
FTY720 Reduces Lipid Accumulation by Upregulating ABCA1 through Liver X Receptor and Sphingosine Kinase 2 Signaling in Macrophages
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
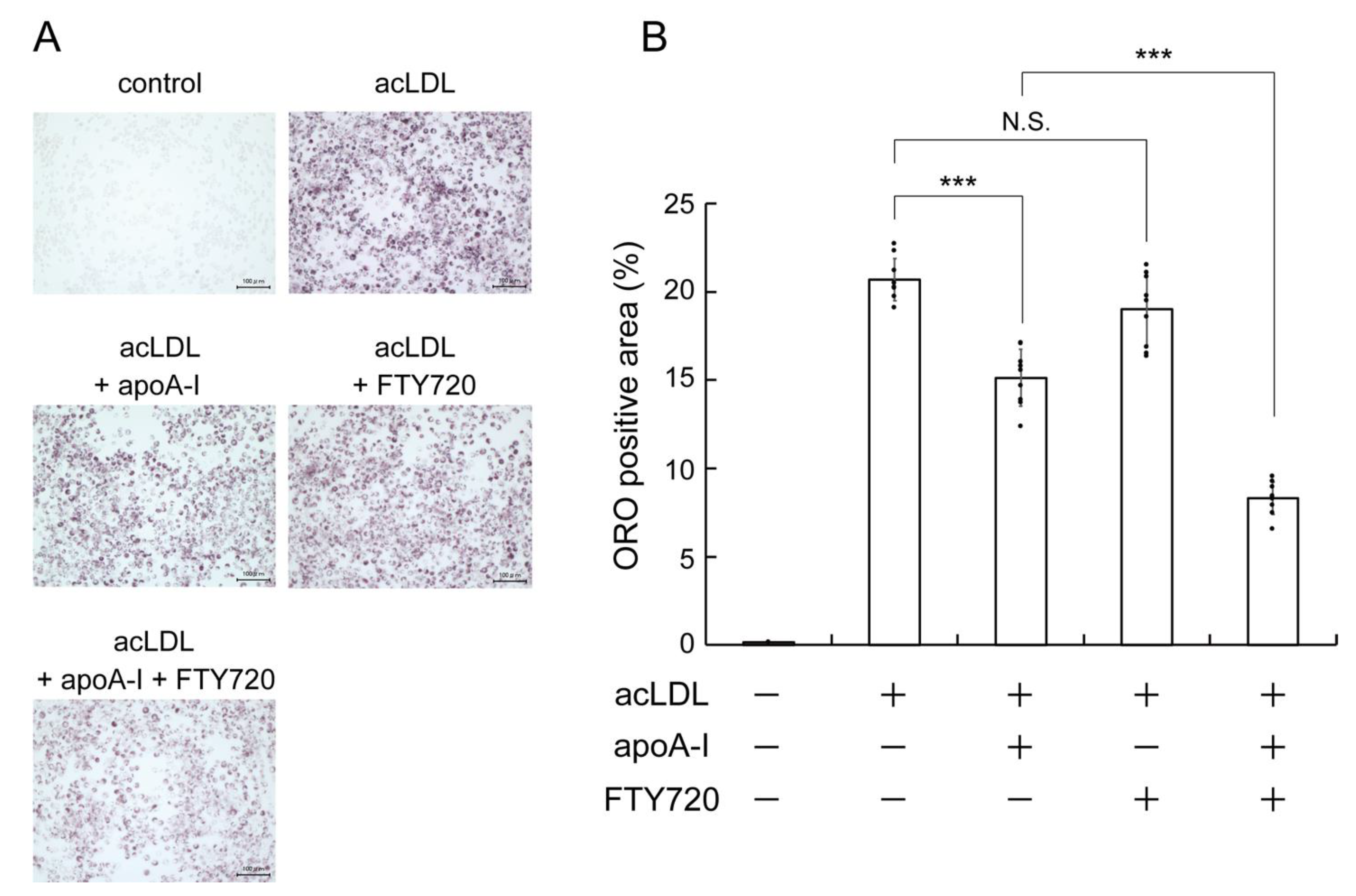
2.1. FTY720 Reduces Lipid Accumulation in J774 Mouse Macrophages
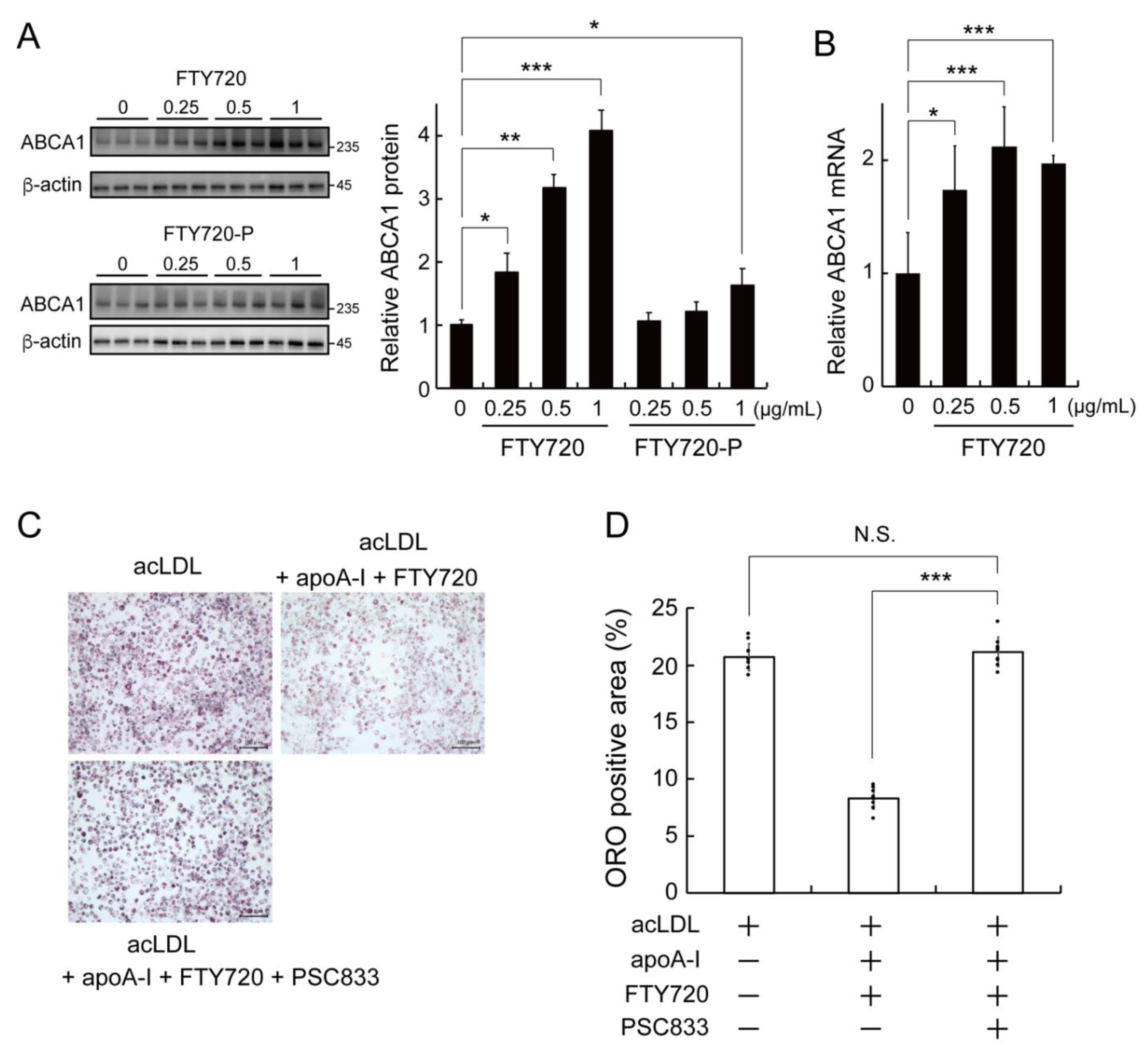
2.2. FTY720 Decreases Lipid Accumulation through Upregulating ABCA1 Expression in J774 Macrophages
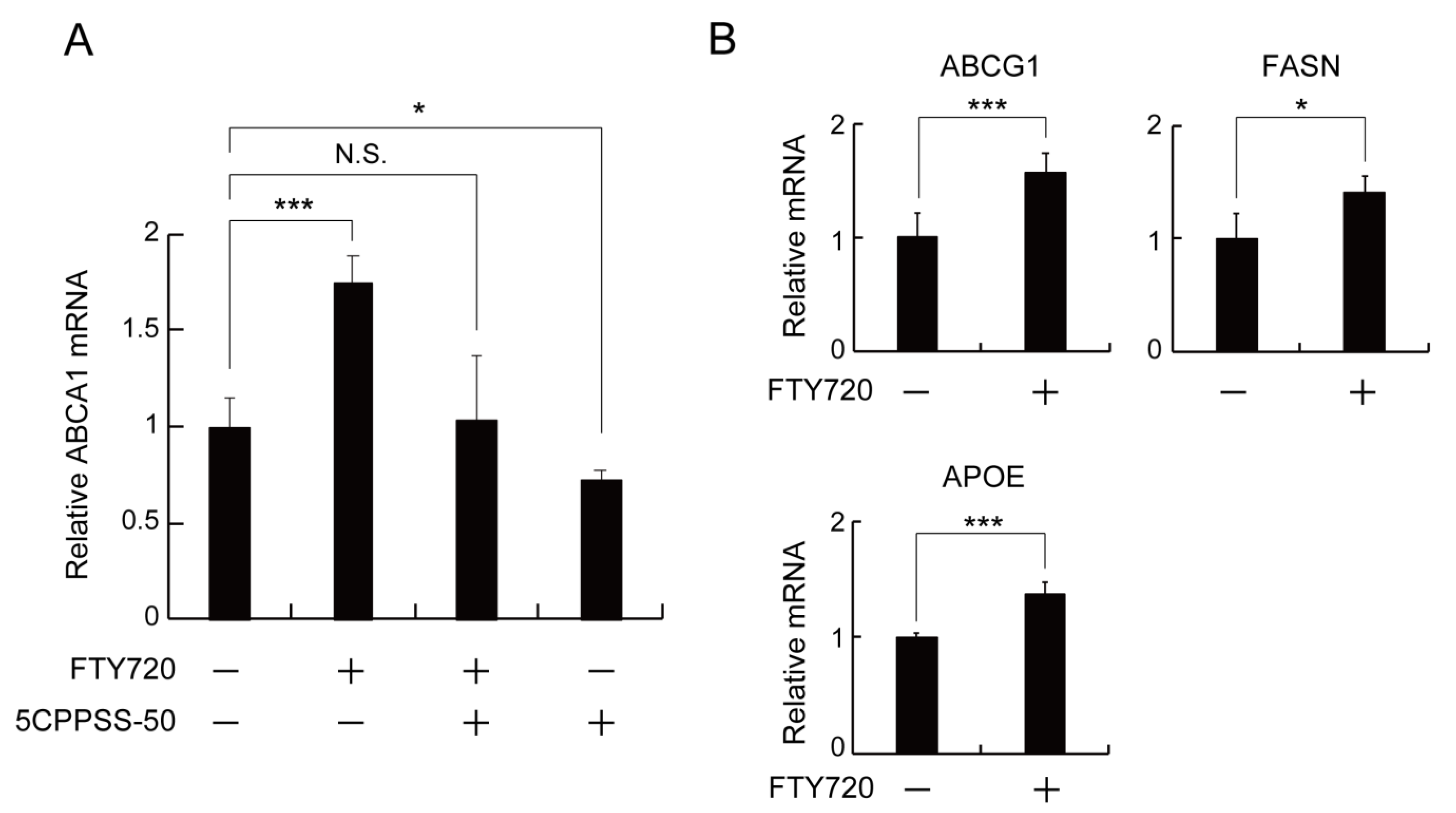
2.3. LXR Activation Is Involved in the FTY720-Induced Upregulation of ABCA1 Expression in J774 Macrophages
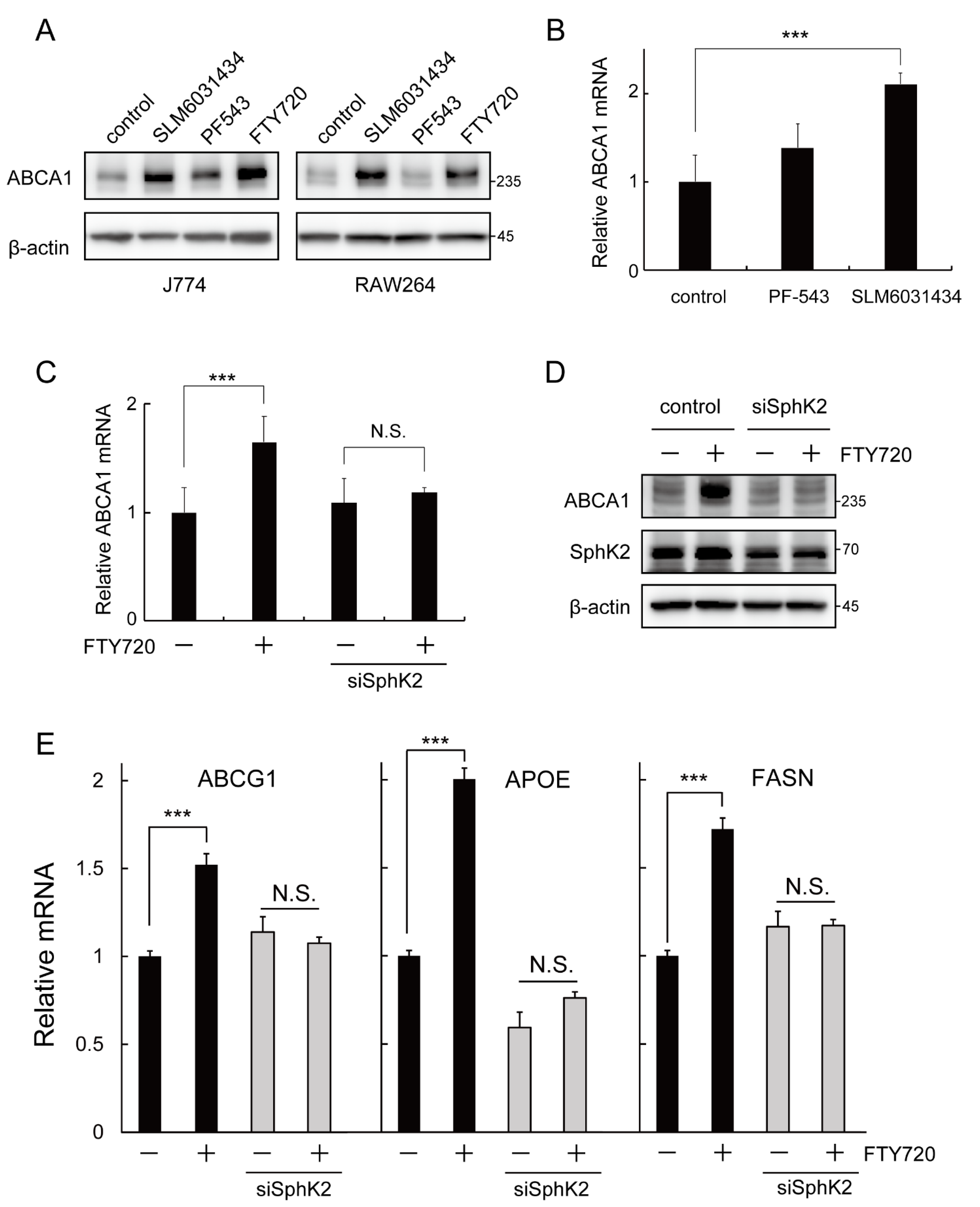
2.4. SphK2 Is Involved in the FTY720-Induced Upregulation of ABCA1 Expression
2.5. FTY720 Enhances ABCA1 Expression by Increasing Histone Acetylation via SphK2
3. Discussion
4. Materials and Methods
4.1. Materials and Reagents
4.2. Cell Culture and siRNA Transfection
4.3. Evaluation of Lipid Accumulation by Oil Red O Staining
4.4. Reverse Transcription-Quantitative Polymerase Chain Reaction (RT-qPCR)
4.5. Western Blot Analysis
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tabas, I.; Bornfeldt, K.E. Macrophage Phenotype and Function in Different Stages of Atherosclerosis. Circ. Res. 2016, 118, 653–667. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, M.L.; Mujawar, Z.; Tamehiro, N. ABC Transporters, Atherosclerosis and Inflammation. Atherosclerosis 2010, 211, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Linton, M.F.; Tao, H.; Linton, E.F.; Yancey, P.G. SR-BI: A Multifunctional Receptor in Cholesterol Homeostasis and Atherosclerosis. Trends Endocrinol. Metab. 2017, 28, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, S. Assembly of High-Density Lipoprotein. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 20–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinecke, J.W. Small HDL Promotes Cholesterol Efflux by the ABCA1 Pathway in Macrophages: Implications for Therapies Targeted to HDL. Circ. Res. 2015, 116, 1101–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muratsu, J.; Koseki, M.; Masuda, D.; Yasuga, Y.; Tomoyama, S.; Ataka, K.; Yagi, Y.; Nakagawa, A.; Hamada, H.; Fujita, S.; et al. Accelerated Atherogenicity in Tangier Disease: A Case Accompanied by Extensive Atherosclerotic Lesions, Leriche Syndrome and Bleeding Tendency, and Review of the Literature. J. Atheroscler. Thromb. 2018, 25, 1076–1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bochem, A.E.; Van Wijk, D.F.; Holleboom, A.E.; Duivenvoorden, R.; Motazacker, M.M.; Dallinga-Thie, G.M.; Groot, E.D.; Kastelein, J.J.P.; Nederveen, A.J.; Hovingh, G.K.; et al. ABCA1 Mutation Carriers with Low High-Density Lipoprotein Cholesterol Are Characterized by a Larger Atherosclerotic Burden. Eur. Heart J. 2013, 34, 286–291. [Google Scholar] [CrossRef] [Green Version]
- Van Eck, M.; Bos, I.S.T.; Kaminski, W.E.; Orsó, E.; Rothe, G.; Twisk, J.; Böttcher, A.; Van Amersfoort, E.S.; Christiansen-Weber, T.A.; Fung-Leung, W.P.; et al. Leukocyte ABCA1 Controls Susceptibility to Atherosclerosis and Macrophage Recruitment into Tissues. Proc. Natl. Acad. Sci. USA 2002, 99, 6298–6303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkateswaran, A.; Laffitte, B.A.; Joseph, S.B.; Mak, P.A.; Wilpitz, D.C.; Edwards, P.A.; Tontonoz, P. Control of Cellular Cholesterol Efflux by the Nuclear Oxysterol Receptor LXRα. Proc. Natl. Acad. Sci. USA 2000, 97, 12097–12102. [Google Scholar] [CrossRef] [Green Version]
- Westerterp, M.; Bochem, A.E.; Yvan-Charvet, L.; Murphy, A.J.; Wang, N.; Tall, A.R. ATP-Binding Cassette Transporters, Atherosclerosis, and Inflammation. Circ. Res. 2014, 114, 157–170. [Google Scholar] [CrossRef]
- Wang, B.; Tontonoz, P. Liver X Receptors in Lipid Signalling and Membrane Homeostasis. Nat. Rev. Endocrinol. 2018, 14, 452–463. [Google Scholar] [CrossRef]
- Brinkmann, V.; Billich, A.; Baumruker, T.; Heining, P.; Schmouder, R.; Francis, G.; Aradhye, S.; Burtin, P. Fingolimod (FTY720): Discovery and Development of an Oral Drug to Treat Multiple Sclerosis. Nat. Rev. Drug Discov. 2010, 9, 883–897. [Google Scholar] [CrossRef]
- Paugh, S.W.; Payne, S.G.; Barbour, S.E.; Milstien, S.; Spiegel, S. The Immunosuppressant FTY720 Is Phosphorylated by Sphingosine Kinase Type 2. FEBS Lett. 2003, 554, 189–193. [Google Scholar] [CrossRef] [Green Version]
- Billich, A.; Bornancin, F.; Dévay, P.; Mechtcheriakova, D.; Urtz, N.; Baumruker, T. Phosphorylation of the Immunomodulatory Drug FTY720 by Sphingosine Kinases. J. Biol. Chem. 2003, 278, 47408–47415. [Google Scholar] [CrossRef] [Green Version]
- Nofer, J.R.; Bot, M.; Brodde, M.; Taylor, P.J.; Salm, P.; Brinkmann, V.; Van Berkel, T.; Assmann, G.; Biessen, E.A.L. FTY720, a Synthetic Sphingosine 1 Phosphate Analogue, Inhibits Development of Atherosclerosis in Low-Density Lipoprotein Receptor-Deficient Mice. Circulation 2007, 115, 501–508. [Google Scholar] [CrossRef] [Green Version]
- Keul, P.; Tölle, M.; Lucke, S.; Von Wnuck Lipinski, K.; Heusch, G.; Schuchardt, M.; Van Der Giet, M.; Levkau, B. The Sphingosine-1-Phosphate Analogue FTY720 Reduces Atherosclerosis in Apolipoprotein E-Deficient Mice. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 607–613. [Google Scholar] [CrossRef] [Green Version]
- Klingenberg, R.; Nofer, J.R.; Rudling, M.; Bea, F.; Blessing, E.; Preusch, M.; Grone, H.J.; Katus, H.A.; Hansson, G.K.; Dengler, T.J. Sphingosine-1-Phosphate Analogue FTY720 Causes Lymphocyte Redistribution and Hypercholesterolemia in ApoE-Deficient Mice. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2392–2399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poti, F.; Costa, S.; Bergonzini, V.; Galletti, M.; Pignatti, E.; Weber, C.; Simoni, M.; Nofer, J.R. Effect of Sphingosine 1-Phosphate (S1P) Receptor Agonists FTY720 and CYM5442 on Atherosclerosis Development in LDL Receptor Deficient (LDL-R−/−) Mice. Vascul. Pharmacol. 2012, 57, 56–64. [Google Scholar] [CrossRef]
- Blumenfeld Kan, S.; Staun-Ram, E.; Golan, D.; Miller, A. HDL-Cholesterol Elevation Associated with Fingolimod and Dimethyl Fumarate Therapies in Multiple Sclerosis. Mult. Scler. J.-Exp. Transl. Clin. 2019, 5, 205521731988272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagao, K.; Maeda, M.; Mañucat, N.B.; Ueda, K. Cyclosporine A and PSC833 Inhibit ABCA1 Function via Direct Binding. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2013, 1831, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.; Hartung, H.P. Mechanism of Action of Oral Fingolimod (FTY720) in Multiple Sclerosis. Clin. Neuropharmacol. 2010, 33, 91–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnute, M.E.; McReynolds, M.D.; Kasten, T.; Yates, M.; Jerome, G.; Rains, J.W.; Hall, T.; Chrencik, J.; Kraus, M.; Cronin, C.N.; et al. Modulation of Cellular S1P Levels with a Novel, Potent and Specific Inhibitor of Sphingosine Kinase-1. Biochem. J. 2012, 444, 79–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kharel, Y.; Morris, E.A.; Congdon, M.D.; Thorpe, S.B.; Tomsig, J.L.; Santos, W.L.; Lynch, K.R. Sphingosine Kinase 2 Inhibition and Blood Sphingosine 1-Phosphate Levels. J. Pharmacol. Exp. Ther. 2015, 355, 23–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hait, N.C.; Allegood, J.; Maceyka, M.; Strub, G.M.; Harikumar, K.B.; Singh, S.K.; Luo, C.; Marmorstein, R.; Kordula, T.; Milstien, S.; et al. Regulation of Histone Acetylation in the Nucleus by Sphingosine-1-Phosphate. Science 2009, 325, 1254–1257. [Google Scholar] [CrossRef] [Green Version]
- Verschuren, L.; de Vries-van der Weij, J.; Zadelaar, S.; Kleemann, R.; Kooistra, T. LXR Agonist Suppresses Atherosclerotic Lesion Growth and Promotes Lesion Regression in ApoE*3Leiden Mice: Time Course and Mechanisms. J. Lipid Res. 2009, 50, 301–311. [Google Scholar] [CrossRef] [Green Version]
- Ma, A.Z.S.; Song, Z.Y.; Zhang, Q. Cholesterol Efflux Is LXRα Isoform-Dependent in Human Macrophages. BMC Cardiovasc. Disord. 2014, 14, 80. [Google Scholar] [CrossRef] [Green Version]
- Diaz Escarcega, R.; McCullough, L.D.; Tsvetkov, A.S. The Functional Role of Sphingosine Kinase 2. Front. Mol. Biosci. 2021, 8, 971–980. [Google Scholar] [CrossRef] [PubMed]
- Hait, N.C.; Wise, L.E.; Allegood, J.C.; O’Brien, M.; Avni, D.; Reeves, T.M.; Knapp, P.E.; Lu, J.; Luo, C.; Miles, M.F.; et al. Active, Phosphorylated Fingolimod Inhibits Histone Deacetylases and Facilitates Fear Extinction Memory. Nat. Neurosci. 2014, 17, 971–980. [Google Scholar] [CrossRef]
- Newton, J.; Hait, N.C.; Maceyka, M.; Colaco, A.; Maczis, M.; Wassif, C.A.; Cougnoux, A.; Porter, F.D.; Milstien, S.; Platt, N.; et al. FTY720/Fingolimod Increases NPC1 and NPC2 Expression and Reduces Cholesterol and Sphingolipid Accumulation in Niemann-Pick Type C Mutant Fibroblasts. FASEB J. 2017, 31, 1719–1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hait, N.C.; Avni, D.; Yamada, A.; Nagahashi, M.; Aoyagi, T.; Aoki, H.; Dumur, C.I.; Zelenko, Z.; Gallagher, E.J.; Leroith, D.; et al. The Phosphorylated Prodrug FTY720 Is a Histone Deacetylase Inhibitor That Reactivates ERα Expression and Enhances Hormonal Therapy for Breast Cancer. Oncogenesis 2015, 4, e156. [Google Scholar] [CrossRef] [PubMed]
- Rohrbach, T.D.; Asgharpour, A.; Maczis, M.A.; Montefusco, D.; Cowart, L.A.; Bedossa, P.; Sanyal, A.J.; Spiegel, S. FTY720/Fingolimod Decreases Hepatic Steatosis and Expression of Fatty Acid Synthase in Diet-Induced Nonalcoholic Fatty Liver Disease in Mice. J. Lipid Res. 2019, 60, 1311–1322. [Google Scholar] [CrossRef] [PubMed]
- Ishimaru, K.; Yoshioka, K.; Kano, K.; Kurano, M.; Saigusa, D.; Aoki, J.; Yatomi, Y.; Takuwa, N.; Okamoto, Y.; Proia, R.L.; et al. Sphingosine Kinase-2 Prevents Macrophage Cholesterol Accumulation and Atherosclerosis by Stimulating Autophagic Lipid Degradation. Sci. Rep. 2019, 9, 18329. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tachibana, K.; Kusumoto, K.; Ogawa, M.; Ando, H.; Shimizu, T.; Ishima, Y.; Ishida, T.; Okuhira, K. FTY720 Reduces Lipid Accumulation by Upregulating ABCA1 through Liver X Receptor and Sphingosine Kinase 2 Signaling in Macrophages. Int. J. Mol. Sci. 2022, 23, 14617. https://doi.org/10.3390/ijms232314617
Tachibana K, Kusumoto K, Ogawa M, Ando H, Shimizu T, Ishima Y, Ishida T, Okuhira K. FTY720 Reduces Lipid Accumulation by Upregulating ABCA1 through Liver X Receptor and Sphingosine Kinase 2 Signaling in Macrophages. International Journal of Molecular Sciences. 2022; 23(23):14617. https://doi.org/10.3390/ijms232314617
Chicago/Turabian StyleTachibana, Koki, Kohshi Kusumoto, Mai Ogawa, Hidenori Ando, Taro Shimizu, Yu Ishima, Tatsuhiro Ishida, and Keiichiro Okuhira. 2022. "FTY720 Reduces Lipid Accumulation by Upregulating ABCA1 through Liver X Receptor and Sphingosine Kinase 2 Signaling in Macrophages" International Journal of Molecular Sciences 23, no. 23: 14617. https://doi.org/10.3390/ijms232314617