
Automated Path Searching Reveals the Mechanism of Hydrolysis Enhancement by T4 Lysozyme Mutants


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Initial Structures and Transition Paths for Three Variants of T4L
2.2. Path Optimization by TAPS
2.2.1. Flowchart of TAPS Optimization
2.2.2. Path Optimization Convergence Check
- (a)
- (b)
- the visualization of the optimization process via the projection of all paths on a low-dimensional space generated by multidimensional scaling (MDS) [66] (more details are provided in the Supplementary Materials and Ref. [34]).
2.3. Free-Energy Calculation
3. Results
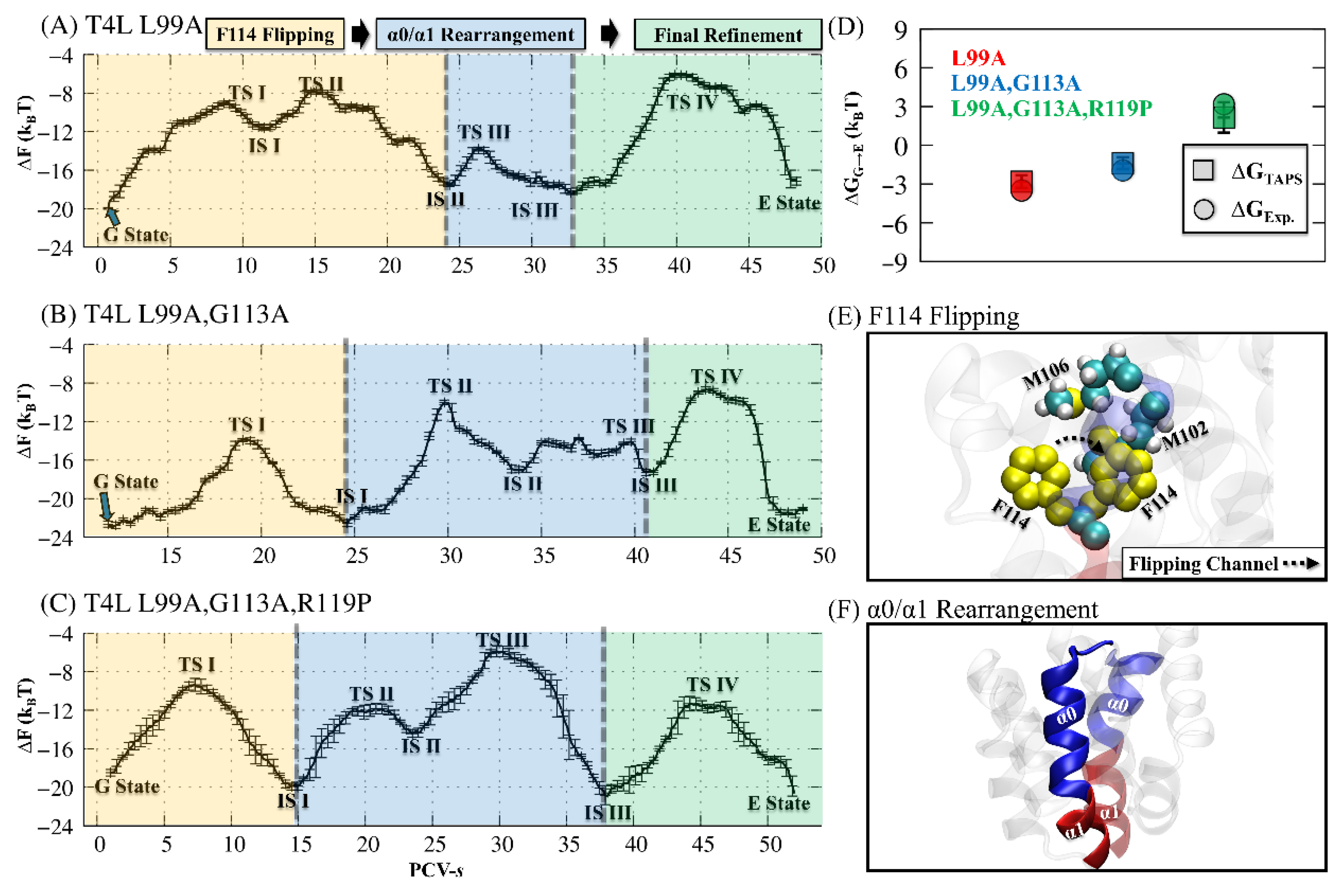
3.1. The Main Activation Mechanism for the Three T4L Variants
3.2. Enhancing the Hydrolysis of T4L through Three Substitutions
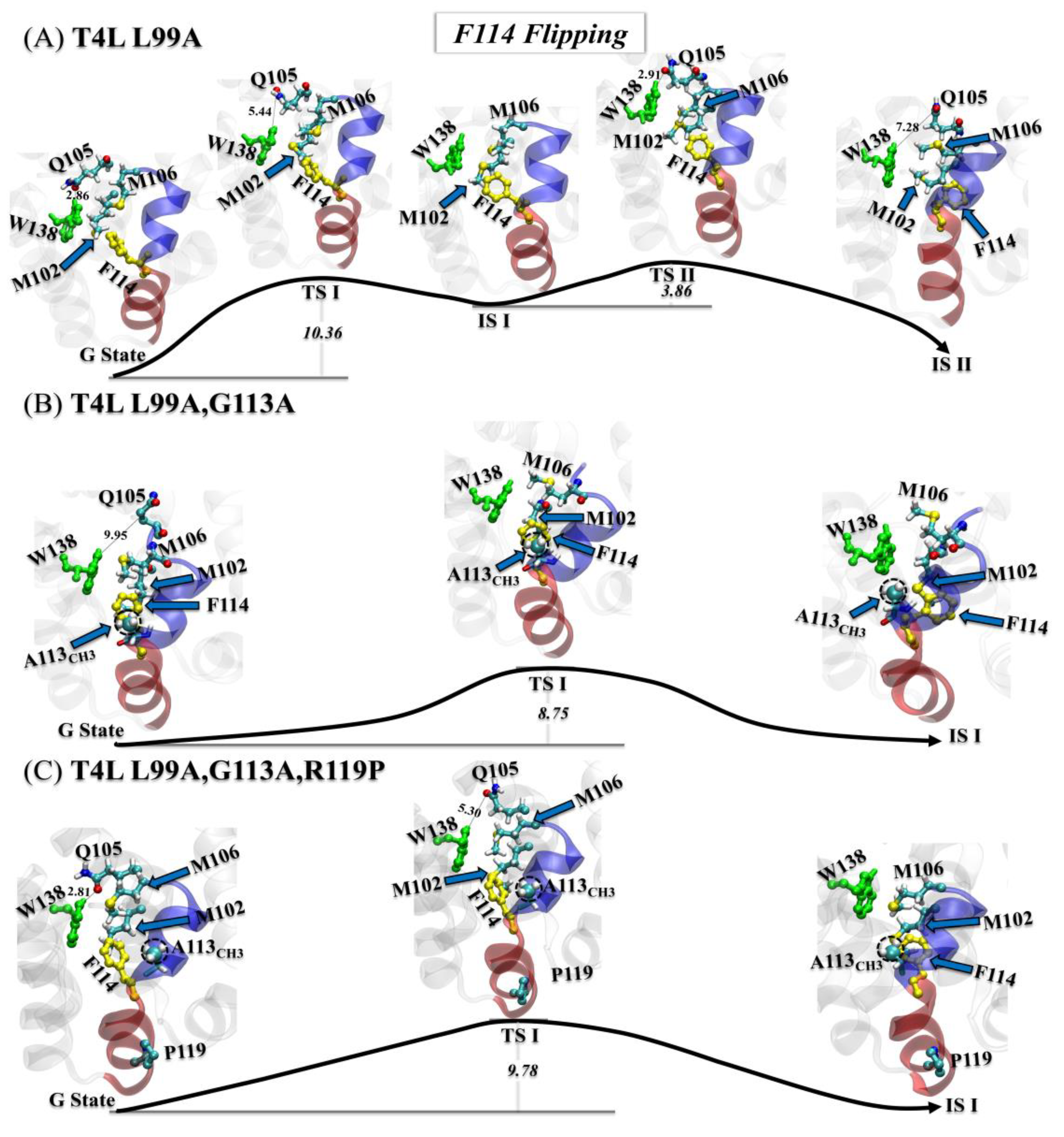
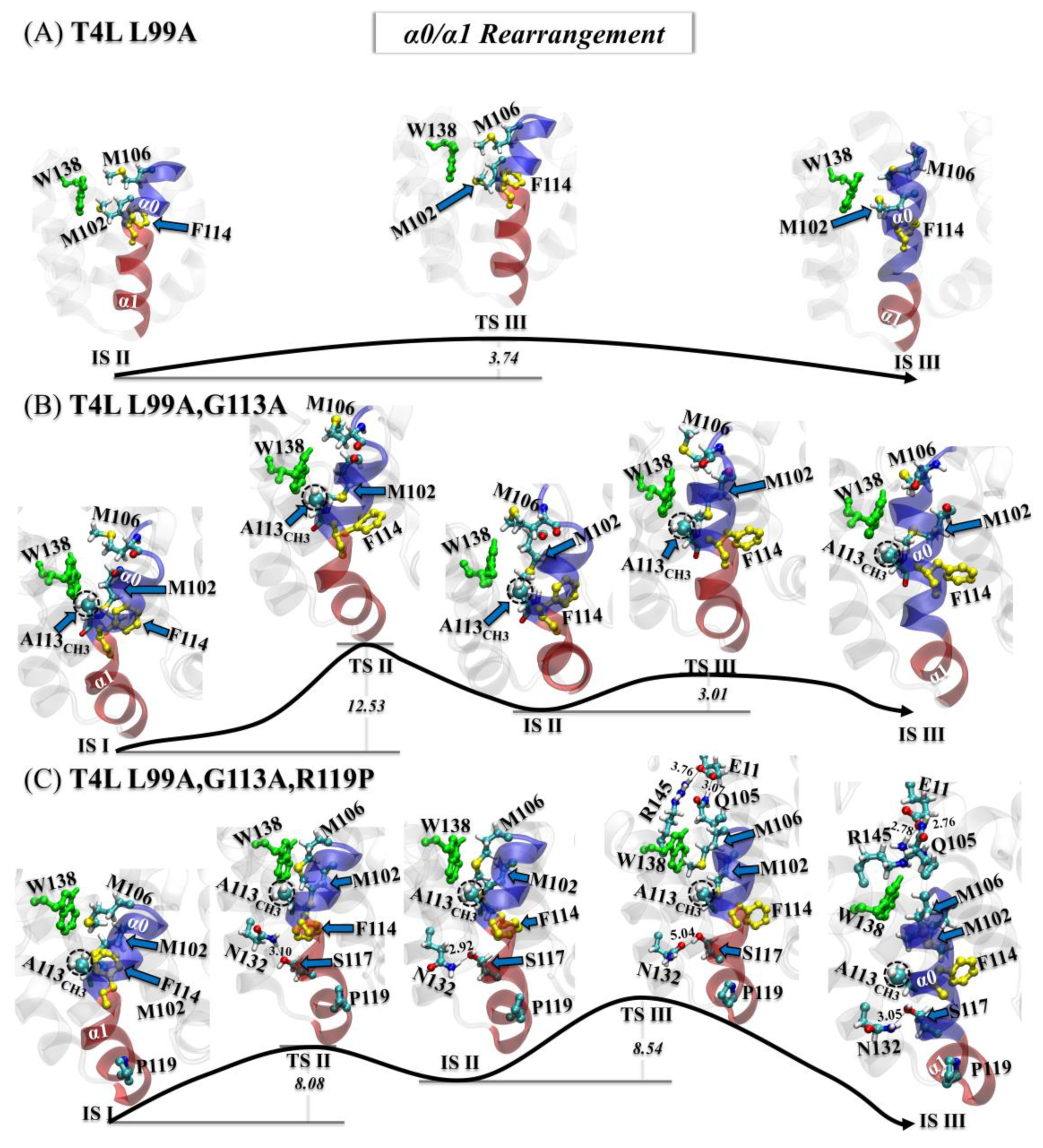
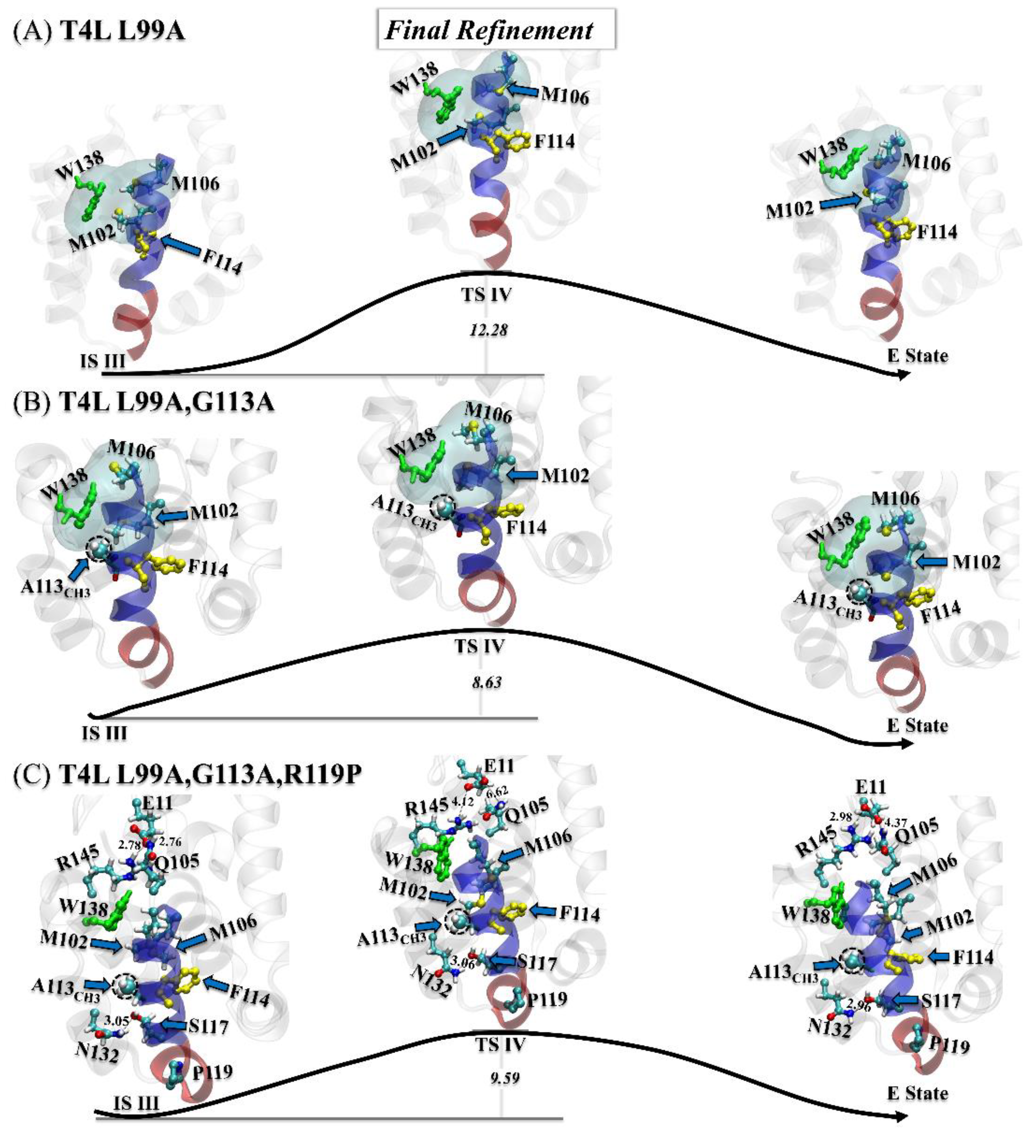
3.3. Transition Mechanism of T4L
3.3.1. Step One: F114 Flipping
3.3.2. Step Two: α0/α1 Rearrangement
3.3.3. Step Three: Final Refinement
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eriksson, A.E.; Baase, W.A.; Wozniak, J.A.; Matthews, B.W. A cavity-containing mutant of T4 lysozyme is stabilized by buried benzene. Nature 1992, 355, 371–373. [Google Scholar] [CrossRef]
- McCarter, J.D.; Withers, G.S. Mechanisms of enzymatic glycoside hydrolysis. Curr. Opin. Struct. Biol. 1994, 4, 885–892. [Google Scholar] [CrossRef]
- Matthews, B.W. Studies on protein stability with T4 lysozyme. Adv. Protein Chem. 1995, 46, 249–278. [Google Scholar] [CrossRef]
- Kuroki, R.; Weaver, L.H.; Matthews, B.W. Structural basis of the conversion of T4 lysozyme into a transglycosidase by reengineering the active site. Proc. Natl. Acad. Sci. USA 1999, 96, 8949–8954. [Google Scholar] [CrossRef] [Green Version]
- Aminlari, L.; Mohammadi Hashemi, M.; Aminlari, M. Modified lysozymes as novel broad spectrum natural antimicrobial agents in foods. J. Food Sci. 2014, 79, R1077–R1090. [Google Scholar] [CrossRef]
- Chien, E.Y.T.; Liu, W.; Zhao, Q.; Katritch, V.; Han, G.W.; Hanson, M.A.; Shi, L.; Newman, A.H.; Javitch, J.A.; Cherezov, V.; et al. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 2010, 330, 1091–1095. [Google Scholar] [CrossRef] [Green Version]
- Granier, S.; Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Weis, W.I.; Kobilka, B.K. Structure of the δ-opioid receptor bound to naltrindole. Nature 2012, 485, 400–404. [Google Scholar] [CrossRef] [Green Version]
- Rosenbaum, D.M.; Cherezov, V.; Hanson, M.A.; Rasmussen, S.G.; Thian, F.S.; Kobilka, T.S.; Choi, H.J.; Yao, X.J.; Weis, W.I.; Stevens, R.C.; et al. GPCR engineering yields high-resolution structural insights into β2-adrenergic receptor function. Science 2007, 318, 1266–1273. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.L.; Chien, E.Y.T.; Mol, C.D.; Fenalti, G.; Liu, W.; Katritch, V.; Abagyan, R.; Brooun, A.; Wells, P.; Christopher Bi, F.; et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 2010, 330, 1066–1071. [Google Scholar] [CrossRef] [Green Version]
- Tong, J.; Yang, H.; Yang, H.; Eom, S.H.; Im, Y.J. Structure of Osh3 reveals a conserved mode of phosphoinositide binding in oxysterol-binding proteins. Structure 2013, 21, 1203–1213. [Google Scholar] [CrossRef]
- Yang, H.; Tong, J.; Lee, C.W.; Ha, S.; Eom, S.H.; Im, Y.J. Structural mechanism of ergosterol regulation by fungal sterol transcription factor Upc2. Nat. Commun. 2015, 6, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Baumlova, A.; Chalupska, D.; Róźycki, B.; Jovic, M.; Wisniewski, E.; Klima, M.; Dubankova, A.; Kloer, D.P.; Nencka, R.; Balla, T.; et al. The crystal structure of the phosphatidylinositol 4-kinase II α. EMBO Rep. 2014, 15, 1085–1092. [Google Scholar] [CrossRef] [Green Version]
- Anderson, D.E.; Lu, J.; McIntosh, L.; Dahlquist, F.W. The folding, stability and dynamics of T4 lysozyme: A perspective using nuclear magnetic resonance. In NMR of Proteins; Clore, G.M., Gronenborn, A.M., Eds.; MacMillan Press: London, UK, 1993; pp. 258–304. [Google Scholar]
- Liu, L.; Baase, W.A.; Matthews, B.W. Halogenated benzenes bound within a non-polar cavity in T4 lysozyme provide examples of I⋯S and I⋯Se halogen-bonding. J. Mol. Biol. 2009, 385, 595–605. [Google Scholar] [CrossRef]
- Bouvignies, G.; Vallurupalli, P.; Hansen, D.F.; Correia, B.E.; Lange, O.; Bah, A.; Vernon, R.M.; Dahlquist, F.W.; Baker, D.; Kay, L.E. Solution structure of a minor and transiently formed state of a T4 lysozyme mutant. Nature 2011, 477, 111–114. [Google Scholar] [CrossRef] [Green Version]
- Skrynnikov, N.R.; Mulder, F.A.; Hon, B.; Dahlquist, F.W.; Kay, L.E. Probing slow time scale dynamics at methyl-containing side chains in proteins by relaxation dispersion NMR measurements: Application to methionine residues in a cavity mutant of T4 lysozyme. J. Am. Chem. Soc. 2001, 123, 4556–4566. [Google Scholar] [CrossRef]
- Vallurupalli, P.; Bouvignies, G.; Kay, L.E. Studying “invisible” excited protein states in slow exchange with a major state conformation. J. Am. Chem. Soc. 2012, 134, 8148–8161. [Google Scholar] [CrossRef]
- Consentius, P.; Gohlke, U.; Loll, B.; Alings, C.; Müller, R.; Heinemann, U.; Kaupp, M.; Wahl, M.; Risse, T. Tracking transient conformational states of T4 lysozyme at room temperature combining X-ray crystallography and site-directed spin labeling. J. Am. Chem. Soc. 2016, 138, 12868–12875. [Google Scholar] [CrossRef]
- Feher, V.A.; Schiffer, J.M.; Mermelstein, D.J.; Mih, N.; Pierce, L.C.; McCammon, J.A.; Amaro, R.E. Mechanisms for benzene dissociation through the excited state of T4 lysozyme L99A mutant. Biophys. J. 2019, 116, 205–214. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, A.E.; Baase, W.A.; Zhang, X.J.; Heinz, D.W.; Blaber, M.P.B.E.; Baldwin, E.P.; Matthews, B.W. Response of a protein structure to cavity-creating mutations and its relation to the hydrophobic effect. Science 1992, 255, 178–183. [Google Scholar] [CrossRef] [Green Version]
- Kuroki, R.; Weaver, L.H.; Matthews, B.W. A covalent enzyme-substrate intermediate with saccharide distortion in a mutant T4 lysozyme. Science 1993, 262, 2030–2033. [Google Scholar] [CrossRef]
- Nucci, N.V.; Fuglestad, B.; Athanasoula, E.A.; Wand, A.J. Role of cavities and hydration in the pressure unfolding of T4 lysozyme. Proc. Nat. Acad. Sci. USA 2014, 111, 13846–13851. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Papaleo, E.; Lindorff-Larsen, K. Mapping transiently formed and sparsely populated conformations on a complex energy landscape. Elife 2016, 5, e17505. [Google Scholar] [CrossRef]
- Sanabria, H.; Rodnin, D.; Hemmen, K.; Peulen, T.O.; Felekyan, S.; Fleissner, M.R.; Dimura, M.; Koberling, F.; Kühnemuth, R.; Hubbell, W.; et al. Resolving dynamics and function of transient states in single enzyme molecules. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Vallurupalli, P.; Chakrabarti, N.; Pomès, R.; Kay, L.E. Atomistic picture of conformational exchange in a T4 lysozyme cavity mutant: An experiment-guided molecular dynamics study. Chem. Sci. 2016, 7, 3602–3613. [Google Scholar] [CrossRef] [Green Version]
- Schiffer, J.M.; Feher, V.A.; Malmstrom, R.D.; Sida, R.; Amaro, R.E. Capturing invisible motions in the transition from ground to rare excited states of T4 lysozyme L99A. Biophys. J. 2016, 111, 1631–1640. [Google Scholar] [CrossRef] [Green Version]
- Smith, Z.; Ravindra, P.; Wang, Y.; Cooley, R.; Tiwary, P. Discovering protein conformational flexibility through artificial-intelligence-aided molecular dynamics. J. Phys. Chem. B 2020, 124, 8221–8229. [Google Scholar] [CrossRef]
- Chodera, J.D.; Singhal, N.; Pande, V.S.; Dill, K.A.; Swope, W.C. Automatic discovery of metastable states for the construction of Markov models of macromolecular conformational dynamics. J. Phys. Chem. 2007, 126, 155101. [Google Scholar] [CrossRef]
- Pan, A.C.; Roux, B. Building Markov state models along pathways to determine free energies and rates of transitions. J. Phys. Chem. 2008, 129, 064107. [Google Scholar] [CrossRef] [Green Version]
- Prinz, J.H.; Wu, H.; Sarich, M.; Keller, B.; Senne, M.; Held, M.; Chodera, J.D.; Schütte, C.; Noé, F. Markov models of molecular kinetics: Generation and validation. J. Phys. Chem. 2011, 134, 174105. [Google Scholar] [CrossRef]
- Malmstrom, R.D.; Lee, C.T.; Van Wart, A.T.; Amaro, R.E. Application of molecular-dynamics based Markov state models to functional proteins. J. Chem. Theory Comput. 2014, 10, 2648–2657. [Google Scholar] [CrossRef]
- Chodera, J.D.; Noé, F. Markov state models of biomolecular conformational dynamics. Curr. Opin. Struct. Biol. 2014, 25, 135–144. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Sheong, F.K.; Zeng, X.; Huang, X. Elucidation of the conformational dynamics of multi-body systems by construction of Markov state models. Phys. Chem. Chem. Phys. 2016, 18, 30228–30235. [Google Scholar] [CrossRef]
- Wang, W.; Cao, S.Q.; Zhu, L.Z.; Huang, X. Constructing Markov State Models to elucidate the functional conformational changes of complex biomolecules. WIREs Comput. Mol. Sci. 2018, 8, e1343. [Google Scholar] [CrossRef]
- Unarta, I.C.; Zhu, L.; Tse, C.K.M.; Cheung, P.P.H.; Yu, J.; Huang, X. Molecular mechanisms of RNA polymerase II transcription elongation elucidated by kinetic network models. Curr. Opin. Struct. Biol. 2018, 49, 54–62. [Google Scholar] [CrossRef]
- Suárez, E.; Wiewiora, R.P.; Wehmeyer, C.; Noé, F.; Chodera, J.D.; Zuckerman, D.M. What Markov state models can and cannot do: Correlation versus path-based observables in protein-folding models. J. Chem. Theory Comput. 2021, 17, 3119–3133. [Google Scholar] [CrossRef]
- Wang, X.; Unarta, I.C.; Cheung, P.P.-H.; Huang, X. Elucidating molecular mechanisms of functional conformational changes of proteins via Markov state models. Curr. Opin. Struct. Biol. 2021, 67, 69–77. [Google Scholar] [CrossRef]
- Unarta, I.C.; Cao, S.; Kubo, S.; Wang, W.; Cheung, P.P.H.; Gao, X.; Takada, S.; Huang, X. Role of bacterial RNA polymerase gate opening dynamics in DNA loading and antibiotics inhibition elucidated by quasi-Markov State Model. Proc. Nat. Acad. Sci. USA 2021, 118, e2024324118. [Google Scholar] [CrossRef]
- Da, L.T.; Wang, D.; Huang, X. Dynamics of pyrophosphate ion release and its coupled trigger loop motion from closed to open state in RNA polymerase II. J. Am. Chem. Soc. 2012, 134, 2399–2406. [Google Scholar] [CrossRef] [Green Version]
- Da, L.T.; Pardo-Avila, F.; Xu, L.; Silva, D.A.; Zhang, L.; Gao, X.; Wang, D.; Huang, X. Bridge helix bending promotes RNA polymerase Ⅱ backtracking through a critical and conserved threonine residue. Nat. Commun. 2016, 7, 11244. [Google Scholar] [CrossRef] [Green Version]
- Da, L.T.; Chao, E.; Shuai, Y.; Wu, S.; Su, X.D.; Yu, J. T7 RNA polymerase translocation is facilitated by a helix opening on the fingers domain that may also prevent backtracking. Nucleic Acids Res. 2017, 45, 7909–7921. [Google Scholar] [CrossRef]
- Da, L.T.; Shi, Y.; Ning, G.; Yu, J. Dynamics of the excised base release in thymine DNA glycosylase during DNA repair process. Nucleic Acids Res. 2018, 46, 568–581. [Google Scholar] [CrossRef] [Green Version]
- Da, L.T.; Yu, J. Base-flipping dynamics from an intrahelical to an extrahelical state exerted by thymine DNA glycosylase during DNA repair process. Nucleic Acids Res. 2018, 46, 5410–5425. [Google Scholar] [CrossRef]
- Tian, J.; Wang, L.; Da, L.T. Atomic resolution of short-range sliding dynamics of thymine DNA glycosylase along DNA minor-groove for lesion recognition. Nucleic Acids Res. 2021, 49, 1278–1293. [Google Scholar] [CrossRef]
- Zhu, L.; Jiang, H.; Cao, S.; Unarta, I.C.; Gao, X.; Huang, X. Critical role of backbone coordination in the mRNA recognition by RNA induced silencing complex. Commun. Biol. 2021, 4, 1–10. [Google Scholar] [CrossRef]
- Xu, H.; Song, K.; Da, L.T. Dynamics of peptide loading into major histocompatibility complex class Ⅰ molecules chaperoned by TAPBPR. Phys. Chem. Chem. Phys. 2022, 24, 12397. [Google Scholar] [CrossRef]
- Bowman, G.R.; Ensign, D.L.; Pande, V.S. Enhanced modeling via network theory: Adaptive sampling of markov state models. J. Chem. Theory Comput. 2010, 6, 787–794. [Google Scholar] [CrossRef] [Green Version]
- Hruska, E.; Balasubramanian, V.; Lee, H.; Jha, S.; Clementi, C. Extensible and scalable adaptive sampling on supercomputers. J. Chem. Theory Comput. 2020, 16, 7915–7925. [Google Scholar] [CrossRef]
- Zhu, L.Z.; Sheong, F.K.; Cao, S.Q.; Liu, S.; Unarta, I.C.; Huang, X. TAPS: A traveling-salesman based automated path searching method for functional conformational changes of biological macromolecules. J. Chem. Phys. 2019, 150, 124105. [Google Scholar] [CrossRef]
- Xi, K.; Hu, Z.; Wu, Q.; Wei, M.; Qian, R.; Zhu, L. Assessing the Performance of Traveling-salesman based Automated Path Searching (TAPS) on Complex Biomolecular Systems. J. Chem. Theory Comput. 2021, 17, 5301–5311. [Google Scholar] [CrossRef]
- Wang, L.; Xi, K.; Zhu, L.; Da, L.T. DNA Deformation Exerted by Regulatory DNA-Binding Motifs in Human Alkyladenine DNA Glycosylase Promotes Base Flipping. J. Chem. Inf. Model. 2022, 62, 3213–3226. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Eswar, N.; Webb, B.; Marti-Renom, M.A.; Madhusudhan, M.S.; Eramian, D.; Shen, M.; Pieper, U.; Sali, A. Comparative protein structure modeling using Modeller. Curr. Prot. Bioinf. 2006, 15, 1–30. [Google Scholar] [CrossRef] [Green Version]
- Li, P.F.; Song, L.F.; Merz, K.M., Jr. Systematic parameterization of monovalent ions employing the nonbonded model. J. Chem. Theory Comput. 2015, 11, 1645–1657. [Google Scholar] [CrossRef]
- Huang, J.; MacKerell, A.D. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Darden, T.; York, D.; Pedersen, L.J. Particle mesh Ewald: An N· log (N) method for Ewald sums in large systems. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Schlitter, J.; Engels, M.; Krüger, P. Targeted molecular dynamics: A new approach for searching pathways of conformational transitions. J. Mol. Graph. 1994, 12, 84–89. [Google Scholar] [CrossRef]
- Branduardi, D.; Gervasio, F.L.; Parrinello, M. From A to B in free energy space. J. Chem. Phys. 2007, 126, 054103. [Google Scholar] [CrossRef]
- Laio, A.; Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef] [Green Version]
- Barducci, A.; Bussi, G.; Parrinello, M. Well-tempered metadynamics: A smoothly converging and tunable free-energy method. Phy. Rev. Lett. 2008, 100, 020603. [Google Scholar] [CrossRef] [Green Version]
- Tribello, G.A.; Bonomi, M.; Branduardi, D.; Camilloni, C.; Bussi, G. PLUMED 2: New feathers for an old bird. Comput. Phys. Commun. 2014, 185, 604–613. [Google Scholar] [CrossRef] [Green Version]
- Applegate, D.L.; Bixby, R.E.; Chvátal, V.; Cook, W.J. The Traveling Salesman Problem: A Computational Study; Princeton University Press: Princeton, NJ, USA, 2006; pp. 1–58. [Google Scholar] [CrossRef] [Green Version]
- Cox, M.A.A.; Cox, T.F. Multidimensional scaling. In Handbook of Data Visualization; Chen, C.-H., Härde, W.K., Unwin, A., Eds.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 315–347. [Google Scholar] [CrossRef]
- Torrie, G.M.; Valleau, J.P. Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comput. Phys. 1977, 23, 187–199. [Google Scholar] [CrossRef]
- Kumar, S.; Rosenberg, J.M.; Bouzida, D.; Swendsen, R.H.; Kollman, P.A. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem. 1992, 13, 1011–1021. [Google Scholar] [CrossRef]
- Souaille, M.; Roux, B. Extension to the weighted histogram analysis method: Combining umbrella sampling with free energy calculations. Comput. Phys. Commun. 2001, 135, 40–57. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Fox, J.C.; Thomas, M.A.; Dishman, A.F.; Larsen, O.; Nakayama, T.; Yoshie, O.; Rosenkilde, M.M.; Volkman, B.F. Structure-function guided modeling of chemokine-GPCR specificity for the chemokine XCL1 and its receptor XCR1. Science 2019, 12, eaat4128. [Google Scholar] [CrossRef]
- Saunders, M.G.; Voth, G.A. Coarse-graining methods for computational biology. Annu. Rev. Biophys. 2013, 42, 73–93. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xi, K.; Zhu, L. Automated Path Searching Reveals the Mechanism of Hydrolysis Enhancement by T4 Lysozyme Mutants. Int. J. Mol. Sci. 2022, 23, 14628. https://doi.org/10.3390/ijms232314628
Xi K, Zhu L. Automated Path Searching Reveals the Mechanism of Hydrolysis Enhancement by T4 Lysozyme Mutants. International Journal of Molecular Sciences. 2022; 23(23):14628. https://doi.org/10.3390/ijms232314628
Chicago/Turabian StyleXi, Kun, and Lizhe Zhu. 2022. "Automated Path Searching Reveals the Mechanism of Hydrolysis Enhancement by T4 Lysozyme Mutants" International Journal of Molecular Sciences 23, no. 23: 14628. https://doi.org/10.3390/ijms232314628
APA StyleXi, K., & Zhu, L. (2022). Automated Path Searching Reveals the Mechanism of Hydrolysis Enhancement by T4 Lysozyme Mutants. International Journal of Molecular Sciences, 23(23), 14628. https://doi.org/10.3390/ijms232314628