

Non-Conserved Amino Acid Residues Modulate the Thermodynamics of Zn(II) Binding to Classical ββα Zinc Finger Domains

 , ,
, ,
Abstract
1. Introduction

2. Results and Discussion
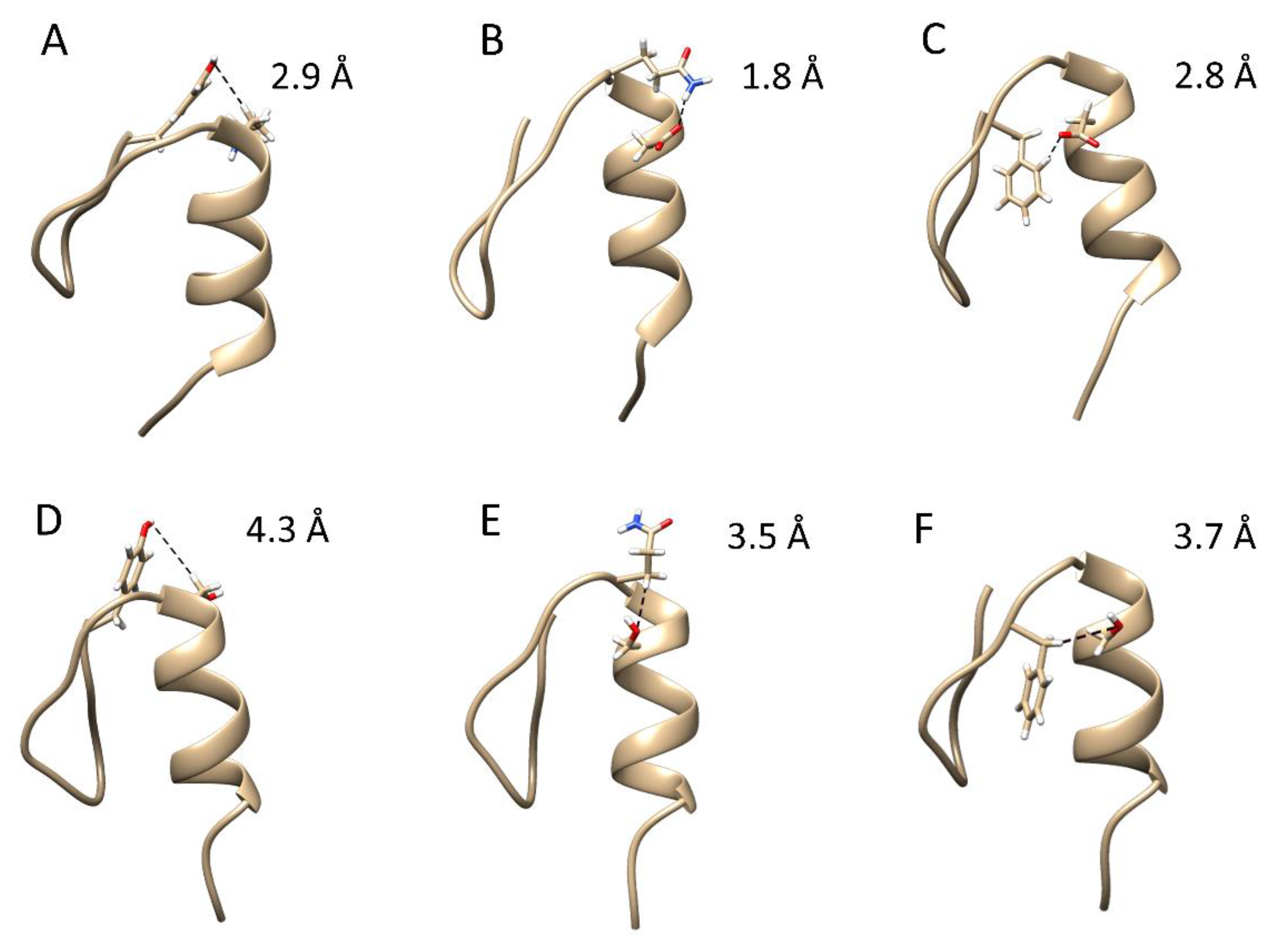
2.1. Classical Molecular Dynamics Simulations
2.2. Steered Molecular Dynamics Simulations
2.3. Spectroscopic Characterization
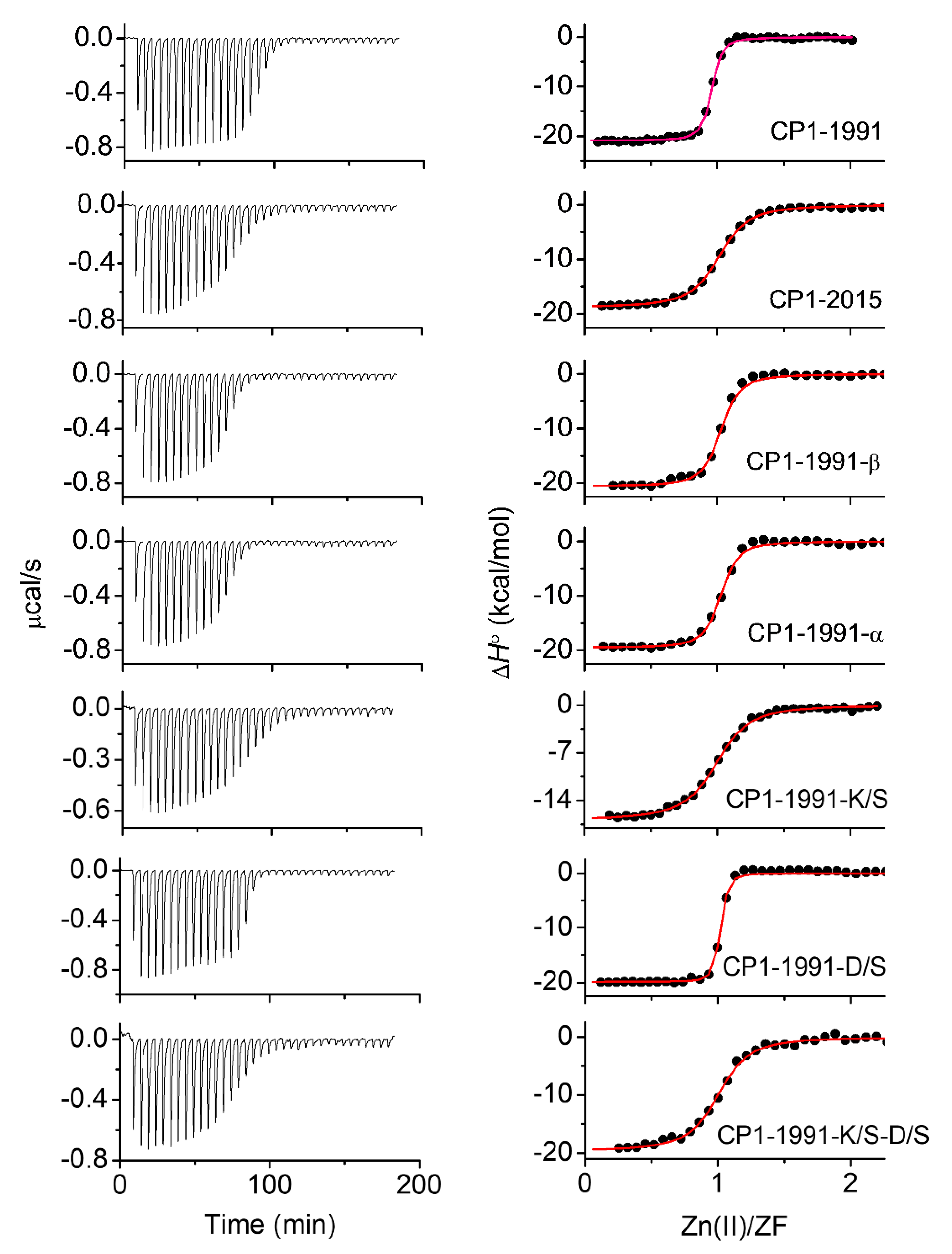
2.4. Isothermal Titration Calorimetry
2.5. Comparison with Previous Reports and Biological Significance
3. Materials and Methods
3.1. Materials
3.2. Computational Studies
3.2.1. Molecular Dynamics (MD) Simulations
3.2.2. Steered Molecular Dynamics (SMD) Simulations
3.3. Zinc Finger Peptide Synthesis
3.4. Determination of pKa Values of ZF Thiols
3.5. Circular Dichroism
3.6. Co(II) Binding to ZF Peptides
3.7. Zinc Finger Competition with Chelators
3.8. Isothermal Titration Calorimetry (ITC)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CD | circular dichroism |
| CCHH | zinc finger with two Cys and two His residues being in Zn(II) coordination sphere |
| CN | contact number |
| CP1 | zinc finger consensus peptide 1 |
| FES | free energy surface |
| ITC | isothermal titration calorimetry |
| MD | molecular dynamics |
| PC | principal component |
| PCA | principal component analysis |
| PDB | protein data bank |
| RMSF | root-mean-square fluctuations |
| SMD | steered molecular dynamics |
| ZF | zinc finger domain |
References
- George, K.L.; Horne, W.S. Heterogeneous-backbone foldamer mimics of zinc finger tertiary structure. J. Am. Chem. Soc. 2017, 139, 7931–7938. [Google Scholar] [CrossRef]
- George, K.L.; Horne, W.S. Foldamer tertiary structure through sequence-guided protein backbone alteration. Acc. Chem. Res. 2018, 51, 1220–1228. [Google Scholar] [CrossRef]
- Thompson, M.; Woodbury, N.W. Fluorescent and photochemical properties of a single zinc finger conjugated to a fluorescent DNA-binding probe. Biochemistry 2000, 39, 4327–4338. [Google Scholar] [CrossRef] [PubMed]
- Gaj, T.; Liu, J.; Anderson, K.E.; Sirk, S.J.; Barbas, C.F., 3rd. Protein delivery using Cys2-His2 zinc-finger domains. ACS Chem. Biol. 2014, 9, 1662–1667. [Google Scholar] [CrossRef]
- Gersbach, C.A.; Gaj, T.; Barbas, C.F., 3rd. Synthetic zinc finger proteins: The advent of targeted gene regulation and genome modification technologies. Acc. Chem. Res. 2014, 47, 2309–2318. [Google Scholar] [CrossRef]
- Hossain, M.A.; Barrow, J.J.; Shen, Y.; Haq, M.I.; Bungert, J. Artificial zinc finger DNA binding domains: Versatile tools for genome engineering and modulation of gene expression. J. Cell Biochem. 2015, 116, 2435–2444. [Google Scholar] [CrossRef]
- Gommans, W.M.; Haisma, H.J.; Rots, M.G. Engineering zinc finger protein transcription factors: The therapeutic relevance of switching endogenous gene expression on or off at command. J. Mol. Biol. 2005, 354, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Sera, T. Zinc-finger-based artificial transcription factors and their applications. Adv. Drug Deliv. Rev. 2009, 61, 513–526. [Google Scholar] [CrossRef] [PubMed]
- Kluska, K.; Adamczyk, J.; Krężel, A. Metal binding properties, stability and reactivity of zinc fingers. Coord. Chem. Rev. 2018, 367, 18–64. [Google Scholar] [CrossRef]
- Padjasek, M.; Kocyła, A.; Kluska, K.; Kerber, O.; Tran, J.B.; Krężel, A. Structural zinc binding sites shaped for greater works: Structure-function relatons in classical zinc finger, hook and clasp domains. J. Inorg. Biochem. 2020, 204, 110955. [Google Scholar] [CrossRef]
- Sénèque, O.; Latour, J.M. Coordination properties of zinc finger peptides revisited: Ligand competition studies reveal higher affinities for zinc and cobalt. J. Am. Chem. Soc. 2010, 132, 17760–17774. [Google Scholar] [CrossRef]
- Miłoch, A.; Krężel, A. Metal binding properties of the zinc finger metallome--insights into variations in stability. Metallomics 2014, 6, 2015–2024. [Google Scholar] [CrossRef] [PubMed]
- Kocyła, A.; Tran, J.B.; Krężel, A. Galvanization of protein-protein interactions in a dynamic zinc interactome. Trends Biochem. Sci. 2021, 46, 64–79. [Google Scholar] [CrossRef]
- Kluska, K.; Adamczyk, J.; Krężel, A. Metal binding properties of zinc fingers with a naturally altered metal binding site. Metallomics 2018, 10, 248–263. [Google Scholar] [CrossRef]
- Shi, Y.; Beger, R.D.; Berg, J.M. Metal binding properties of single amino acid deletion mutants of zinc finger peptides: Studies using cobalt(II) as a spectroscopic probe. Biophys. J. 1993, 64, 749–753. [Google Scholar] [CrossRef]
- Krizek, B.A.; Amann, B.T.; Kilfoil, V.J.; Merkle, D.L.; Berg, J.M. A consensus zinc finger peptide: Design, high-affinity metal binding, a pH-dependent structure, and a His to Cys sequence variant. J. Am. Chem. Soc. 1991, 113, 4518–4523. [Google Scholar] [CrossRef]
- Kim, C.A.; Berg, J.M. A 2.2 A resolution crystal structure of a designed zinc finger protein bound to DNA. Nat. Struct. Biol. 1996, 3, 940–945. [Google Scholar] [CrossRef]
- Besold, A.N.; Widger, L.R.; Namuswe, F.; Michalek, J.L.; Michel, S.L.; Goldberg, D.P. Revisiting and re-engineering the classical zinc finger peptide: Consensus peptide-1 (CP-1). Mol. Biosyst. 2016, 12, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, J.; Wang, J.; Wang, W. Metal-coupled folding of Cys2His2 zinc-finger. J. Am. Chem. Soc. 2008, 130, 892–900. [Google Scholar] [CrossRef] [PubMed]
- Papaleo, E.; Mereghetti, P.; Fantucci, P.; Grandori, R.; De Gioia, L. Free-energy landscape, principal component analysis, and structural clustering to identify representative conformations from molecular dynamics simulations: The myoglobin case. J. Mol. Graph. Model. 2009, 27, 889–899. [Google Scholar] [CrossRef]
- Brandt, E.G.; Hellgren, M.; Brinck, T.; Bergman, T.; Edholm, O. Molecular dynamics study of zinc binding to cysteines in a peptide mimic of the alcohol dehydrogenase structural zinc site. Phys. Chem. Chem. Phys. 2009, 11, 975–983. [Google Scholar] [CrossRef]
- Tjörnhammar, R.; Edholm, O. Molecular dynamics simulations of Zn2+ coordination in protein binding sites. J. Chem. Phys. 2010, 132, 205101. [Google Scholar] [CrossRef] [PubMed]
- Peris-Díaz, M.D.; Guran, R.; Domene, C.; de Los Rios, V.; Zitka, O.; Adam, V.; Krężel, A. An integrated mass spectrometry and molecular dynamics simulations approach reveals the spatial organization impact of metal-binding sites on the stability of metal-depleted metallothionein-2 species. J. Am. Chem. Soc. 2021, 143, 16486–16501. [Google Scholar] [CrossRef] [PubMed]
- Perišić, O.; Wriggers, W. Mechanism for the unfolding of the TOP7 protein in steered molecular dynamics simulations as revealed by mutual information analysis. Front. Mol. Biosci. 2021, 8, 696609. [Google Scholar] [CrossRef]
- Isralewitz, B.; Gao, M.; Schulten, K. Steered molecular dynamics and mechanical functions of proteins. Curr. Opin. Struct. Biol. 2001, 11, 224–230. [Google Scholar] [CrossRef]
- Zhu, G.P.; Xu, C.; Teng, M.K.; Tao, L.M.; Zhu, X.Y.; Wu, C.J.; Hang, J.; Niu, L.W.; Wang, Y.Z. Increasing the thermostability of D-xylose isomerase by introduction of a proline into the turn of a random coil. Protein Eng. 1999, 12, 635–638. [Google Scholar] [CrossRef]
- Muslin, E.H.; Clark, S.E.; Henson, C.A. The effect of proline insertions on the thermostability of a barley alpha-glucosidase. Protein Eng. 2002, 15, 29–33. [Google Scholar] [CrossRef]
- Watanabe, K.; Masuda, T.; Ohashi, H.; Mihara, H.; Suzuki, Y. Multiple proline substitutions cumulatively thermostabilize Bacillus cereus ATCC7064 oligo-1,6-glucosidase. Irrefragable proof supporting the proline rule. Eur. J. Biochem. 1994, 226, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Vijayakumar, M.; Qian, H.; Zhou, H.X. Hydrogen bonds between short polar side chains and peptide backbone: Prevalence in proteins and effects on helix-forming propensities. Proteins 1999, 34, 497–507. [Google Scholar] [CrossRef]
- Song, B.; Bomar, M.G.; Kibler, P.; Kodukula, K.; Galande, A.K. The serine-proline turn: A novel hydrogen-bonded template for designing peptidomimetics. Org. Lett. 2012, 14, 732–735. [Google Scholar] [CrossRef]
- Rich, A.M.; Bombarda, E.; Schenk, A.D.; Lee, P.E.; Cox, E.H.; Spuches, A.M.; Hudson, L.D.; Kieffer, B.; Wilcox, D.E. Thermodynamics of Zn2+ binding to Cys2His2 and Cys2HisCys zinc fingers and a Cys4 transcription factor site. J. Am. Chem. Soc. 2012, 134, 10405–10418. [Google Scholar] [CrossRef]
- Blasie, C.A.; Berg, J.M. Structure-based thermodynamic analysis of a coupled metal binding-protein folding reaction involving a zinc finger peptide. Biochemistry 2002, 41, 15068–15073. [Google Scholar] [CrossRef] [PubMed]
- Reddi, A.R.; Guzman, T.R.; Breece, R.M.; Tierney, D.L.; Gibney, B.R. Deducing the energetic cost of protein folding in zinc finger proteins using designed metallopeptides. J. Am. Chem. Soc. 2007, 129, 12815–12827. [Google Scholar] [CrossRef] [PubMed]
- Lachenmann, M.J.; Ladbury, J.E.; Qian, X.; Huang, K.; Singh, R.; Weiss, M.A. Solvation and the hidden thermodynamics of a zinc finger probed by nonstandard repair of a protein crevice. Protein Sci. 2004, 13, 3115–3126. [Google Scholar] [CrossRef]
- Lachenmann, M.J.; Ladbury, J.; Phillips, N.B.; Narayana, N.; Qian, X.; Weiss, M.A. The hidden thermodynamics of a zinc finger. J. Mol. Biol. 2002, 316, 969–989. [Google Scholar] [CrossRef]
- Qian, X.; Weiss, M.A. Two-dimensional NMR studies of the zinc finger motif: Solution structures and dynamics of mutant ZFY domains containing aromatic substitutions in the hydrophobic core. Biochemistry 1992, 31, 7463–7476. [Google Scholar] [CrossRef]
- Krężel, A.; Maret, W. Zinc-buffering capacity of a eukaryotic cell at physiological pZn. J. Biol. Inorg. Chem. 2006, 11, 1049–1062. [Google Scholar] [CrossRef] [PubMed]
- Maret, W. Zinc in cellular regulation: The nature and significance of “zinc signals”. Int. J. Mol. Sci. 2017, 18, 2285. [Google Scholar] [CrossRef]
- Wang, Y.; Weisenhorn, E.; MacDiarmid, C.W.; Andreini, C.; Bucci, M.; Taggartm, J.; Banci, L.; Russell, J.; Coon, J.J.; Eide, D.J. The cellular economy of the Saccharomyces cerevisiae zinc proteome. Metallomics 2018, 10, 1755–1776. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Macchiagodena, M.; Pagliai, M.; Andreini, C.; Rosato, A.; Procacci, P. Upgrading and validation of the AMBER force field for histidine and cysteine zinc(II)-binding residues in sites with four protein ligands. J. Chem. Inf. Model. 2019, 59, 3803–3816. [Google Scholar] [CrossRef] [PubMed]
- Grant, B.J.; Rodrigues, A.P.; ElSawy, K.M.; McCammon, J.A.; Caves, L.S. Bio3d: An R package for the comparative analysis of protein structures. Bioinformatics 2006, 22, 2695–2696. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. Available online: https://ggplot2.tidyverse.org (accessed on 1 March 2019).
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- PLUMED Consortium. Promoting transparency and reproducibility in enhanced molecular simulations. Nat. Methods 2019, 16, 670–673. [Google Scholar] [CrossRef]
- Krężel, A.; Wójcik, J.; Maciejczyk, M.; Bal, W. Zn(II) complexes of glutathione disulfide: Structural basis of elevated stabilities. Inorg. Chem. 2011, 50, 72–85. [Google Scholar] [CrossRef][Green Version]
- Kochańczyk, T.; Nowakowski, M.; Wojewska, D.; Kocyła, A.; Ejchart, A.; Koźmiński, W.; Krężel, A. Metal-coupled folding as the driving force for the extreme stability of Rad50 zinc hook dimer assembly. Sci. Rep. 2016, 6, 36346. [Google Scholar] [CrossRef] [PubMed]
- Krężel, A.; Latajka, R.; Bujacz, G.D.; Bal, W. Coordination properties of tris(2-carboxyethyl)phosphine, a newly introduced thiol reductant, and its oxide. Inorg. Chem. 2003, 42, 1994–2003. [Google Scholar] [CrossRef] [PubMed]
- Kochańczyk, T.; Jakimowicz, P.; Krężel, A. Femtomolar Zn(II) affinity of minimal zinc hook peptides--a promising small tag for protein engineering. Chem. Commun. 2013, 49, 1312–1314. [Google Scholar] [CrossRef]
- Alderighi, L.; Gans, P.; Ienco, A.; Peters, D.; Sabatini, A.; Vacca, A. Hyperquad simulation and speciation (HySS): A utility program for the investigation of equilibria involving soluble and partially soluble species. Coord. Chem. Rev. 1999, 184, 311–318. [Google Scholar] [CrossRef]
- Krężel, A.; Maret, W. The biological inorganic chemistry of zinc ions. Arch, Biochem. Biophys. 2016, 611, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Pomorski, A.; Kochańczyk, T.; Miłoch, A.; Krężel, A. Method for accurate determination of dissociation constants of optical ratiometric systems: Chemical probes, genetically encoded sensors, and interacting molecules. Anal. Chem. 2013, 85, 11479–11486. [Google Scholar] [CrossRef] [PubMed]
- Fukada, H.; Takahashi, K. Enthalpy and heat capacity changes for the proton dissociation of various buffer components in 0.1 M potassium chloride. Proteins 1998, 33, 159–166. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ZF Peptide | −logKd7.4 | Kd7.4 (fM) | ∆(−logKd7.4) |
|---|---|---|---|
| CP1-1991 | 14.49 ± 0.05 | 3.2 ± 0.4 | 0 |
| CP1-2015 | 12.30 ± 0.02 | 501 ± 23 | 2.19 |
| CP1-1991-β | 14.40 ± 0.04 | 4.0 ± 0.4 | 0.09 |
| CP1-1991-α | 13.50 ± 0.05 | 32 ± 3 | 0.99 |
| CP1-1991-K/S | 14.04 ± 0.05 | 9.1 ± 0.9 | 0.45 |
| CP1-1991-D/S | 14.29 ± 0.04 | 5.1 ± 0.4 | 0.20 |
| CP1-1991-K/S-D/S | 13.52 ± 0.04 | 30 ± 3 | 0.97 |
| ZF Peptide | ΔG° | ΔΔG° | −TΔS° | ΔHITC | ΔH° | ΔHCysH | ΔHZn-pep | ΔHfolding | nH |
|---|---|---|---|---|---|---|---|---|---|
| (kcal/mol) | |||||||||
| CP-1991 | −19.77 ± 0.05 | 0 | −6.71 | −20.87 ± 0.19 | −13.06 | 13.23 | −26.29 | −6.29 | 1.56 |
| CP-2015 | −16.78 ± 0.02 | 2.99 | −6.20 | −18.83 ± 0.39 | −10.58 | 13.97 | −24.44 | −4.55 | 1.64 |
| CP-1991-β | −19.64 ± 0.04 | 0.13 | −7.02 | −20.61 ± 0.40 | −12.62 | 13.53 | −26.15 | −6.15 | 1.59 |
| CP-1991-α | −18.42 ± 0.05 | 1.35 | −7.31 | −19.53 ± 0.43 | −11.10 | 14.27 | −25.37 | −5.37 | 1.68 |
| CP-1991-K/S | −19.15 ± 0.05 | 0.62 | −10.78 | −16.82 ± 0.20 | −8.37 | 14.31 | −22.68 | −2.68 | 1.68 |
| CP-1991-D/S | −19.49 ± 0.04 | 0.28 | −7.89 | −20.24 ± 0.35 | −11.60 | 14.01 | −25.62 | −5.62 | 1.65 |
| CP-1991-K/S-D/S | −18.44 ± 0.04 | 1.33 | −7.23 | −19.68 ± 0.47 | −11.21 | 14.34 | −25.55 | −5.55 | 1.69 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kluska, K.; Chorążewska, A.; Peris-Díaz, M.D.; Adamczyk, J.; Krężel, A. Non-Conserved Amino Acid Residues Modulate the Thermodynamics of Zn(II) Binding to Classical ββα Zinc Finger Domains. Int. J. Mol. Sci. 2022, 23, 14602. https://doi.org/10.3390/ijms232314602
Kluska K, Chorążewska A, Peris-Díaz MD, Adamczyk J, Krężel A. Non-Conserved Amino Acid Residues Modulate the Thermodynamics of Zn(II) Binding to Classical ββα Zinc Finger Domains. International Journal of Molecular Sciences. 2022; 23(23):14602. https://doi.org/10.3390/ijms232314602
Chicago/Turabian StyleKluska, Katarzyna, Aleksandra Chorążewska, Manuel David Peris-Díaz, Justyna Adamczyk, and Artur Krężel. 2022. "Non-Conserved Amino Acid Residues Modulate the Thermodynamics of Zn(II) Binding to Classical ββα Zinc Finger Domains" International Journal of Molecular Sciences 23, no. 23: 14602. https://doi.org/10.3390/ijms232314602
APA StyleKluska, K., Chorążewska, A., Peris-Díaz, M. D., Adamczyk, J., & Krężel, A. (2022). Non-Conserved Amino Acid Residues Modulate the Thermodynamics of Zn(II) Binding to Classical ββα Zinc Finger Domains. International Journal of Molecular Sciences, 23(23), 14602. https://doi.org/10.3390/ijms232314602