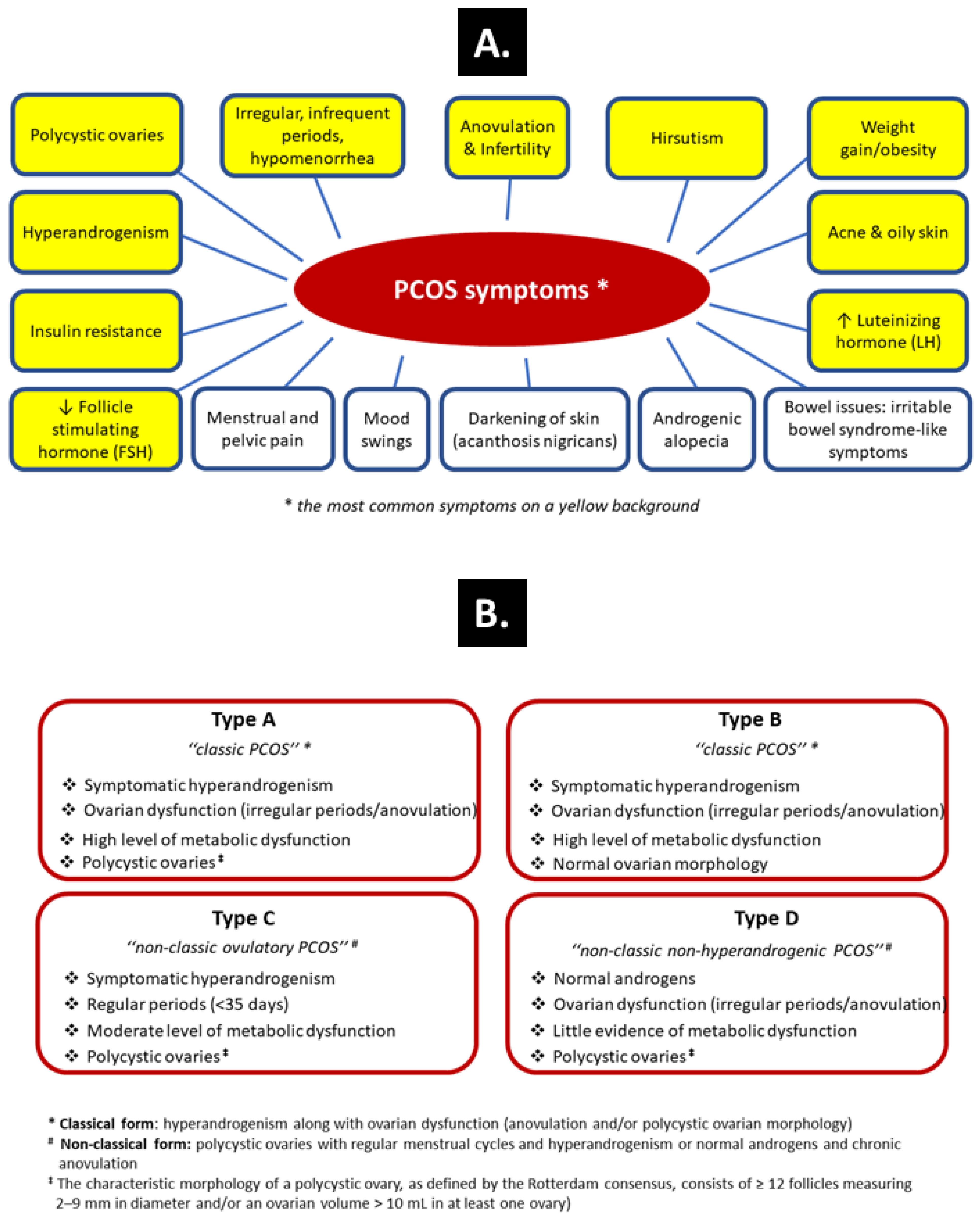


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tissue | Exemplary Loci with Differential Methylation | References |
|---|---|---|
| Peripheral blood | LY6G6F, KCTD21, ADCY9, RABL2B, ZNF611, VASH1, FST, LMNA, PPARGC1A, L-1, TMSB15B, RPF1, DNA2, EPHA8, LHCGR, EPHX1, JAML, KBTBD12, SLC29A1, GPR176, MYOZ2, PIGT, C2CD4B, PCDHA7, HMGA1, PCDH18 | Sang et al., 2013, 2014 [156,157]; Shen et al., 2013 [158]; Ting et al., 2013 [159]; Wang et al., 2014 [160]; Li et al., 2017 [161]; Sagvekar et al., 2017 [162]; Zhao et al., 2017 [163] |
| CLCA4, LECT1, CXCR1, HDAC4, IGFR1, LEPR, ABCG1, SH3RF3, MAN2C1 | Arpon et al., 2019 [164] | |
| SLC19A1, EFNA2 | Day et al., 2017 [165] | |
| LETM1, RBM20, IRS2, MAN2A2, 1q25.3, FCRL6, SLAMF1, APOBEC3H, 15q26.1 | Liu et al., 2019 [166] | |
| ABCG1, PHOSPHO1, SOCS3, SREBF1, TXNIP | Chambers et al., 2015 [167] | |
| ABCG1, PHOSPHO1 | Dayeh et al., 2016 [168] | |
| ABCG1, PHOSPHO1, SREBF, NFATC2IP, KLHL18, FTH1P20 | Wahl et al., 2017 [169] | |
| ABCG1, SREBF1, TXNIP, PROC, SLC43A1, PHGDH, MAN2A2 | Cardona et al., 2019 [170] | |
| Alu element repeats methylation | Zhao et al., 2012 [171] | |
| SLC7A11, SLC1A5, SLC43A1, PHGDH, PSORS1C1, SREBF1, ABCG1 | Ma et al., 2019 [172] | |
| SLC7A11, SLC43A1, SLC1A5, PHGDH, PSORS1C1, SREBF1, ANKS3 | Nano et al., 2017 [173] | |
| Umbilical cord blood | PRKN, PAX6, B4GALT7, MEST, CACNA2D2, RGMA, PRDM10, ESR1, APP, RBPMS LHCGR, CASP10, SPHK1, PCSK6, ARHGAP45, MIB2 | Lambertini et al., 2017 [174] |
| Whole ovarian tissue | FBN1, NAV2, PRDM1, RNF213, SSBP2, TNIK, ZFAND3, ZNF503, SLC2A8, NRIP1, IGF2BP2, CYP19A1, AMHR2, SNURF, SUMO3, PNMA6A, ADRA1D, SCML1, C2CD6, NR0B1, INSR, AMH, SPANXD, TUBA3E, FAM47B, MAB21L1, RBM3 | Yu et al., 2013, 2015 [175,176]; Wang et al., 2014 [177] |
| Granulosa cells | MATN4, DLGAP2, CDH13, GAREM2, GSC, ANKRD34C, ATP8B2 and PPARG, L-1, LHCGR, SMG6, CCR5, LHB, NTN1, ARFGAP1, MDGA1, NCOR1, YAP1, CD9, NR4A1, EDN2, BNIP3, LIF | Qu et al., 2012 [178]; Wang et al., 2014 [160]; Xu et al., 2016 [179]; Jiang et al., 2017 [180]; Sagvekar et al., 2017 [162] |
| Liver | GRB10, PPP1R1A, IGFBP2, ABCC3, MOGAT1, PRDM16 | Nilsson et al., 2015 [181] |
| PRKCE, PDGFA | Kirchner et al., 2016 [182] | |
| PDGFA | Abderrahmani et al., 2018 [183] | |
| SYT7, LTBR, CATSPER2, LPAL2, NCALD, ZDHHC11, LGTN, OXT, PRSS21 | Barajas-Olmos et al., 2018 [154] | |
| IGFBP2, IGF1, PRKCE, PGC1A, SREBF2, FOXA1, FOXA2, ZNF274 | Hotta et al., 2018 [184] | |
| FGFR2, MAT1A, CASP1, COL1A1, COL1A2, COL4A1, COL4A2, LAMA4, LAMB1, CTGF, PDGFA, CCR7, CCL5, STAT1, TNFAIP8 | Murphy et al., 2013 [185] | |
| PPARGC1A, DNMT1, HDAC9, ALKBH5, LDHB, COL4A1, ARL4C, SEMA3E, ITGB4 | de Mello et al., 2017 [186] | |
| E2F1, TFAP2A NFKB1, HNF4A, HNF1A, SREBF1, TCF4, ETS1 | Bysani et al., 2017 [187] | |
| FGFR2, IGF1, MTHFD2, PTGFRN, ZBTB38, MGMT, FBLIM1, CYR61, NQO1 | Hotta et al., 2018 [184] | |
| Subcutaneous adipose tissue | ZZEF1, TPT1, STUB1, DMAP1, RAB5B, PPARG, SVEP1, SAV1, RORA, RAB6A, CNST, PUM1, DIP2C, SNX8, SRGAP3, ZFHX3, OR52W1, BBX | Kokosar et al., 2016 [188] |
| Skeletal muscle | CST3, SPRTN, COL1A1, SCMH1, VAT1, CSPP1, ERP29, ADK, KLF10, HEATR3, HJV, MAP2K6, FOXO3 | Nilsson et al., 2018 [189] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szukiewicz, D.; Trojanowski, S.; Kociszewska, A.; Szewczyk, G. Modulation of the Inflammatory Response in Polycystic Ovary Syndrome (PCOS)—Searching for Epigenetic Factors. Int. J. Mol. Sci. 2022, 23, 14663. https://doi.org/10.3390/ijms232314663
Szukiewicz D, Trojanowski S, Kociszewska A, Szewczyk G. Modulation of the Inflammatory Response in Polycystic Ovary Syndrome (PCOS)—Searching for Epigenetic Factors. International Journal of Molecular Sciences. 2022; 23(23):14663. https://doi.org/10.3390/ijms232314663
Chicago/Turabian StyleSzukiewicz, Dariusz, Seweryn Trojanowski, Anna Kociszewska, and Grzegorz Szewczyk. 2022. "Modulation of the Inflammatory Response in Polycystic Ovary Syndrome (PCOS)—Searching for Epigenetic Factors" International Journal of Molecular Sciences 23, no. 23: 14663. https://doi.org/10.3390/ijms232314663
APA StyleSzukiewicz, D., Trojanowski, S., Kociszewska, A., & Szewczyk, G. (2022). Modulation of the Inflammatory Response in Polycystic Ovary Syndrome (PCOS)—Searching for Epigenetic Factors. International Journal of Molecular Sciences, 23(23), 14663. https://doi.org/10.3390/ijms232314663