

Gαq Is the Specific Mediator of PAR-1 Transactivation of Kinase Receptors in Vascular Smooth Muscle Cells

 , ,
, , 
 , and
, and 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
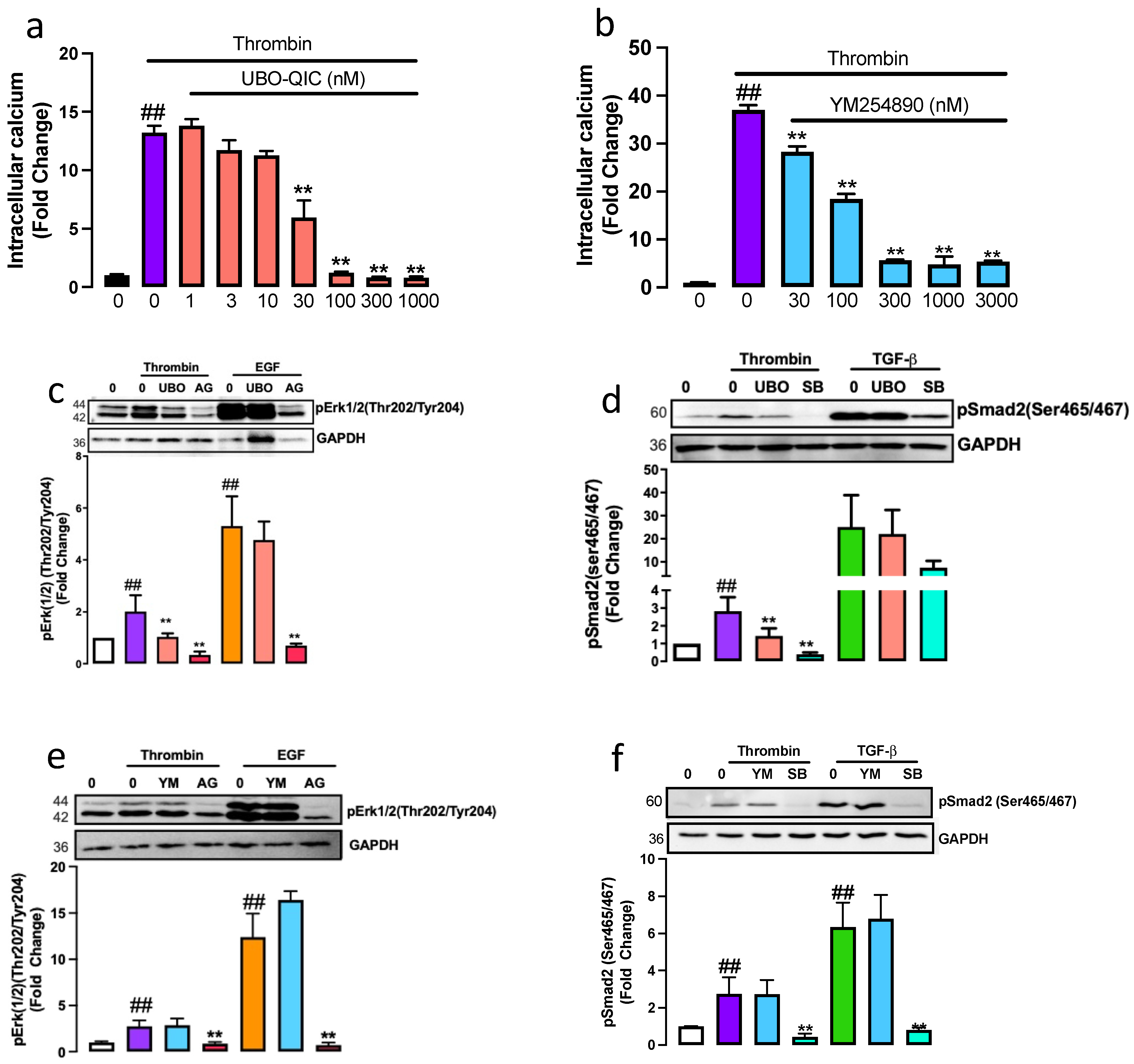
2.1. The Gαq Is Involved in Thrombin-Induced PAR-1 Transactivation of the TGFBR1 and EGFR
2.2. Gαq Is Involved in PAR-1 Mediated mRNA Expression of GAG Enzymes and GAG Chain Elongation
2.3. Thrombin-Mediated GAG Chain Elongation Is Regulated by Gαq but Not Gα11
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Quantification of Intracellular Calcium Release
4.4. Western Blot Analysis
4.5. Quantitative Real-Time-Polymerase Chain Reaction Analysis
4.6. Quantification of Radiolabel Incorporation into Proteoglycans
4.7. Nucleofection Using siRNA
4.8. Immunofluorescent Imaging
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sriram, K.; Insel, P.A. G Protein-Coupled Receptors as Targets for Approved Drugs: How Many Targets and How Many Drugs? Mol. Pharmacol. 2018, 93, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Kamato, D.; Rostam, M.A.; Bernard, R.; Piva, T.J.; Mantri, N.; Guidone, D.; Zheng, W.; Osman, N.; Little, P.J. The expansion of GPCR transactivation-dependent signalling to include serine/threonine kinase receptors represents a new cell signalling frontier. Cell. Mol. Life Sci. 2015, 72, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Kamato, D.; Burch, M.L.; Osman, N.; Zheng, W.; Little, P.J. Therapeutic implications of endothelin and thrombin G-protein-coupled receptor transactivation of tyrosine and serine/threonine kinase cell surface receptors. J. Pharm. Pharmacol. 2012, 65, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Little, P.J.; Burch, M.L.; Al-Aryahi, S.; Zheng, W. The paradigm of g protein receptor transactivation: A mechanistic definition and novel example. Sci. World J. 2011, 11, 709–714. [Google Scholar] [CrossRef] [PubMed]
- Daub, H.; Weiss, F.U.; Wallasch, C.; Ullrich, A. Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature 1996, 379, 557–560. [Google Scholar] [CrossRef]
- Burch, M.L.; Ballinger, M.L.; Yang, S.N.; Getachew, R.; Itman, C.; Loveland, K.; Osman, N.; Little, P.J. Thrombin stimulation of proteoglycan synthesis in vascular smooth muscle is mediated by protease-activated receptor-1 transactivation of the transforming growth factor beta type I receptor. J. Biol. Chem. 2010, 285, 26798–26805. [Google Scholar] [CrossRef]
- Kamato, D.; Ta, H.; Afroz, R.; Xu, S.; Osman, N.; Little, P.J. Mechanisms of PAR-1 mediated kinase receptor transactivation: Smad linker region phosphorylation. J. Cell Commun. Signal. 2019, 13, 539–548. [Google Scholar] [CrossRef]
- Kamato, D.; Thach, L.; Getachew, R.; Burch, M.; Hollenberg, M.D.; Zheng, W.; Little, P.J.; Osman, N. Protease activated receptor-1 mediated dual kinase receptor transactivation stimulates the expression of glycosaminoglycan synthesizing genes. Cell Signal 2016, 28, 110–119. [Google Scholar] [CrossRef]
- Kamato, D.; Bhaskarala, V.V.; Mantri, N.; Oh, T.G.; Ling, D.; Janke, R.; Zheng, W.; Little, P.J.; Osman, N. RNA sequencing to determine the contribution of kinase receptor transactivation to G protein coupled receptor signalling in vascular smooth muscle cells. PLoS ONE 2017, 12, e0180842. [Google Scholar] [CrossRef]
- George, A.J.; Purdue, B.W.; Gould, C.M.; Thomas, D.W.; Handoko, Y.; Qian, H.; Quaife-Ryan, G.A.; Morgan, K.A.; Simpson, K.J.; Thomas, W.G.; et al. A functional siRNA screen identifies genes modulating angiotensin II-mediated EGFR transactivation. J. Cell Sci. 2013, 126, 5377–5390. [Google Scholar] [CrossRef]
- Prenzel, N.; Zwick, E.; Daub, H.; Leserer, M.; Abraham, R.; Wallasch, C.; Ullrich, A. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature 1999, 402, 884–888. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Chen, Y.-G. Regulation of TGF-β receptor activity. Cell Biosci. 2012, 2, 9. [Google Scholar] [CrossRef] [PubMed]
- Nigro, J.; Osman, N.; Dart, A.M.; Little, P.J. Insulin Resistance and Atherosclerosis. Endocr. Rev. 2006, 27, 242–259. [Google Scholar] [CrossRef]
- Afroz, R.; Cao, Y.; Rostam, M.A.; Ta, H.; Xu, S.; Zheng, W.; Osman, N.; Kamato, D.; Little, P.J. Signalling pathways regulating galactosaminoglycan synthesis and structure in vascular smooth muscle: Implications for lipoprotein binding and atherosclerosis. Pharmacol. Ther. 2018, 187, 88–97. [Google Scholar] [CrossRef]
- Burch, M.L.; Getachew, R.; Osman, N.; Febbraio, M.A.; Little, P.J. Thrombin-mediated proteoglycan synthesis utilizes both protein-tyrosine kinase and serine/threonine kinase receptor transactivation in vascular smooth muscle cells. J. Biol. Chem. 2013, 288, 7410–7419. [Google Scholar] [CrossRef] [PubMed]
- Syrovatkina, V.; Alegre, K.O.; Dey, R.; Huang, X.Y. Regulation, Signaling, and Physiological Functions of G-Proteins. J. Mol. Biol. 2016, 428, 3850–3868. [Google Scholar] [CrossRef]
- Kamato, D.; Thach, L.; Bernard, R.; Chan, V.; Zheng, W.; Kaur, H.; Brimble, M.; Osman, N.; Little, P.J. Structure, Function, Pharmacology, and Therapeutic Potential of the G Protein, Galpha/q,11. Front. Cardiovasc. Med. 2015, 2, 14. [Google Scholar] [CrossRef]
- Kamato, D.; Mitra, P.; Davis, F.; Osman, N.; Chaplin, R.; Cabot, P.J.; Afroz, R.; Thomas, W.; Zheng, W.; Kaur, H.; et al. Gaq proteins: Molecular pharmacology and therapeutic potential. Cell. Mol. Life Sci. 2017, 74, 1379–1390. [Google Scholar] [CrossRef]
- Zaima, K.; Deguchi, J.; Matsuno, Y.; Kaneda, T.; Hirasawa, Y.; Morita, H. Vasorelaxant effect of FR900359 from Ardisia crenata on rat aortic artery. J. Nat. Med. 2013, 67, 196–201. [Google Scholar] [CrossRef]
- Kawasaki, T.; Taniguchi, M.; Moritani, Y.; Hayashi, K.; Saito, T.; Takasaki, J.; Nagai, K.; Inagaki, O.; Shikama, H. Antithrombotic and thrombolytic efficacy of YM-254890, a G q/11 inhibitor, in a rat model of arterial thrombosis. Thromb. Haemost. 2003, 90, 406–413. [Google Scholar] [CrossRef]
- Gao, Z.G.; Jacobson, K.A. On the selectivity of the Gαq inhibitor UBO-QIC: A comparison with the Gαi inhibitor pertussis toxin. Biochem. Pharmacol. 2016, 107, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Kukkonen, J.P. G-protein inhibition profile of the reported Gq/11 inhibitor UBO-QIC. Biochem. Biophys. Res. Commun. 2016, 469, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Alqahtani, S.; Nasrullah, M.Z.A.; Shen, J. Functional evidence for biased inhibition of G protein signaling by YM-254890 in human coronary artery endothelial cells. Eur. J. Pharmacol. 2021, 891, 173706. [Google Scholar] [CrossRef] [PubMed]
- Chaplin, R.; Thach, L.; Hollenberg, M.D.; Cao, Y.; Little, P.J.; Kamato, D. Insights into cellular signalling by G protein coupled receptor transactivation of cell surface protein kinase receptors. J. Cell Commun. Signal. 2017, 11, 117–125. [Google Scholar] [CrossRef]
- Mohamed, R.; Janke, R.; Guo, W.; Cao, Y.; Zhou, Y.; Zheng, W.; Babaahmadi-Rezaei, H.; Xu, S.; Kamato, D.; Little, P.J. GPCR transactivation signalling in vascular smooth muscle cells: Role of NADPH oxidases and reactive oxygen species. Vasc. Biol. 2019, 1, R1–R11. [Google Scholar] [CrossRef]
- Neylon, C.B.; Nickashin, A.; Little, P.J.; Tkachuk, V.A.; Bobik, A. Thrombin-induced Ca2+ mobilization in vascular smooth muscle utilizes a slowly ribosylating pertussis toxin-sensitive G protein. Evidence for the involvement of a G protein in inositol trisphosphate-dependent Ca2+ release. J. Biol. Chem. 1992, 267, 7295–7302. [Google Scholar] [CrossRef]
- Pierce, K.L.; Tohgo, A.; Ahn, S.; Field, M.E.; Luttrell, L.M.; Lefkowitz, R.J. Epidermal Growth Factor (EGF) Receptor-dependent ERK Activation by G Protein-coupled Receptors: A Co-Culture System for Identifying Intermediates Upstream and Downstream of Heparin-Binding EGF Shedding. J. Biol. Chem. 2001, 276, 23155–23160. [Google Scholar] [CrossRef]
- Zhou, Y.; Little, P.J.; Cao, Y.; Ta, H.T.; Kamato, D. Lysophosphatidic acid receptor 5 transactivation of TGFBR1 stimulates the mRNA expression of proteoglycan synthesizing genes XYLT1 and CHST3. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118848. [Google Scholar] [CrossRef]
- Jenkins, R.G.; Su, X.; Su, G.; Scotton, C.J.; Camerer, E.; Laurent, G.J.; Davis, G.E.; Chambers, R.C.; Matthay, M.A.; Sheppard, D. Ligation of protease-activated receptor 1 enhances alpha(v)beta6 integrin-dependent TGF-beta activation and promotes acute lung injury. J. Clin. Investig. 2006, 116, 1606–1614. [Google Scholar] [CrossRef]
- Tatler, A.L.; John, A.E.; Jolly, L.; Habgood, A.; Porte, J.; Brightling, C.; Knox, A.J.; Pang, L.; Sheppard, D.; Huang, X. Integrin αvβ5-mediated TGF-β activation by airway smooth muscle cells in asthma. J. Immunol. 2011, 187, 6094–6107. [Google Scholar] [CrossRef]
- Cattaneo, F.; Guerra, G.; Parisi, M.; De Marinis, M.; Tafuri, D.; Cinelli, M.; Ammendola, R. Cell-surface receptors transactivation mediated by g protein-coupled receptors. Int. J. Mol. Sci. 2014, 15, 19700–19728. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, Y.; Fujii, H.; Sumiyoshi, S.; Wight, T.N.; Sueishi, K. Early human atherosclerosis: Accumulation of lipid and proteoglycans in intimal thickenings followed by macrophage infiltration. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1159–1165. [Google Scholar] [CrossRef] [PubMed]
- Babaahmadi-Rezaei, H.; Little, P.J.; Mohamed, R.; Zadeh, G.M.; Kheirollah, A.; Mehr, R.N.; Kamato, D.; Dayati, P. Endothelin-1 mediated glycosaminoglycan synthesizing gene expression involves NOX-dependent transactivation of the transforming growth factor-beta receptor. Mol. Cell. Biochem. 2022, 441, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Little, P.J.; Burch, M.L.; Getachew, R.; Al-aryahi, S.; Osman, N. Endothelin-1 stimulation of proteoglycan synthesis in vascular smooth muscle is mediated by endothelin receptor transactivation of the transforming growth factor-[beta] type I receptor. J. Cardiovasc. Pharmacol. 2010, 56, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Little, P.J.; Xu, S.; Kamato, D. Curcumin Inhibits Lysophosphatidic Acid Mediated MCP-1 Expression via Blocking ROCK Signalling. Molecules 2021, 26, 2320. [Google Scholar] [CrossRef]
- Tietze, D.; Kaufmann, D.; Tietze, A.A.; Voll, A.; Reher, R.; König, G.; Hausch, F. Structural and Dynamical Basis of G Protein Inhibition by YM-254890 and FR900359: An Inhibitor in Action. J. Chem. Inf. Model. 2019, 59, 4361–4373. [Google Scholar] [CrossRef]
- Schrage, R.; Schmitz, A.L.; Gaffal, E.; Annala, S.; Kehraus, S.; Wenzel, D.; Bullesbach, K.M.; Bald, T.; Inoue, A.; Shinjo, Y.; et al. The experimental power of FR900359 to study Gq-regulated biological processes. Nat. Commun. 2015, 6, 10156. [Google Scholar] [CrossRef]
- Boesgaard, M.W.; Harpsøe, K.; Malmberg, M.; Underwood, C.R.; Inoue, A.; Mathiesen, J.M.; König, G.M.; Kostenis, E.; Gloriam, D.E.; Bräuner-Osborne, H. Delineation of molecular determinants for FR900359 inhibition of Gq/11 unlocks inhibition of Gαs. J. Biol. Chem. 2020, 295, 13850–13861. [Google Scholar] [CrossRef]
- Taboubi, S.; Garrouste, F.; Parat, F.; Pommier, G.; Faure, E.; Monferran, S.; Kovacic, H.; Lehmann, M. Gq-coupled purinergic receptors inhibit insulin-like growth factor-I/phosphoinositide 3-kinase pathway-dependent keratinocyte migration. Mol. Biol. Cell 2010, 21, 946–955. [Google Scholar] [CrossRef]
- Harada, T.; Horinouchi, T.; Higa, T.; Hoshi, A.; Higashi, T.; Terada, K.; Mai, Y.; Nepal, P.; Horiguchi, M.; Hatate, C.; et al. Endothelin-1 activates extracellular signal-regulated kinases 1/2 via transactivation of platelet-derived growth factor receptor in rat L6 myoblasts. Life Sci. 2014, 104, 24–31. [Google Scholar] [CrossRef]
- Barauna, V.G.; Magalhaes, F.C.; Campos, L.C.; Reis, R.I.; Kunapuli, S.P.; Costa-Neto, C.M.; Miyakawa, A.A.; Krieger, J.E. Shear stress-induced Ang II AT1 receptor activation: G-protein dependent and independent mechanisms. Biochem. Biophys. Res. Commun. 2013, 434, 647–652. [Google Scholar] [CrossRef]
- O’Brien, S.L.; Johnstone, E.K.M.; Devost, D.; Conroy, J.; Reichelt, M.E.; Purdue, B.W.; Ayoub, M.A.; Kawai, T.; Inoue, A.; Eguchi, S.; et al. BRET-based assay to monitor EGFR transactivation by the AT1R reveals Gq/11 protein-independent activation and AT1R-EGFR complexes. Biochem. Pharmacol. 2018, 158, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.Y.; Porte, J.; Knox, A.J.; Weinreb, P.H.; Maher, T.M.; Violette, S.M.; McAnulty, R.J.; Sheppard, D.; Jenkins, G. Lysophosphatidic acid induces alphavbeta6 integrin-mediated TGF-beta activation via the LPA2 receptor and the small G protein G alpha(q). Am. J. Pathol. 2009, 174, 1264–1279. [Google Scholar] [CrossRef]
- Tanski, W.J.; Roztocil, E.; Hemady, E.A.; Williams, J.A.; Davies, M.G. Role of Galphaq in smooth muscle cell proliferation. J. Vasc. Surg. 2004, 39, 639–644. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mukai, H.; Munekata, E.; Higashijima, T. G protein antagonists. A novel hydrophobic peptide competes with receptor for G protein binding. J. Biol. Chem. 1992, 267, 16237–16243. [Google Scholar] [CrossRef]
- Jiang, Y.-P.; Ballou, L.M.; Lu, Z.; Li, W.; Kelly, D.J.; Cohen, I.S.; Lin, R.Z. Reversible Heart Failure in Gαq Transgenic Mice. J. Biol. Chem. 2006, 281, 29988–29992. [Google Scholar] [CrossRef] [PubMed]
- Uemura, T.; Kawasaki, T.; Taniguchi, M.; Moritani, Y.; Hayashi, K.; Saito, T.; Takasaki, J.; Uchida, W.; Miyata, K. Biological properties of a specific Galpha q/11 inhibitor, YM-254890, on platelet functions and thrombus formation under high-shear stress. Br. J. Pharmacol. 2006, 148, 61–69. [Google Scholar] [CrossRef]
- Kawasaki, T.; Taniguchi, M.; Moritani, Y.; Uemura, T.; Shigenaga, T.; Takamatsu, H.; Hayashi, K.; Takasaki, J.; Saito, T.; Nagai, K. Pharmacological properties of YM-254890, a specific G(alpha)q/11 inhibitor, on thrombosis and neointima formation in mice. Thromb. Haemost. 2005, 94, 184–192. [Google Scholar] [CrossRef]
- Neylon, C.B.; Little, P.J.; Cragoe, E.J., Jr.; Bobik, A. Intracellular pH in human arterial smooth muscle. Regulation by Na+/H+ exchange and a novel 5-(N-ethyl-N-isopropyl)amiloride-sensitive Na(+)- and HCO3(-)-dependent mechanism. Circ. Res. 1990, 67, 814–825. [Google Scholar] [CrossRef] [PubMed]
- Nigro, J.; Dilley, R.J.; Little, P.J. Differential effects of gemfibrozil on migration, proliferation and proteoglycan production in human vascular smooth muscle cells. Atherosclerosis 2002, 162, 119–129. [Google Scholar] [CrossRef]
- Rostam, M.A.; Shajimoon, A.; Kamato, D.; Mitra, P.; Piva, T.; Getachew, R.; Cao, Y.; Zheng, W.; Osman, N.; Little, P.J. Flavopiridol inhibits TGF-beta-stimulated biglycan synthesis by blocking linker region phosphorylation and nuclear translocation of Smad2. J. Pharmacol. Exp. Ther. 2018, 365, 156–164. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamato, D.; Gabr, M.; Kumarapperuma, H.; Chia, Z.J.; Zheng, W.; Xu, S.; Osman, N.; Little, P.J. Gαq Is the Specific Mediator of PAR-1 Transactivation of Kinase Receptors in Vascular Smooth Muscle Cells. Int. J. Mol. Sci. 2022, 23, 14425. https://doi.org/10.3390/ijms232214425
Kamato D, Gabr M, Kumarapperuma H, Chia ZJ, Zheng W, Xu S, Osman N, Little PJ. Gαq Is the Specific Mediator of PAR-1 Transactivation of Kinase Receptors in Vascular Smooth Muscle Cells. International Journal of Molecular Sciences. 2022; 23(22):14425. https://doi.org/10.3390/ijms232214425
Chicago/Turabian StyleKamato, Danielle, Mai Gabr, Hirushi Kumarapperuma, Zheng J. Chia, Wenhua Zheng, Suowen Xu, Narin Osman, and Peter J. Little. 2022. "Gαq Is the Specific Mediator of PAR-1 Transactivation of Kinase Receptors in Vascular Smooth Muscle Cells" International Journal of Molecular Sciences 23, no. 22: 14425. https://doi.org/10.3390/ijms232214425
APA StyleKamato, D., Gabr, M., Kumarapperuma, H., Chia, Z. J., Zheng, W., Xu, S., Osman, N., & Little, P. J. (2022). Gαq Is the Specific Mediator of PAR-1 Transactivation of Kinase Receptors in Vascular Smooth Muscle Cells. International Journal of Molecular Sciences, 23(22), 14425. https://doi.org/10.3390/ijms232214425