
19S Proteasome Subunits as Oncogenes and Prognostic Biomarkers in FLT3-Mutated Acute Myeloid Leukemia (AML)


 , , , ,
, , , , 
Abstract
:1. Introduction
2. Results
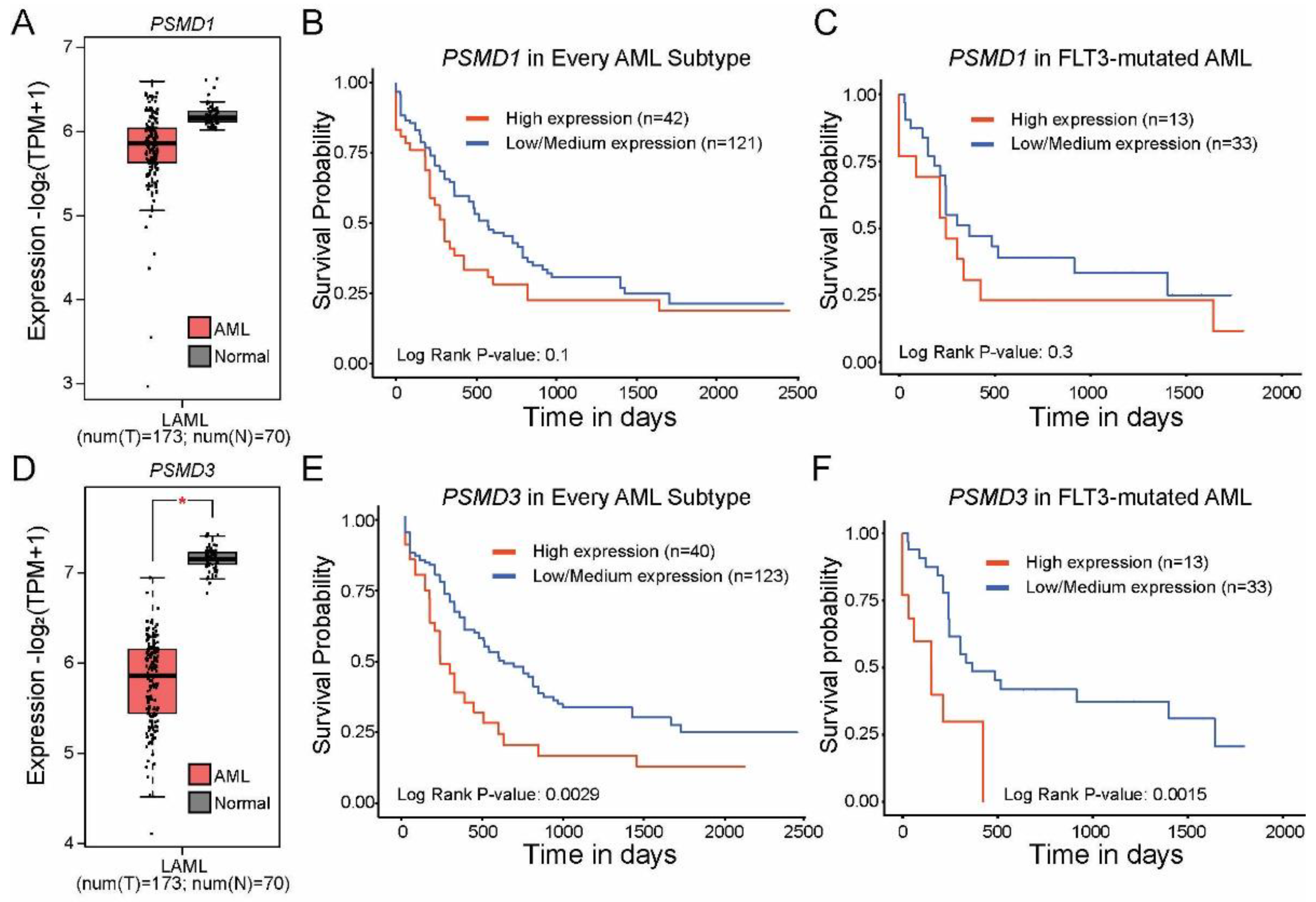
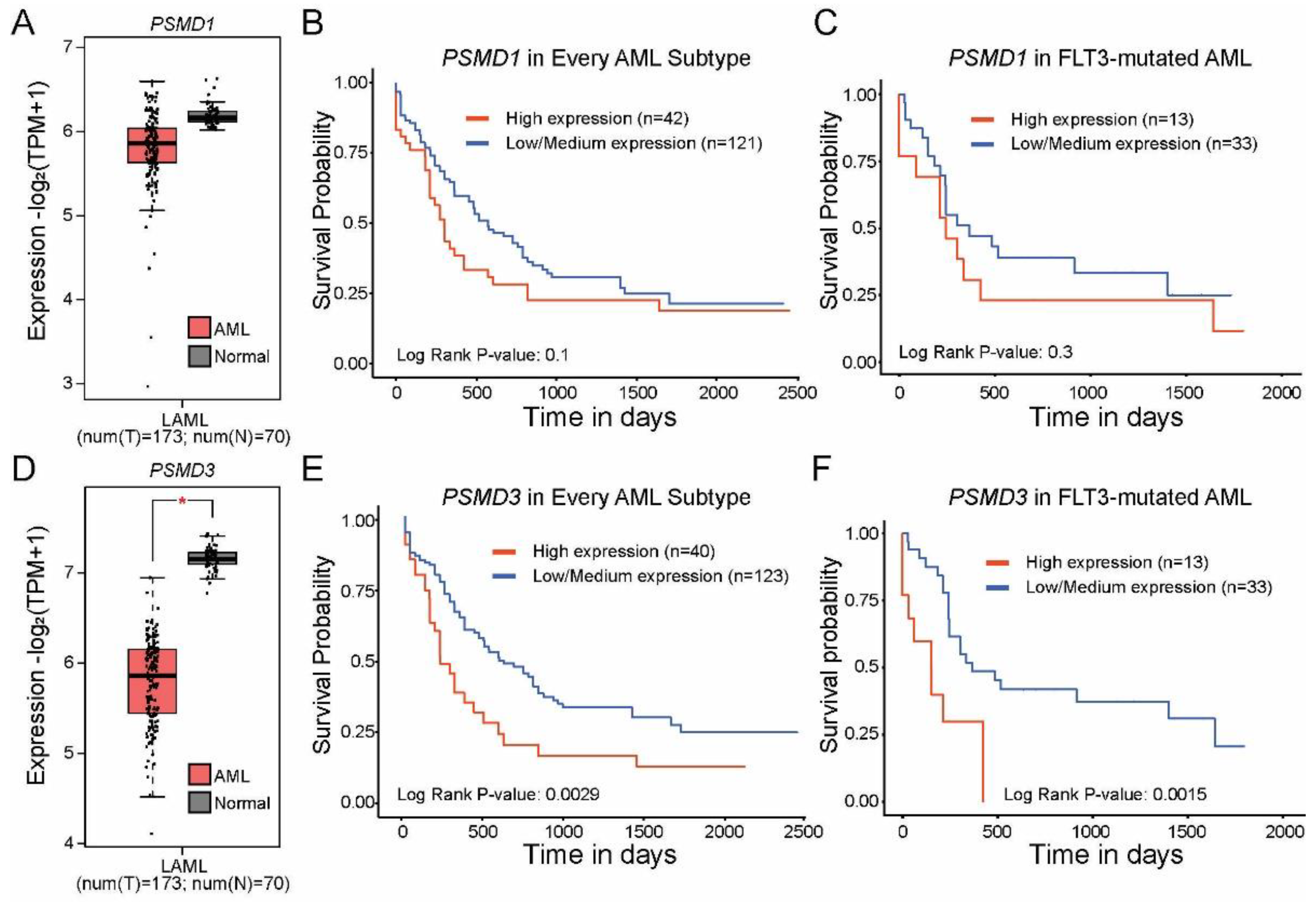
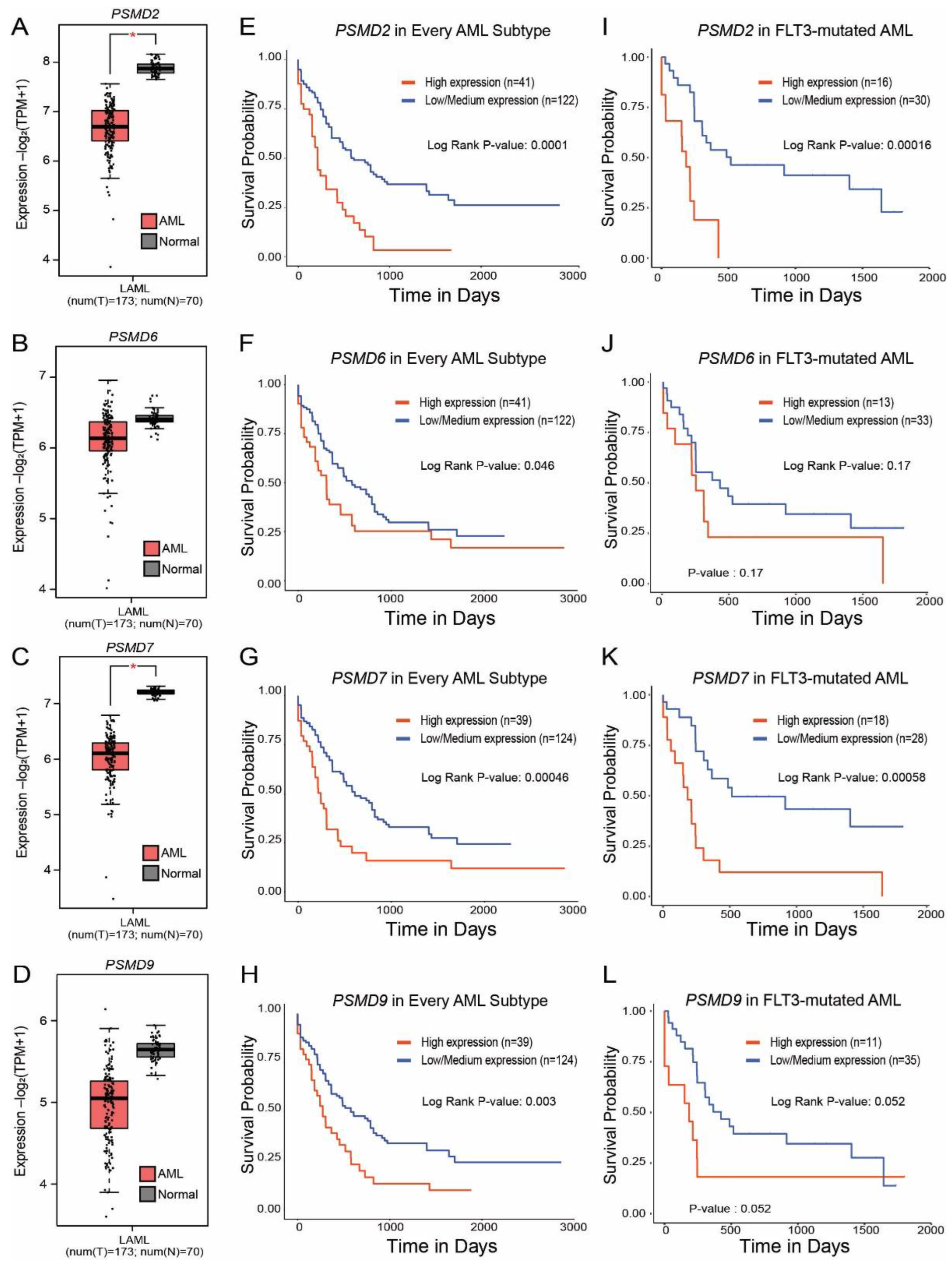
2.1. High Levels of PSMD3 but Not PSMD1 mRNA Expression Correlated with Worse OS in AML Patients with Mutated versus Wild-Type FLT3
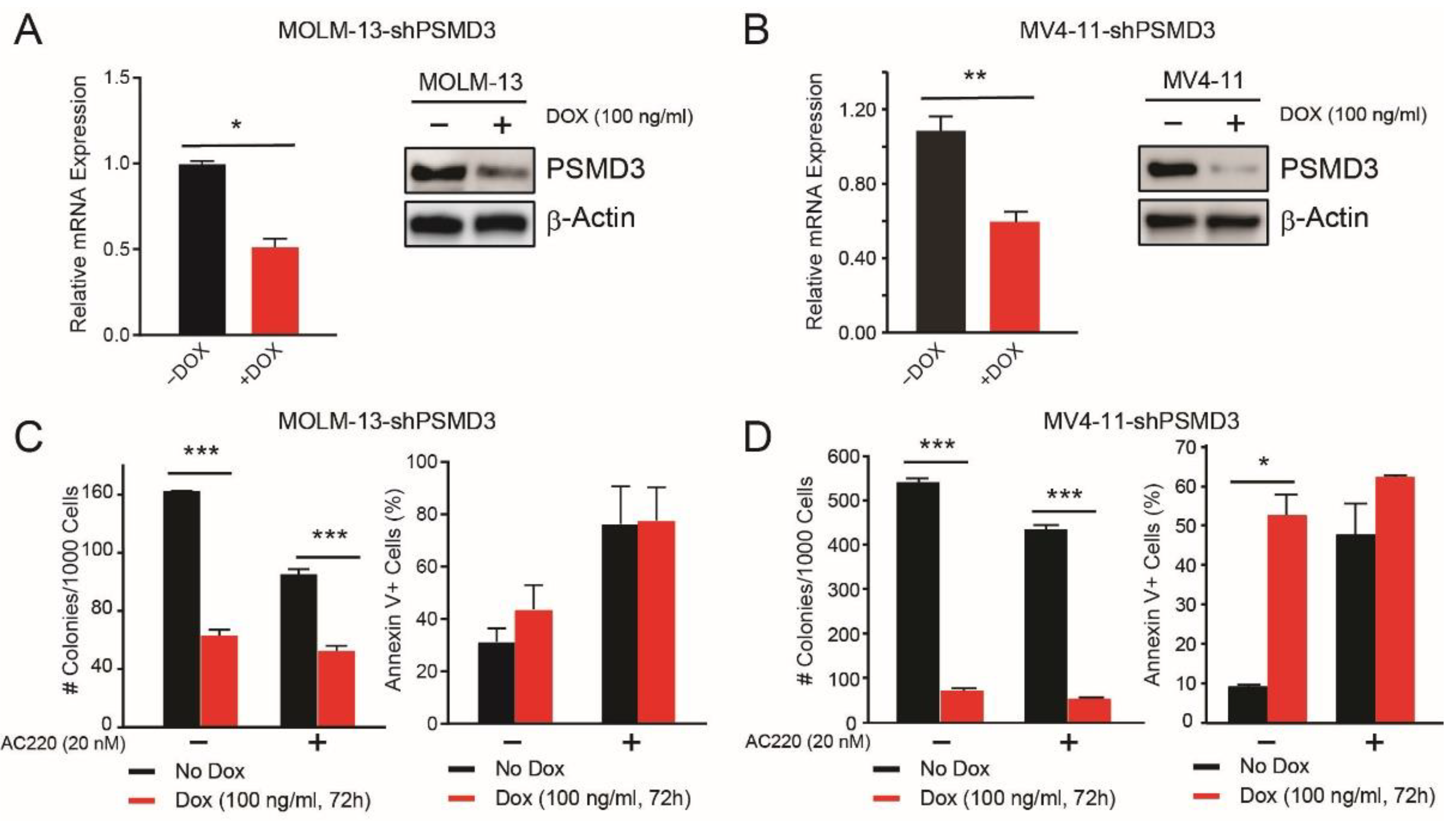
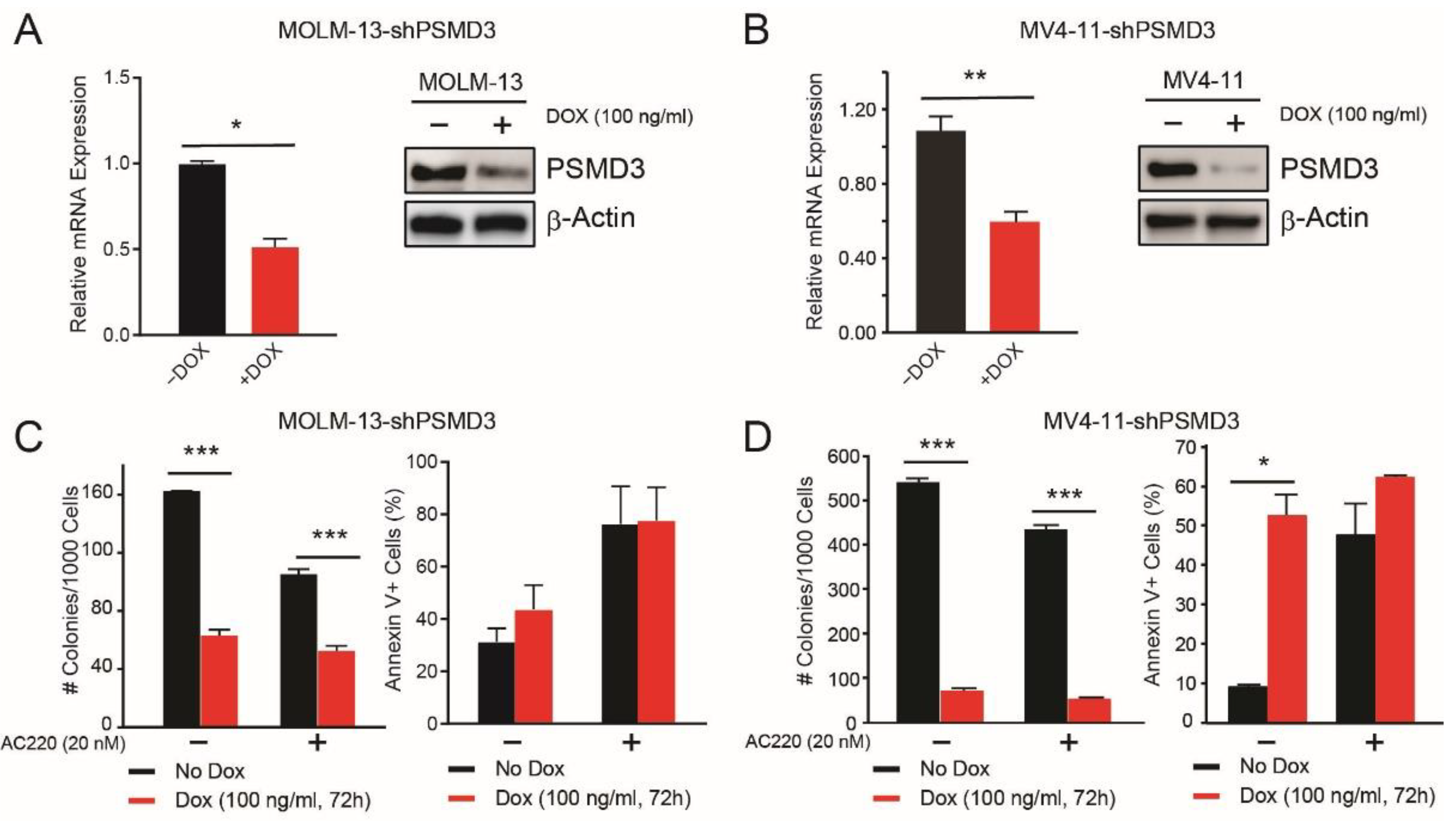
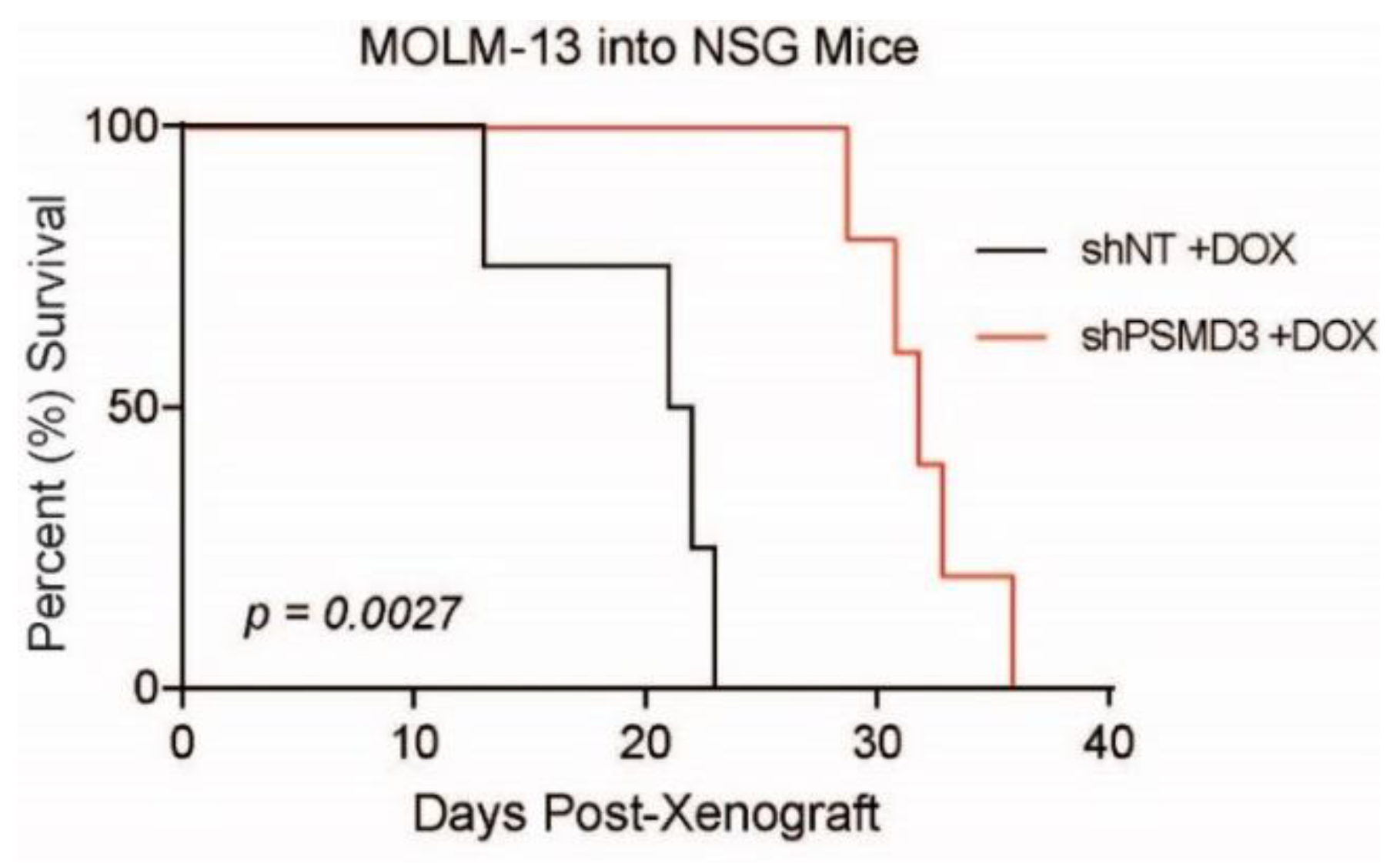
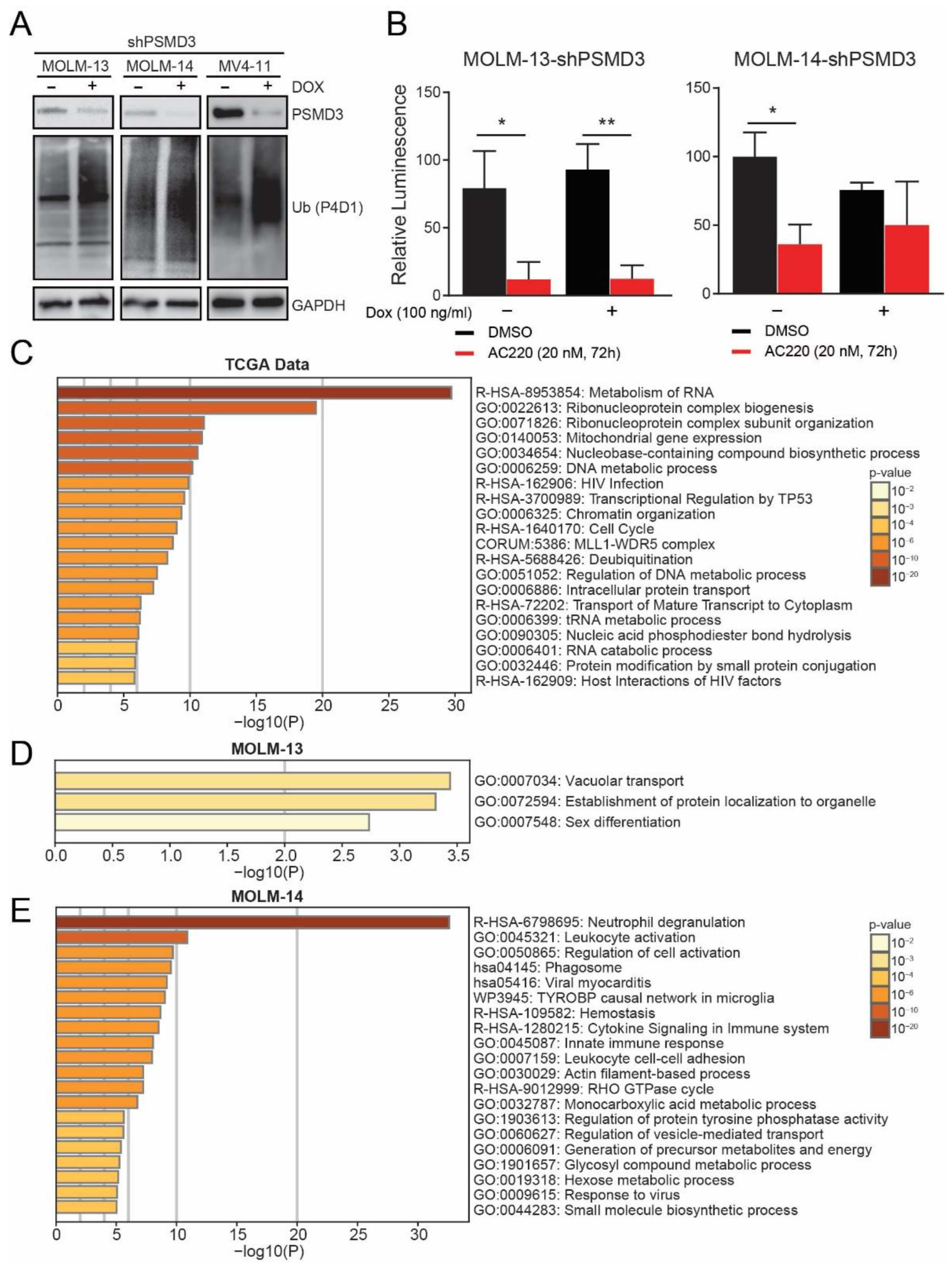
2.2. PSMD3 Knockdown Reduced Survival of FLT3-Mutated AML Cell Lines In Vitro and In Vivo
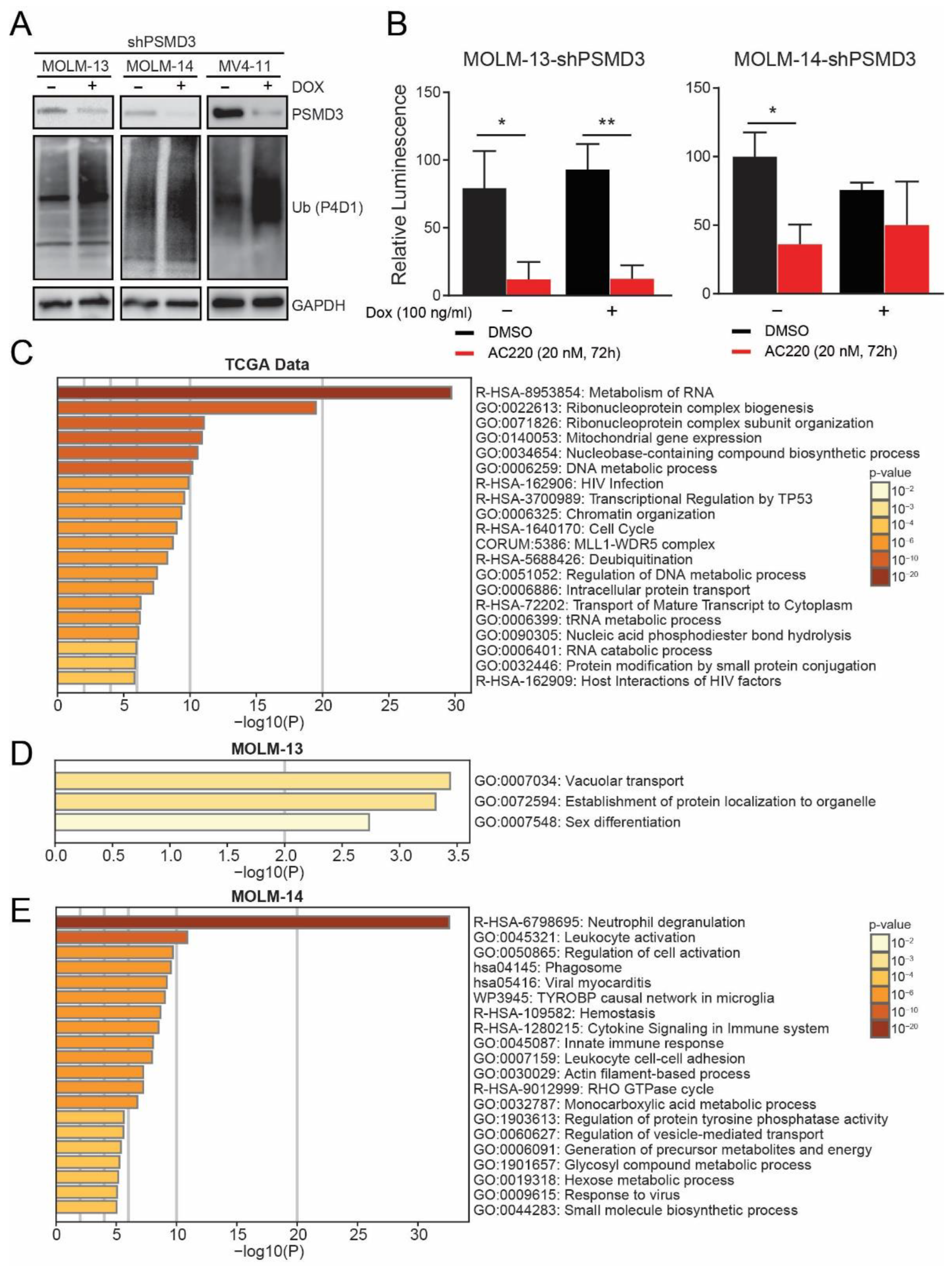
2.3. PSMD3 Knockdown in FLT3+ AML Resulted in An Increase of Global Ubiquitylated Proteins, but Had Little Effect on NF-κB Luciferase Reporter Activity
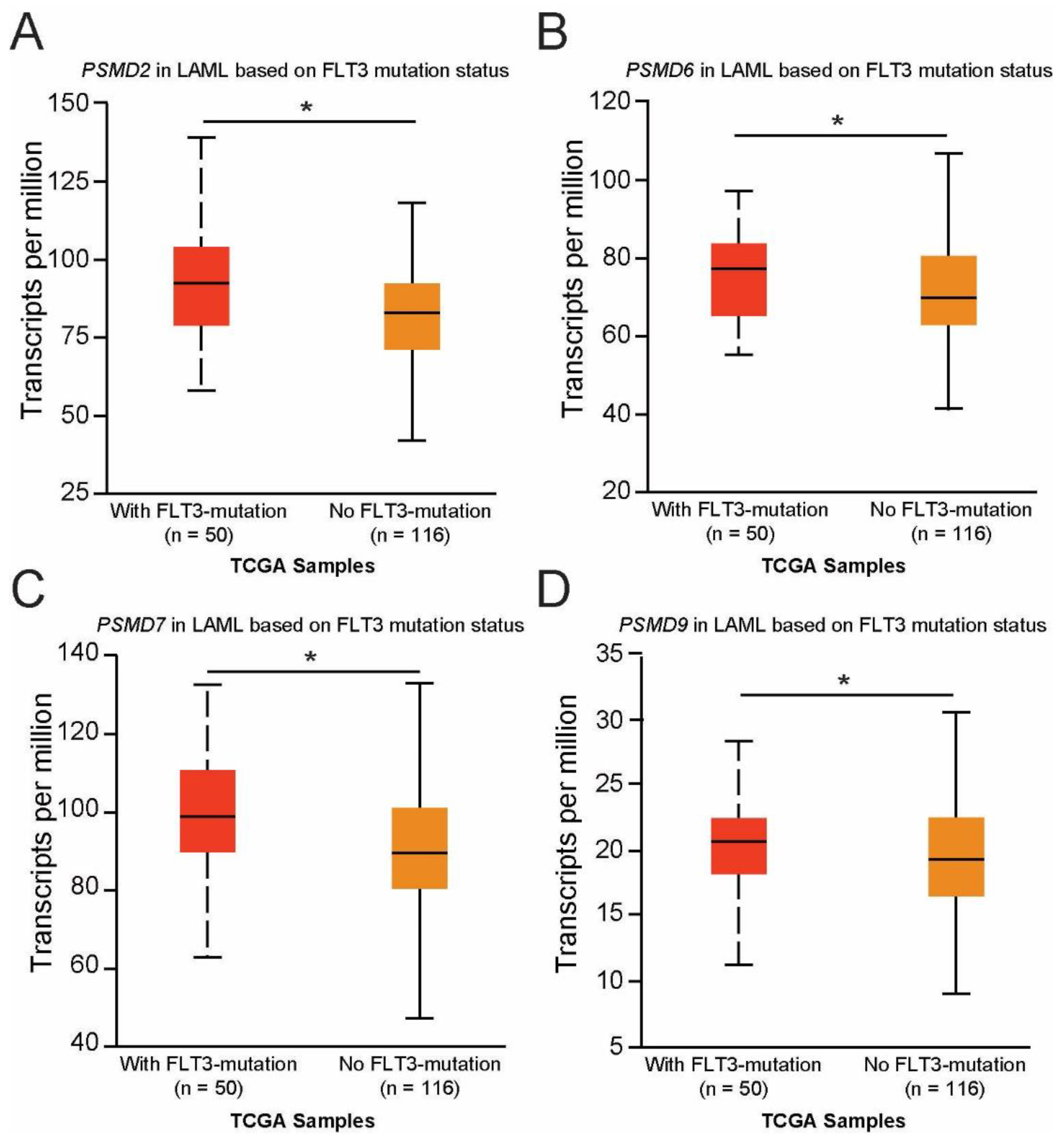
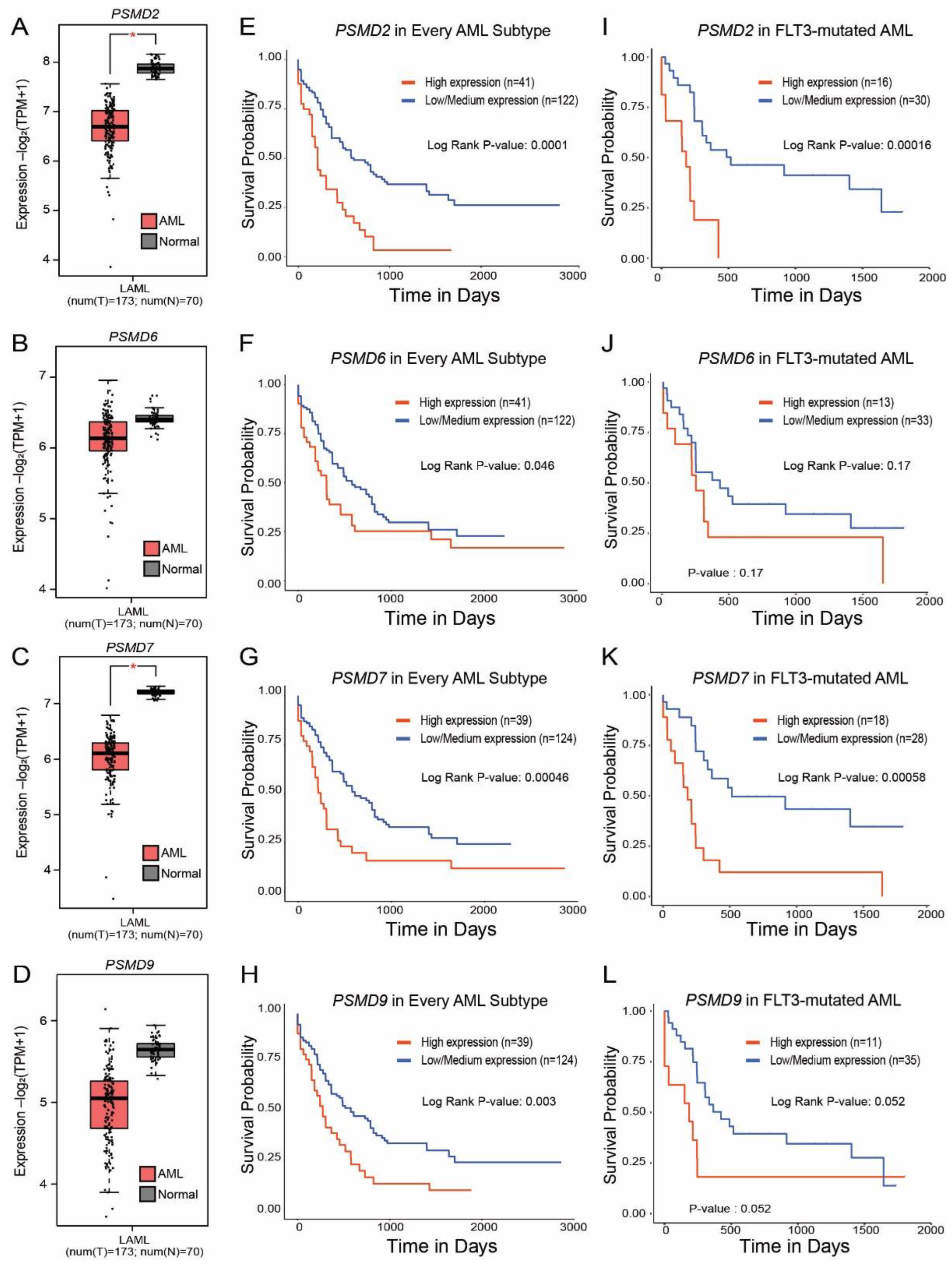
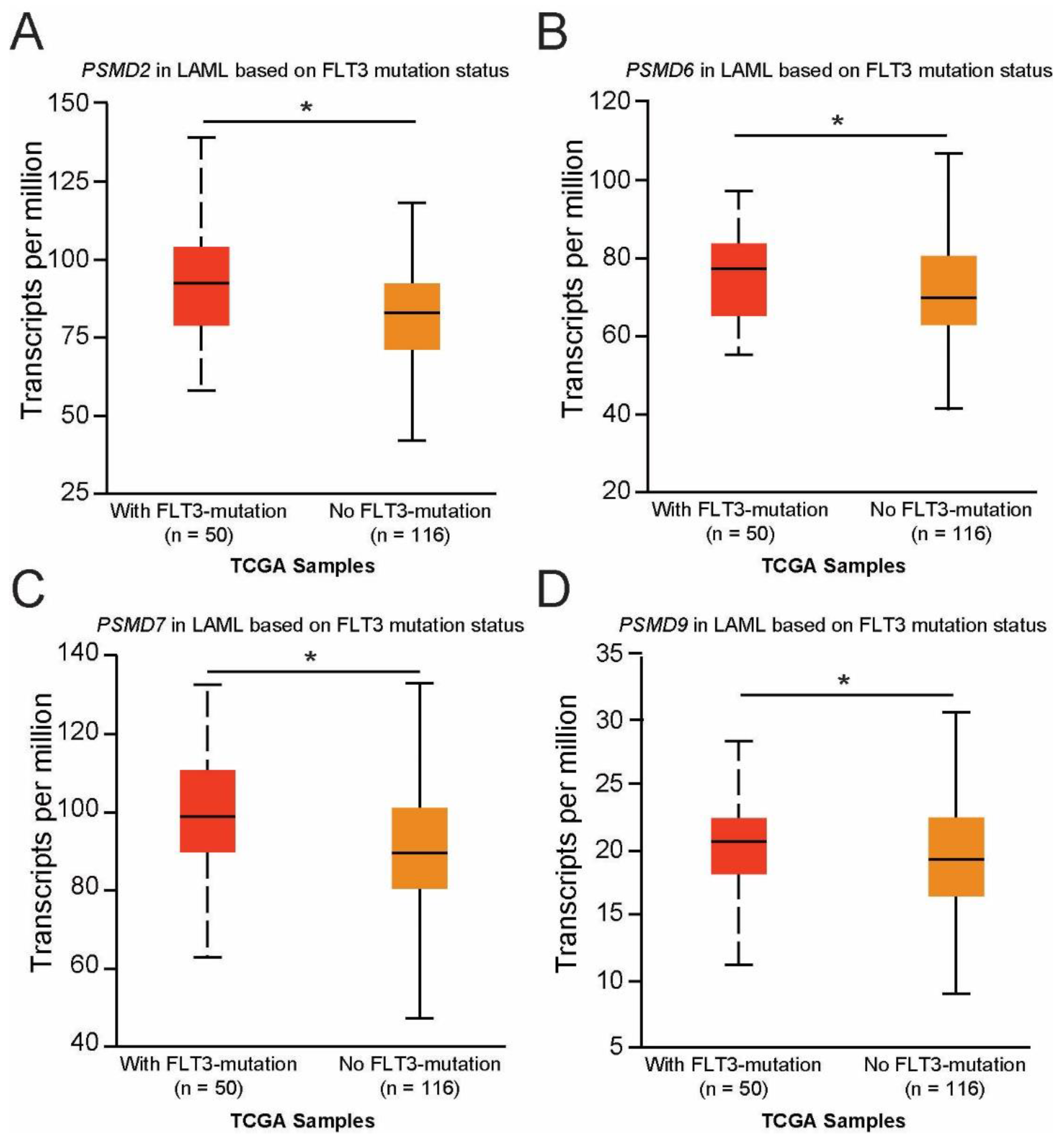
2.4. Expression of PSMD2, PSMD6, PSMD7, and PSMD9 Are Elevated in AML Patients with Mutated versus Wild-Type FLT3, Which Correlated with Worse Outcomes
3. Discussion
4. Materials and Methods
4.1. Analysis of 19S Regulatory Subunit Expression in AML Patients Using Publicly Available Databases
4.2. Cell Lines
4.3. Lentiviral shRNA Constructs
4.4. Analysis of PSMD3 mRNA Expression Using Reverse Transcription-Quantitative Polymerase Chain Reaction (RT-qPCR)
4.5. Immunoblot to Confirm Knockdown of PSMD3 at the Protein Level
4.6. Apoptosis Assays of FLT3+ AML Cells Expressing shPSMD3
4.7. Colony Formation Assays on FLT3+ AML Cells Expressing shPSMD3
4.8. NOD-Scid Il2rgammanull (NSG) AML Xenografts
4.9. NF-κB Luciferase Reporter Assays
4.10. Co-Expression Network of PSMD3 and Functional Enrichment Analysis
4.11. Proteomics Sample Preparation and Analyses
4.12. Genomic Alterations of PSMD Subunits in AML
4.13. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Daver, N.; Schlenk, R.F.; Russell, N.H.; Levis, M.J. Targeting FLT3 mutations in AML: Review of current knowledge and evidence. Leukemia 2019, 33, 299–312. [Google Scholar] [CrossRef] [Green Version]
- McKenna, H.J.; Stocking, K.L.; Miller, R.E.; Brasel, K.; De Smedt, T.; Maraskovsky, E.; Maliszewski, C.R.; Lynch, D.H.; Smith, J.; Pulendran, B.; et al. Mice lacking flt3 ligand have deficient hematopoiesis affecting hematopoietic progenitor cells, dendritic cells, and natural killer cells. Blood 2000, 95, 3489–3497. [Google Scholar] [CrossRef] [PubMed]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Döhner, K.; Marcucci, G.; et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Perl, A.E.; Martinelli, G.; Cortes, J.E.; Neubauer, A.; Berman, E.; Paolini, S.; Montesinos, P.; Baer, M.R.; Larson, R.A.; Ustun, C.; et al. Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML. N. Engl. J. Med. 2019, 381, 1728–1740. [Google Scholar] [CrossRef]
- Ambinder, A.J.; Levis, M. Potential targeting of FLT3 acute myeloid leukemia. Haematologica 2021, 106, 671–681. [Google Scholar] [CrossRef]
- Grundler, R.; Thiede, C.; Miething, C.; Steudel, C.; Peschel, C.; Duyster, J. Sensitivity toward tyrosine kinase inhibitors varies between different activating mutations of the FLT3 receptor. Blood 2003, 102, 646–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagrintseva, K.; Geisenhof, S.; Kern, R.; Eichenlaub, S.; Reindl, C.; Ellwart, J.W.; Hiddemann, W.; Spiekermann, K. FLT3-ITD-TKD dual mutants associated with AML confer resistance to FLT3 PTK inhibitors and cytotoxic agents by overexpression of Bcl-x(L). Blood 2005, 105, 3679–3685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levis, M.; Pham, R.; Smith, B.D.; Small, D. In vitro studies of a FLT3 inhibitor combined with chemotherapy: Sequence of administration is important to achieve synergistic cytotoxic effects. Blood 2004, 104, 1145–1150. [Google Scholar] [CrossRef] [Green Version]
- Cerchione, C.; Peleteiro Raíndo, A.; Mosquera Orgueira, A.; Mosquera Torre, A.; Bao Pérez, L.; Marconi, G.; Isidori, A.; Pérez Encinas, M.M.; Martinelli, G. Safety of FLT3 inhibitors in patients with acute myeloid leukemia. Expert. Rev. Hematol. 2021, 14, 851–865. [Google Scholar] [CrossRef]
- Zhou, J.; Chng, W.J. Resistance to FLT3 inhibitors in acute myeloid leukemia: Molecular mechanisms and resensitizing strategies. World J. Clin. Oncol. 2018, 9, 90–97. [Google Scholar] [CrossRef]
- Rubio, A.J.; Bencomo-Alvarez, A.E.; Young, J.E.; Velazquez, V.V.; Lara, J.J.; Gonzalez, M.A.; Eiring, A.M. 26S proteasome non-ATPase regulatory subunits 1 (PSMD1) and 3 (PSM3) as putative targets for cancer prognosis and therapy. Cells 2021, 10, 2390. [Google Scholar] [CrossRef]
- Liu, C.W.; Jacobson, A.D. Functions of the 19S complex in proteasomal degradation. Trends Biochem. Sci. 2013, 38, 103–110. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K. The proteasome: Overview of structure and functions. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2009, 85, 12–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dou, Q.P.; Zonder, J.A. Overview of proteasome inhibitor-based anti-cancer therapies: Perspective on bortezomib and second generation proteasome inhibitors versus future generation inhibitors of ubiquitin-proteasome system. Curr. Cancer Drug Targets 2014, 14, 517–536. [Google Scholar] [CrossRef] [Green Version]
- Cloos, J.; Roeten, M.S.; Franke, N.E.; van Meerloo, J.; Zweegman, S.; Kaspers, G.J.; Jansen, G. (Immuno)proteasomes as therapeutic target in acute leukemia. Cancer Metastasis. Rev. 2017, 36, 599–615. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Reyes, R.G.; Quinet, G.; Gonzalez-Santamarta, M.; Larrue, C.; Sarry, J.E.; Rodriguez, M.S. Inhibition of the proteasome and proteaphagy enhances apoptosis in FLT3-ITD-driven acute myeloid leukemia. FEBS Open Bio 2021, 11, 48–60. [Google Scholar] [CrossRef]
- Larrue, C.; Saland, E.; Boutzen, H.; Vergez, F.; David, M.; Joffre, C.; Hospital, M.A.; Tamburini, J.; Delabesse, E.; Manenti, S.; et al. Proteasome inhibitors induce FLT3-ITD degradation through autophagy in AML cells. Blood 2016, 127, 882–892. [Google Scholar] [CrossRef] [Green Version]
- Oerlemans, R.; Franke, N.E.; Assaraf, Y.G.; Cloos, J.; van Zantwijk, I.; Berkers, C.R.; Scheffer, G.L.; Debipersad, K.; Vojtekova, K.; Lemos, C.; et al. Molecular basis of bortezomib resistance: Proteasome subunit beta5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood 2008, 112, 2489–2499. [Google Scholar] [CrossRef] [Green Version]
- Cetin, G.; Klafack, S.; Studencka-Turski, M.; Kruger, E.; Ebstein, F. The Ubiquitin-Proteasome System in Immune Cells. Biomolecules 2021, 11, 60. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Shi, K.Z.; Liao, X.Y.; Li, S.J.; Bao, D.; Qian, Y.; Li, D.J. The Silence of PSMC6 Inhibits Cell Growth and Metastasis in Lung Adenocarcinoma. BioMed. Res. Int. 2021, 2021, 9922185. [Google Scholar] [CrossRef] [PubMed]
- Bencomo-Alvarez, A.E.; Rubio, A.J.; Olivas, I.M.; Gonzalez, M.A.; Ellwood, R.; Fiol, C.R.; Eide, C.A.; Lara, J.J.; Barreto-Vargas, C.; Jave-Suarez, L.F.; et al. Proteasome 26S subunit, non-ATPases 1 (PSMD1) and 3 (PSMD3), play an oncogenic role in chronic myeloid leukemia by stabilizing nuclear factor-kappa B. Oncogene 2021, 40, 2697–2710. [Google Scholar] [CrossRef]
- Fararjeh, A.S.; Chen, L.C.; Ho, Y.S.; Cheng, T.C.; Liu, Y.R.; Chang, H.L.; Chang, H.W.; Wu, C.H.; Tu, S.H. Proteasome 26S Subunit, non-ATPase 3 (PSMD3) Regulates Breast Cancer by Stabilizing HER2 from Degradation. Cancers 2019, 11, 527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okumura, T.; Ikeda, K.; Ujihira, T.; Okamoto, K.; Horie-Inoue, K.; Takeda, S.; Inoue, S. Proteasome 26S subunit PSMD1 regulates breast cancer cell growth through p53 protein degradation. J. Biochem. 2018, 163, 19–29. [Google Scholar] [CrossRef]
- Dai, Y.J.; Hu, F.; He, S.Y.; Wang, Y.Y. Epigenetic landscape analysis of lncRNAs in acute myeloid leukemia with DNMT3A mutations. Ann. Transl. Med. 2020, 8, 318. [Google Scholar] [CrossRef]
- Perl, A.E.; Larson, R.A.; Podoltsev, N.A.; Strickland, S.; Wang, E.S.; Atallah, E.; Schiller, G.J.; Martinelli, G.; Neubauer, A.; Sierra, J.; et al. Follow-up of patients with R/R FLT3-mutation-positive AML treated with gilteritinib in the phase 3 ADMIRAL trial. Blood 2022, 139, 3366–3375. [Google Scholar] [CrossRef] [PubMed]
- Numan, Y.; Abdel Rahman, Z.; Grenet, J.; Boisclair, S.; Bewersdorf, J.P.; Collins, C.; Barth, D.; Fraga, M.; Bixby, D.L.; Zeidan, A.M.; et al. Gilteritinib clinical activity in relapsed/refractory FLT3 mutated acute myeloid leukemia previously treated with FLT3 inhibitors. Am. J. Hematol. 2022, 97, 322–328. [Google Scholar] [CrossRef]
- Dennis, M.; Thomas, I.F.; Ariti, C.; Upton, L.; Burnett, A.K.; Gilkes, A.; Radia, R.; Hemmaway, C.; Mehta, P.; Knapper, S.; et al. Randomized evaluation of quizartinib and low-dose ara-C vs low-dose ara-C in older acute myeloid leukemia patients. Blood Adv. 2021, 5, 5621–5625. [Google Scholar] [CrossRef] [PubMed]
- Larson, R.A.; Mandrekar, S.J.; Huebner, L.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Döhner, K.; et al. Midostaurin reduces relapse in FLT3-mutant acute myeloid leukemia: The Alliance CALGB 10603/RATIFY trial. Leukemia 2021, 35, 2539–2551. [Google Scholar] [CrossRef]
- Mo, H.M.; Wu, Q.Y.; Han, D.Y.; Liu, R.; Ma, X.; Zhou, P.; Xu, K.L. Effects of PSMB5 on proliferation and bortezomib chemo-resistance in human myeloma cells and its related molecular mechanisms. Zhonghua Xue Ye Xue Za Zhi 2017, 38, 1053–1057. [Google Scholar]
- Anchoori, R.K.; Karanam, B.; Peng, S.; Wang, J.W.; Jiang, R.; Tanno, T.; Orlowski, R.Z.; Matsui, W.; Zhao, M.; Rudek, M.A.; et al. A bis-benzylidine piperidone targeting proteasome ubiquitin receptor RPN13/ADRM1 as a therapy for cancer. Cancer Cell 2013, 24, 791–805. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Ray, A.; Li, S.; Das, D.S.; Tai, Y.T.; Carrasco, R.D.; Chauhan, D.; Anderson, K.C. Targeting proteasome ubiquitin receptor Rpn13 in multiple myeloma. Leukemia 2016, 30, 1877–1886. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Nowicka, U.; Sridharan, V.; Liu, F.; Randles, L.; Hymel, D.; Dyba, M.; Tarasov, S.G.; Tarasova, N.I.; Zhao, X.Z.; et al. Structure of the Rpn13-Rpn2 complex provides insights for Rpn13 and Uch37 as anticancer targets. Nat. Commun. 2017, 8, 15540. [Google Scholar] [CrossRef] [PubMed]
- VanderLinden, R.T.; Hemmis, C.W.; Yao, T.; Robinson, H.; Hill, C.P. Structure and energetics of pairwise interactions between proteasome subunits RPN2, RPN13, and ubiquitin clarify a substrate recruitment mechanism. J. Biol. Chem. 2017, 292, 9493–9504. [Google Scholar] [CrossRef] [Green Version]
- Hemmis, C.W.; Heard, S.C.; Hill, C.P. Phosphorylation of Tyr-950 in the proteasome scaffolding protein RPN2 modulates its interaction with the ubiquitin receptor RPN13. J. Biol. Chem. 2019, 294, 9659–9665. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Park, P.M.C.; Wu, L.; Ray, A.; Picaud, S.; Li, D.; Wimalasena, V.K.; Du, T.; Filippakopoulos, P.; Anderson, K.C.; et al. Development and preclinical validation of a novel covalent ubiquitin receptor Rpn13 degrader in multiple myeloma. Leukemia 2019, 33, 2685–2694. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Du, T.; Ray, A.; Chauhan, K.; Samur, M.; Munshi, N.; Chauhan, D.; Anderson, K.C. Identification of novel anti-tumor therapeutic target via proteomic characterization of ubiquitin receptor ADRM1/Rpn13. Blood Cancer J. 2021, 11, 13. [Google Scholar] [CrossRef]
- Dickson, P.; Abegg, D.; Vinogradova, E.; Takaya, J.; An, H.; Simanski, S.; Cravatt, B.F.; Adibekian, A.; Kodadek, T. Physical and Functional Analysis of the Putative Rpn13 Inhibitor RA190. Cell Chem. Biol. 2020, 27, 1371–1382.e6. [Google Scholar] [CrossRef]
- Osei-Amponsa, V.; Sridharan, V.; Tandon, M.; Evans, C.N.; Klarmann, K.; Cheng, K.T.; Lack, J.; Chari, R.; Walters, K.J. Impact of Losing hRpn13 Pru or UCHL5 on Proteasome Clearance of Ubiquitinated Proteins and RA190 Cytotoxicity. Mol. Cell. Biol. 2020, 40, 18. [Google Scholar] [CrossRef]
- Soong, R.S.; Anchoori, R.K.; Roden, R.B.S.; Cho, R.L.; Chen, Y.C.; Tseng, S.C.; Huang, Y.L.; Liao, P.C.; Shyu, Y.C. Bis-benzylidine Piperidone RA190 treatment of hepatocellular carcinoma via binding RPN13 and inhibiting NF-kappaB signaling. BMC Cancer 2020, 20, 386. [Google Scholar] [CrossRef]
- Yu, G.Y.; Wang, X.; Zheng, S.S.; Gao, X.M.; Jia, Q.A.; Zhu, W.W.; Lu, L.; Jia, H.L.; Chen, J.H.; Dong, Q.Z.; et al. RA190, a Proteasome Subunit ADRM1 Inhibitor, Suppresses Intrahepatic Cholangiocarcinoma by Inducing NF-KB-Mediated Cell Apoptosis. Cell Physiol. Biochem. 2018, 47, 1152–1166. [Google Scholar] [CrossRef]
- Salah Fararjeh, A.; Al-Khader, A.; Al-Saleem, M.; Abu Qauod, R. The Prognostic Significance of Proteasome 26S Subunit, Non-ATPase (PSMD) Genes for Bladder Urothelial Carcinoma Patients. Cancer Inform. 2021, 20, 11769351211067692. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Guo, Y. A Ubiquitin-Proteasome Gene Signature for Predicting Prognosis in Patients With Lung Adenocarcinoma. Front. Genet. 2022, 13, 893511. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Lu, G. Prognostic Implication and Immunological Role of PSMD2 in Lung Adenocarcinoma. Front. Genet. 2022, 13, 905581. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yang, X.; Xu, X.; Zhang, J.; Zhang, L.; Xu, H.; Miao, Z.; Li, D.; Wang, S. Deubiquitinase PSMD7 regulates cell fate and is associated with disease progression in breast cancer. Am. J. Transl. Res. 2020, 12, 5433–5448. [Google Scholar] [PubMed]
- Xu, X.; Xuan, X.; Zhang, J.; Xu, H.; Yang, X.; Zhang, L.; Zhao, Y.; Xu, H.; Li, D. PSMD7 downregulation suppresses lung cancer progression by regulating the p53 pathway. J. Cancer 2021, 12, 4945–4957. [Google Scholar] [CrossRef]
- Zhang, S.; Yu, S.; Wang, J.; Cheng, Z. Identification of PSMD7 as a prognostic factor correlated with immune infiltration in head and neck squamous cell carcinoma. Biosci. Rep. 2021, 41, 3. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, D.; Zhao, G. Transcriptional activation of Proteasome 26S non-ATPase subunit 7 by forkhead box P3 participates in gastric cancer cell proliferation and apoptosis. Bioengineered 2022, 13, 2525–2536. [Google Scholar] [CrossRef]
- Ud Din Farooqee, S.B.; Christie, J.; Venkatraman, P. PSMD9 ribosomal protein network maintains nucleolar architecture and WT p53 levels. Biochem. Biophys. Res. Commun. 2021, 563, 105–112. [Google Scholar] [CrossRef]
- Lieschke, G.J.; Grail, D.; Hodgson, G.; Metcalf, D.; Stanley, E.; Cheers, C.; Fowler, K.J.; Basu, S.; Zhan, Y.F.; Dunn, A.R. Mice lacking granulocyte colony-stimulating factor have chronic neutropenia, granulocyte and macrophage progenitor cell deficiency, and impaired neutrophil mobilization. Blood 1994, 84, 1737–1746. [Google Scholar] [CrossRef]
- Okada, Y.; Kamatani, Y.; Takahashi, A.; Matsuda, K.; Hosono, N.; Ohmiya, H.; Daigo, Y.; Yamamoto, K.; Kubo, M.; Nakamura, Y.; et al. Common variations in PSMD3-CSF3 and PLCB4 are associated with neutrophil count. Hum. Mol. Genet. 2010, 19, 2079–2085. [Google Scholar] [CrossRef]
- Wang, J.; Liu, R.; Mo, H.; Xiao, X.; Xu, Q.; Zhao, W. Deubiquitinase PSMD7 promotes the proliferation, invasion, and cisplatin resistance of gastric cancer cells by stabilizing RAD23B. Int. J. Biol. Sci. 2021, 17, 3331–3342. [Google Scholar] [CrossRef] [PubMed]
- Kao, T.J.; Wu, C.C.; Phan, N.N.; Liu, Y.H.; Ta, H.D.K.; Anuraga, G.; Wu, Y.F.; Lee, K.H.; Chuang, J.Y.; Wang, C.Y. Prognoses and genomic analyses of proteasome 26S subunit, ATPase (PSMC) family genes in clinical breast cancer. Aging 2021, 13, 17970. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Qi, F.; Wei, C.; Liu, J.; Zhang, Y.; Luan, W.; Gu, J. PSMC2 knockdown suppressed tumor progression of skin cutaneous melanoma. Cell Death Discov. 2021, 7, 323. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Xiong, H.; Zuo, Y.; Hu, S.; Zhu, C.; Min, A. PSMC2 knockdown inhibits the progression of oral squamous cell carcinoma by promoting apoptosis via PI3K/Akt pathway. Cell Cycle 2022, 21, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, J.; Chen, H.; Bianba, T.; Pan, Y.; Wang, X.; Jiang, Y.; Yang, Z. PSMC2 promotes the progression of gastric cancer via induction of RPS15A/mTOR pathway. Oncogenesis 2022, 11, 12. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Wang, Y.; Wang, D.; Wan, J.; Qin, X.; Mu, Z.; Hu, N. PSMC2 is overexpressed in glioma and promotes proliferation and anti-apoptosis of glioma cells. World J. Surg. Oncol. 2022, 20, 84. [Google Scholar] [CrossRef]
- He, Z.; Yang, X.; Huang, L.; Zhou, L.; Zhang, S.; Sun, J.; Zheng, M.; Ma, J.; Feng, B.; Zang, L. PSMC5 Promotes Proliferation and Metastasis of Colorectal Cancer by Activating Epithelial-Mesenchymal Transition Signaling and Modulating Immune Infiltrating Cells. Front. Cell Dev. Biol. 2021, 9, 657917. [Google Scholar] [CrossRef]
- Shi, C.X.; Kortüm, K.M.; Zhu, Y.X.; Bruins, L.A.; Jedlowski, P.; Votruba, P.G.; Luo, M.; Stewart, R.A.; Ahmann, J.; Braggio, E.; et al. CRISPR Genome-Wide Screening Identifies Dependence on the Proteasome Subunit PSMC6 for Bortezomib Sensitivity in Multiple Myeloma. Mol. Cancer Ther. 2017, 16, 2862–2870. [Google Scholar] [CrossRef] [Green Version]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- Khorashad, J.S.; Eiring, A.M.; Mason, C.C.; Gantz, K.C.; Bowler, A.D.; Redwine, H.M.; Yu, F.; Kraft, I.L.; Pomicter, A.D.; Reynolds, K.R.; et al. shRNA library screening identifies nucleocytoplasmic transport as a mediator of BCR-ABL1 kinase-independent resistance. Blood 2015, 125, 1772–1781. [Google Scholar] [CrossRef] [Green Version]
- Eiring, A.M.; Page, B.D.; Kraft, I.L.; Mason, C.C.; Vellore, N.A.; Resetca, D.; Zabriskie, M.S.; Zhang, T.Y.; Khorashad, J.S.; Engar, A.J.; et al. Combined STAT3 and BCR-ABL1 inhibition induces synthetic lethality in therapy-resistant chronic myeloid leukemia. Leukemia 2015, 29, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Alias | Full Gene Name | Chromosome Location |
|---|---|---|---|
| PSMD1 | Rpn2 | Proteasome 26S Subunit, Non-ATPase 1 | chr2:231,056,845–231,173,116 |
| PSMD2 | Rpn1 | Proteasome 26S Subunit Ubiquitin Receptor, Non-ATPase 2 | chr3:184,299,198–184,309,050 |
| PSMD3 | Rpn3 | Proteasome 26S Subunit, Non-ATPase 3 | chr17:39,980,807–39,997,959 |
| PSMD4 | Rpn10 | Proteasome 26S Ubiquitin Receptor, Non-ATPase 4 | chr1:151,254,709–151,267,479 |
| PSMD5 | KIAA0072, S5B | Proteasome 26S Subunit, Non-ATPase 5 | chr9:120,815,496–120,842,951 |
| PSMD6 | Rpn7 | Proteasome 26S Subunit, Non-ATPase 6 | chr3: 64,010,550–64,024,010 |
| PSMD7 | Rpn8 | Proteasome 26S Subunit, Non-ATPase 7 | chr16:74,296,814–74,306,288 |
| PSMD8 | Rpn12 | Proteasome 26S Subunit, Non-ATPase 8 | chr19:38,374,550–38,383,824 |
| PSMD9 | Rpn4 | Proteasome 26S Subunit, Non-ATPase 9 | chr12:121,888,732–121,918,297 |
| PSMD10 | Gankyrin, P28 | Proteasome 26S Subunit, Non-ATPase 10 | chrX:108,084,207–108,091,549 |
| PSMD11 | Rpn6 | Proteasome 26S Subunit, Non-ATPase 11 | chr17:32,444,379–32,483,319 |
| PSMD12 | Rpn5 | Proteasome 26S Subunit, Non-ATPase 12 | chr17:67,337,916–67,366,605 |
| PSMD13 | Rpn9 | Proteasome 26S Subunit, Non-ATPase 13 | chr11:236,966–252,984 |
| PSMD14 | Rpn11 | Proteasome 26S Subunit, Non-ATPase 14 | chr2:161,308,425–161,411,717 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lara, J.J.; Bencomo-Alvarez, A.E.; Gonzalez, M.A.; Olivas, I.M.; Young, J.E.; Lopez, J.L.; Velazquez, V.V.; Glovier, S.; Keivan, M.; Rubio, A.J.; et al. 19S Proteasome Subunits as Oncogenes and Prognostic Biomarkers in FLT3-Mutated Acute Myeloid Leukemia (AML). Int. J. Mol. Sci. 2022, 23, 14586. https://doi.org/10.3390/ijms232314586
Lara JJ, Bencomo-Alvarez AE, Gonzalez MA, Olivas IM, Young JE, Lopez JL, Velazquez VV, Glovier S, Keivan M, Rubio AJ, et al. 19S Proteasome Subunits as Oncogenes and Prognostic Biomarkers in FLT3-Mutated Acute Myeloid Leukemia (AML). International Journal of Molecular Sciences. 2022; 23(23):14586. https://doi.org/10.3390/ijms232314586
Chicago/Turabian StyleLara, Joshua J., Alfonso E. Bencomo-Alvarez, Mayra A. Gonzalez, Idaly M. Olivas, James E. Young, Jose L. Lopez, Vanessa V. Velazquez, Steven Glovier, Mehrshad Keivan, Andres J. Rubio, and et al. 2022. "19S Proteasome Subunits as Oncogenes and Prognostic Biomarkers in FLT3-Mutated Acute Myeloid Leukemia (AML)" International Journal of Molecular Sciences 23, no. 23: 14586. https://doi.org/10.3390/ijms232314586
APA StyleLara, J. J., Bencomo-Alvarez, A. E., Gonzalez, M. A., Olivas, I. M., Young, J. E., Lopez, J. L., Velazquez, V. V., Glovier, S., Keivan, M., Rubio, A. J., Dang, S. K., Solecki, J. P., Allen, J. C., Tapia, D. N., Tychhon, B., Astudillo, G. E., Jordan, C., Chandrashekar, D. S., & Eiring, A. M. (2022). 19S Proteasome Subunits as Oncogenes and Prognostic Biomarkers in FLT3-Mutated Acute Myeloid Leukemia (AML). International Journal of Molecular Sciences, 23(23), 14586. https://doi.org/10.3390/ijms232314586