
Long-Term In Vitro Assessment of Biodegradable Radiopaque Composites for Fiducial Marker Fabrication



Abstract
1. Introduction
2. Results
2.1. Fabrication of Experimental Samples
2.2. Degradation Experiment
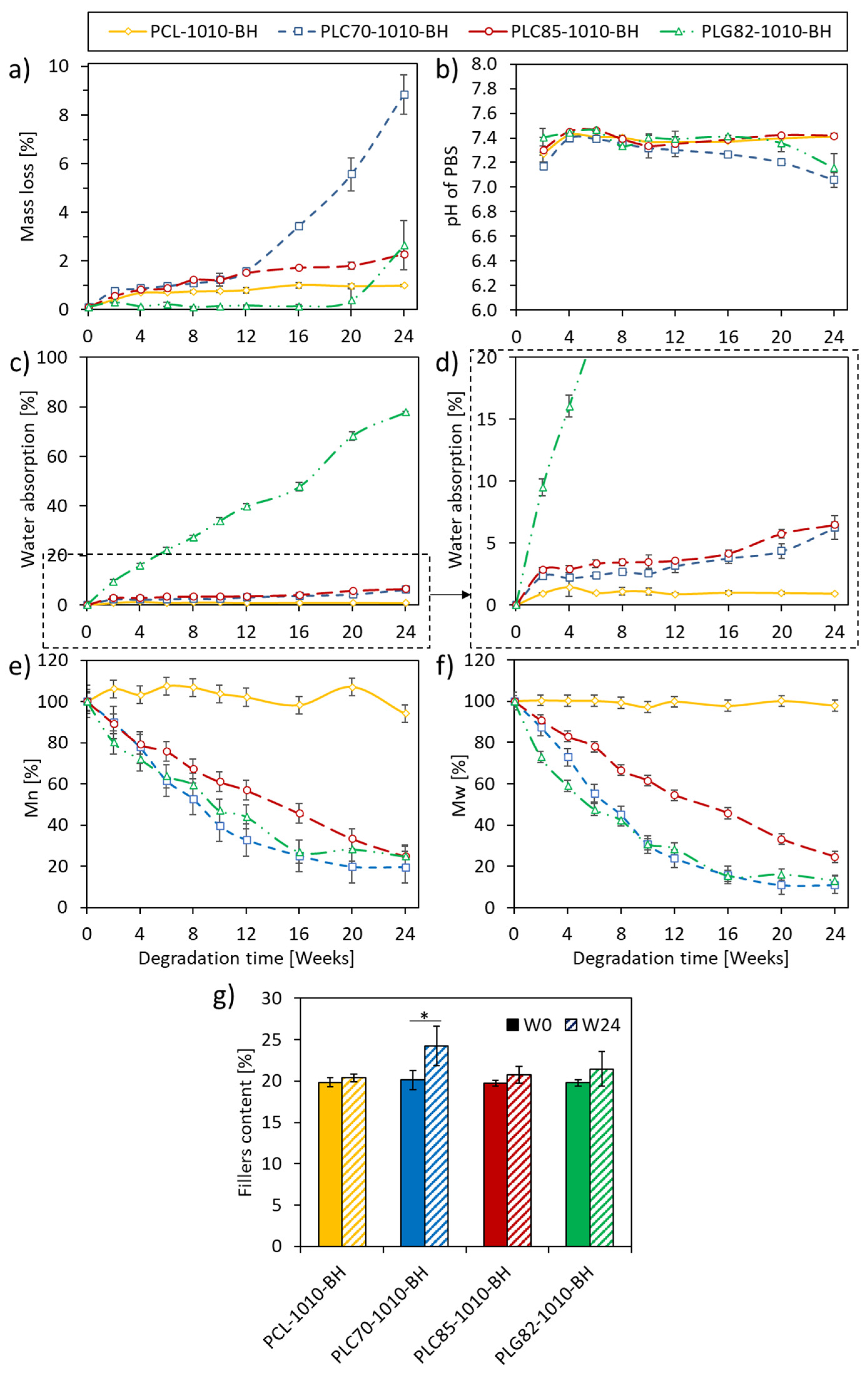
2.3. Mass Loss
2.4. pH of PBS
2.5. Water Absorption
2.6. MW Loss
2.7. Filler Content
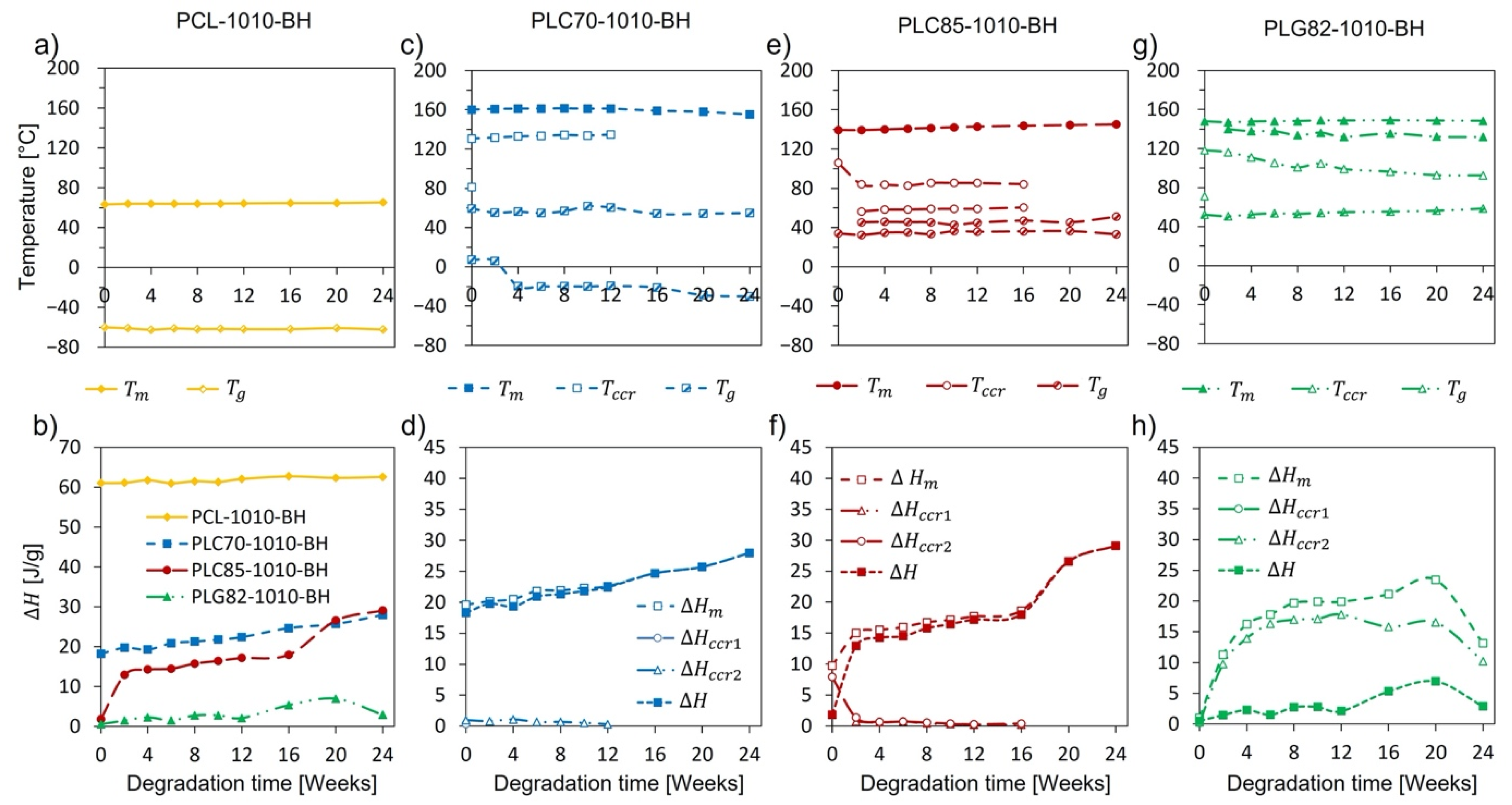
2.8. Thermal Properties
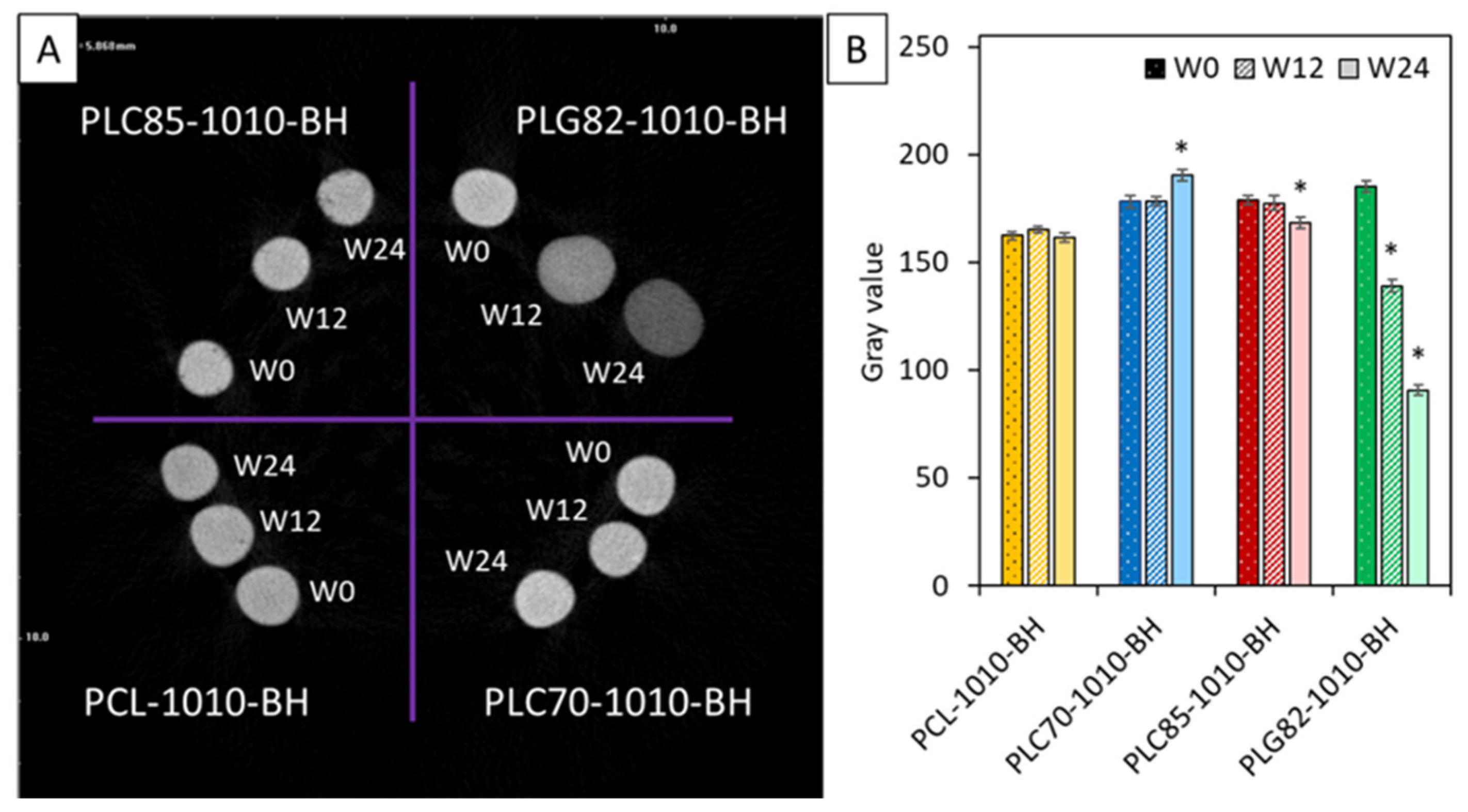
2.9. Contrasting Properties
2.10. Mechanical Properties
2.11. Surface Properties
3. Discussion
3.1. PCL-Based Composite
3.2. PLC70-Based Composite
3.3. PLC85-Based Composite
3.4. PLG82-Based Composite
4. Materials and Methods
4.1. Materials Characterisation
4.1.1. Incubation in PBS
4.1.2. MicroCT
4.1.3. Mechanical Testing
4.1.4. Water Contact Angle Measurements
4.1.5. SEM
4.1.6. GPC
4.1.7. TGA
4.1.8. DSC
4.1.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kong, S.-H.; Haouchine, N.; Soares, R.; Klymchenko, A.S.; Andreiuk, B.; Marques, B.; Shabat, G.; Piechaud, T.; Diana, M.; Cotin, S.; et al. Robust augmented reality registration method for localization of solid organs’ tumors using CT-derived virtual biomechanical model and fluorescent fiducials. Surg. Endosc. 2017, 31, 2863–2871. [Google Scholar] [CrossRef] [PubMed]
- Hansen, A.E.; Henriksen, J.R.; Jølck, R.I.; Fliedner, F.P.; Bruun, L.M.; Scherman, J.; Jensen, A.I.; Rosenschöld, P.M.A.; Moorman, L.; Kurbegovic, S.; et al. Multimodal soft tissue markers for bridging high-resolution diagnostic imaging with therapeutic intervention. Sci. Adv. 2020, 6, abb5353. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.H.; Gil, M.S.; Lee, D.S.; Han, Y.; Park, H.C.; Sohn, J.W.; Kim, H.Y.; Shin, E.H.; Yu, J.I.; Noh, J.M.; et al. Preclinical investigation for developing injectable fiducial markers using a mixture of BaSO4 and biodegradable polymer for proton therapy. Med. Phys. 2015, 42, 2626–2637. [Google Scholar] [CrossRef] [PubMed]
- De Roover, R.; Crijns, W.; Poels, K.; Peeters, R.; Draulans, C.; Haustermans, K.; Depuydt, T. Characterization of a novel liquid fiducial marker for multimodal image guidance in stereotactic body radiotherapy of prostate cancer. Med. Phys. 2018, 45, 2205–2217. [Google Scholar] [CrossRef]
- Ng, M.; Brown, E.; Williams, A.; Chao, M.; Lawrentschuk, N.; Chee, R. Fiducial markers and spacers in prostate radiotherapy: Current applications. Br. J. Urol. 2014, 113, 13–20. [Google Scholar] [CrossRef]
- Górecka, Ż.; Teichmann, J.; Nitschke, M.; Chlanda, A.; Choińska, E.; Werner, C.; Święszkowski, W. Biodegradable fiducial markers for X-ray imaging—Soft tissue integration and biocompatibility. J. Mater. Chem. B 2016, 4, 5700–5712. [Google Scholar] [CrossRef]
- Lee, J.H.; Jung, H.W.; Kang, I.K.; Lee, H.B. Cell behaviour on polymer surfaces with different functional groups. Biomaterials 1994, 15, 705–711. [Google Scholar] [CrossRef]
- Chausse, V.; Schieber, R.; Raymond, Y.; Ségry, B.; Sabaté, R.; Kolandaivelu, K.; Ginebra, M.-P.; Pegueroles, M. Solvent-cast direct-writing as a fabrication strategy for radiopaque stents. Addit. Manuf. 2021, 48, 102392. [Google Scholar] [CrossRef]
- Chan, W.; Bini, T.; Venkatraman, S.S.; Boey, F.; Venkatraman, S. Effect of radio-opaque filler on biodegradable stent properties. J. Biomed. Mater. Res. Part A 2006, 79, 47–52. [Google Scholar] [CrossRef]
- Zada, M.H.; Gallimidi, Z.; Schlesinger−Laufer, M.; Nyska, A.; Domb, A.J. Biodegradable Breast Tissue Marker Clip. ACS Appl. Bio. Mater. 2020, 3, 7439–7453. [Google Scholar] [CrossRef]
- Wang, Q.; Yu, X.; Chen, X.; Gao, J.; Shi, D.; Shen, Y.; Tang, J.; He, J.; Li, A.; Yu, L.; et al. A Facile Composite Strategy to Prepare a Biodegradable Polymer Based Radiopaque Raw Material for “Visualizable” Biomedical Implants. ACS Appl. Mater. Interfaces 2022, 14, 24197–24212. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-C.; Hsu, L.-H.; Tsai, Y.-F.; Sumi, S.; Yang, K.-C. Enhancement of biodegradation and osseointegration of poly(ε-caprolactone)/calcium phosphate ceramic composite screws for osteofixation using calcium sulfate. Biomed. Mater. 2016, 11, 025012. [Google Scholar] [CrossRef] [PubMed]
- Górecka, Z.; Grzelecki, D.; Paskal, W.; Choińska, E.; Gilewicz, J.; Wrzesień, R.; Macherzyński, W.; Tracz, M.; Budzińska-Wrzesień, E.; Bedyńska, M.; et al. Biodegradable Fiducial Markers for Bimodal Near-Infrared Fluorescence- and X-ray-Based Imaging. ACS Biomater. Sci. Eng. 2022, 8, 859–870. [Google Scholar] [CrossRef]
- Ashokan, A.; Menon, D.; Nair, S.; Koyakutty, M. A molecular receptor targeted, hydroxyapatite nanocrystal based multi-modal contrast agent. Biomaterials 2010, 31, 2606–2616. [Google Scholar] [CrossRef] [PubMed]
- Ashokan, A.; Chandran, P.; Sadanandan, A.R.; Koduri, C.K.; Retnakumari, A.P.; Menon, D.; Nair, S.; Koyakutty, M. Development and haematotoxicological evaluation of doped hydroxyapatite based multimodal nanocontrast agent for near-infrared, magnetic resonance and X-ray contrast imaging. Nanotoxicology 2012, 6, 652–666. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.-C.; Tang, C.-M.; Lu, C.-C.; Chuang, C.-C. Evaluating the feasibility of applying cobalt-hydroxyapatite ingots as radiotherapy markers. Mater. Today Commun. 2020, 24, 101162. [Google Scholar] [CrossRef]
- Ahola, N.; Männistö, N.; Veiranto, M.; Karp, M.; Rich, J.; Efimov, A.; Seppälä, J.; Kellomäki, M. An in vitro study of composites of poly(L-lactide-co-ε-caprolactone), β-tricalcium phosphate and ciprofloxacin intended for local treatment of osteomyelitis. Biomatter 2013, 3, e23162. [Google Scholar] [CrossRef][Green Version]
- Choi, S.Y.; Hur, W.; Kim, B.K.; Shasteen, C.; Kim, M.H.; Choi, L.M.; Lee, S.H.; Park, C.G.; Park, M.; Min, H.S.; et al. Bioabsorbable bone fixation plates for X-ray imaging diagnosis by a radiopaque layer of barium sulfate and poly(lactic-co-glycolic acid). J. Biomed. Mater. Res. Part B Appl. Biomater. 2015, 103, 596–607. [Google Scholar] [CrossRef]
- Gleadall, A.; Pan, J.; Kruft, M.-A.; Kellomäki, M. Degradation mechanisms of bioresorbable polyesters. Part 2. Effects of initial molecular weight and residual monomer. Acta Biomater. 2014, 10, 2233–2240. [Google Scholar] [CrossRef]
- Nair, L.S.; Laurencin, C.T. Biodegradable polymers as biomaterials. Prog. Polym. Sci. 2007, 32, 762–798. [Google Scholar] [CrossRef]
- Ang, H.Y.; Toong, D.; Chow, W.S.; Seisilya, W.; Wu, W.; Wong, P.; Venkatraman, S.S.; Foin, N.; Huang, Y. Radiopaque Fully Degradable Nanocomposites for Coronary Stents. Sci. Rep. 2018, 8, 17409. [Google Scholar] [CrossRef] [PubMed]
- Hoekstra, J.W.M.; van den Beucken, J.J.J.P.; Leeuwenburgh, S.C.G.; Bronkhorst, E.M.; Meijer, G.J.; Jansen, J.A. Tantalum oxide and barium sulfate as radiopacifiers in injectable calcium phosphate-poly(lactic-co-glycolic acid) cements for monitoring in vivo degradation. J. Biomed. Mater. Res. Part A 2014, 102, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Samuel, R.; Girard, E.; Chagnon, G.; Dejean, S.; Favier, D.; Coudane, J.; Nottelet, B. Radiopaque poly(ε-caprolactone) as additive for X-ray imaging of temporary implantable medical devices. RSC Adv. 2015, 5, 84125–84133. [Google Scholar] [CrossRef][Green Version]
- Walejewska, E.; Idaszek, J.; Heljak, M.; Chlanda, A.; Choinska, E.; Hasirci, V.; Swieszkowski, W. The effect of introduction of filament shift on degradation behaviour of PLGA- and PLCL-based scaffolds fabricated via additive manufacturing. Polym. Degrad. Stab. 2020, 171, 109030. [Google Scholar] [CrossRef]
- Idaszek, J.; Bruinink, A.; Święszkowski, W. Ternary composite scaffolds with tailorable degradation rate and highly improved colonization by human bone marrow stromal cells. J. Biomed. Mater. Res. Part A 2015, 103, 2394–2404. [Google Scholar] [CrossRef] [PubMed]
- Rinoldi, C.; Fallahi, A.; Yazdi, I.K.; Campos Paras, J.; Kijeńska-Gawrońska, E.; Trujillo-de Santiago, G.; Tuoheti, A.; Demarchi, D.; Annabi, N.; Khademhosseini, A.; et al. Mechanical and Biochemical Stimulation of 3D Multilayered Scaffolds for Tendon Tissue Engineering. ACS Biomater. Sci. Eng. 2019, 5, 2953–2964. [Google Scholar] [CrossRef]
- Kijeńska, E.; Prabhakaran, M.P.; Swieszkowski, W.; Kurzydlowski, K.J.; Ramakrishna, S. Electrospun bio-composite P(LLA-CL)/collagen I/collagen III scaffolds for nerve tissue engineering. J. Biomed. Mater. Res. Part B Appl. Biomater. 2012, 100, 1093–1102. [Google Scholar] [CrossRef]
- Park, J.-W.; Hwang, J.-U.; Back, J.-H.; Jang, S.-W.; Kim, H.-J.; Kim, P.-S.; Shin, S.; Kim, T. High strength PLGA/Hydroxyapatite composites with tunable surface structure using PLGA direct grafting method for orthopedic implants. Compos. Part B Eng. 2019, 178, 107449. [Google Scholar] [CrossRef]
- Amestoy, H.; Diego, P.; Meaurio, E.; Muñoz, J.; Sarasua, J.-R. Crystallization Behavior and Mechanical Properties of Poly(ε-caprolactone) Reinforced with Barium Sulfate Submicron Particles. Materials 2021, 14, 2368. [Google Scholar] [CrossRef]
- Liuyun, J.; Chengdong, X.; Lixin, J.; Lijuan, X. Effect of hydroxyapatite with different morphology on the crystallization behavior, mechanical property and in vitro degradation of hydroxyapatite/poly(lactic-co-glycolic) composite. Compos. Sci. Technol. 2014, 93, 61–67. [Google Scholar] [CrossRef]
- Slagowski, J.M.; Colbert, L.; Cazacu, I.M.; Singh, B.S.; Martin, R.; Koay, E.J.; Taniguchi, C.M.; Koong, A.C.; Bhutani, M.S.; Herman, J.M.; et al. Evaluation of the Visibility and Artifacts of 11 Common Fiducial Markers for Image Guided Stereotactic Body Radiation Therapy in the Abdomen. Pract. Radiat. Oncol. 2020, 10, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, N.; Maeda, K.; Abo, D.; Morita, R.; Takao, S.; Matsuura, T.; Katoh, N.; Umegaki, K.; Shimizu, S.; Shirato, H. Quantitative evaluation of image recognition performance of fiducial markers in real-time tumor-tracking radiation therapy. Phys. Med. Eur. J. Med. Phys. 2019, 65, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Samarasena, J.B.; Chang, K.; Topazian, M. Endoscopic Ultrasound and Fine-Needle Aspiration for Pancreatic and Biliary Disorders. In Clinical Gastrointestinal Endoscopy, 3rd ed.; Chandrasekhara, V., Elmunzer, B.J., Khashab, M.A., Muthusamy, V.R., Eds.; Elsevier: Philadelphia, PA, USA, 2019; Chapter 51; pp. 571–591.e5. [Google Scholar] [CrossRef]
- Zhai, Z.; Morthomas, J.; Fusco, C.; Perez, M.; Lame, O. Crystallization and Molecular Topology of Linear Semicrystalline Polymers: Simulation of Uni- and Bimodal Molecular Weight Distribution Systems. Macromolecules 2019, 52, 4196–4208. [Google Scholar] [CrossRef]
- Sivalingam, G.; Madras, G. Thermal degradation of binary physical mixtures and copolymers of poly(ε-caprolactone), poly(d, l-lactide), poly(glycolide). Polym. Degrad. Stab. 2004, 84, 393–398. [Google Scholar] [CrossRef]
- Capone, C.; Di Landro, L.; Inzoli, F.; Penco, M.; Sartore, L. Thermal and mechanical degradation during polymer extrusion processing. Polym. Eng. Sci. 2007, 47, 1813–1819. [Google Scholar] [CrossRef]
- Saw, L.T.; Zainuddin, F.; Viet, C.X.; Lan, D.N.U. Effect of filler size and loading on thermo-mechanical degradation of polypropylene-ethylene/wollastonite composite. IOP Conf. Ser. Mater. Sci. Eng. 2020, 864, 012114. [Google Scholar] [CrossRef]
- Grayson, A.C.R.; Cima, M.J.; Langer, R. Size and temperature effects on poly(lactic-co-glycolic acid) degradation and microreservoir device performance. Biomaterials 2005, 26, 2137–2145. [Google Scholar] [CrossRef]
- Garkhal, K.; Verma, S.; Jonnalagadda, S.; Kumar, N. Fast degradable poly(L-lactide-co-ɛ-caprolactone) microspheres for tissue engineering: Synthesis, characterization, and degradation behavior. J. Polym. Sci. Part A Polym. Chem. 2007, 45, 2755–2764. [Google Scholar] [CrossRef]
- Rom, M.; Fabia, J.; Ślusarczyk, C.; Janicki, J.; Kasperczyk, J.; Dobrzynski, P. Structural transformation of terpolymer poly(L-lactide-glycolide-trimethylene carbonate) with shape memory effect during the degradation process. Polimery/Polymers 2014, 59, 562–568. [Google Scholar] [CrossRef]
- Huang, M.-H.; Li, S.; Hutmacher, D.W.; Coudane, J.; Vert, M. Degradation characteristics of poly(ε-caprolactone)-based copolymers and blends. J. Appl. Polym. Sci. 2006, 102, 1681–1687. [Google Scholar] [CrossRef]
- Chan, M.F.; Cohen, G.; Deasy, J. Qualitative Evaluation of Fiducial Markers for Radiotherapy Imaging. Technol. Cancer Res. Treat. 2015, 14, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Attia, M.F.; Brummel, B.R.; Lex, T.R.; Van Horn, B.A.; Whitehead, D.C.; Alexis, F. Recent Advances in Polyesters for Biomedical Imaging. Adv. Healthc. Mater. 2018, 7, e1800798. [Google Scholar] [CrossRef]
- Fernández, J.; Larrañaga, A.; Etxeberria, A.; Wang, W.; Sarasua, J.R. A new generation of poly(lactide/ε-caprolactone) polymeric biomaterials for application in the medical field. J. Biomed. Mater. Res. Part A 2014, 102, 3573–3584. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishna, S.; Mayer, J.; Wintermantel, E.; Leong, K.W. Biomedical applications of polymer-composite materials: A review. Compos. Sci. Technol. 2001, 61, 1189–1224. [Google Scholar] [CrossRef]
- Singh, B.; Kumar, R.; Chohan, J.S. Polymer matrix composites in 3D printing: A state of art review. Mater. Today Proc. 2020, 33, 1562–1567. [Google Scholar] [CrossRef]
- Sadaba, N.; Martini, R.; Barthelat, F.; de Arenaza, I.M.; Larrañaga, A.; Sarasua, J.; Zuza, E. Understanding the toughness mechanism prompted by submicron rigid particles in polylactide/barium sulfate composites. Polym. Test. 2018, 69, 340–349. [Google Scholar] [CrossRef]
- Górecka, Z.; Idaszek, J.; Kołbuk, D.; Choińska, E.; Chlanda, A.; Święszkowski, W. The effect of diameter of fibre on formation of hydrogen bonds and mechanical properties of 3D-printed PCL. Mater. Sci. Eng. C 2020, 114, 111072. [Google Scholar] [CrossRef]
- Rae, T. Tolerance of Mouse Macrophages in Vitro to Barium Sulfate Used in Orthopedic Bone Cement. J. Biomed. Mater. Res. 1977, 11, 839–846. [Google Scholar] [CrossRef]
- Konduru, N.; Keller, J.; Ma-Hock, L.; Gröters, S.; Landsiedel, R.; Donaghey, T.C.; Brain, J.D.; Wohlleben, W.; Molina, R.M. Biokinetics and Effects of Barium Sulfate Nanoparticles. Part Fibre Toxicol 2014, 11. [Google Scholar] [CrossRef]
- Moreno, E.C.; Gregory, T.M.; Brown, W.E. Preparation and Solubility of Hydroxyapatite. J. Res. Nat. Bur. Stand. A Phys. Chem. 1968, 72A, 1–15. [Google Scholar] [CrossRef]
- Laukkarinen, J.; Lämsä, T.; Nordback, I.; Mikkonen, J.; Sand, J. A Novel Biodegradable Pancreatic Stent for Human Pancreatic Applications: A Preclinical Safety Study in a Large Animal Model. Gastrointest. Endosc. 2008, 67, 1106–1112. [Google Scholar] [CrossRef]
- Motskin, M.; Wright, D.M.; Muller, K.; Kyle, N.; Gard, T.G.; Porter, A.E.; Skepper, J.N. Hydroxyapatite Nano and Microparticles: Correlation of Particle Properties with Cytotoxicity and Biostability. Biomaterials 2009, 30, 3307–3317. [Google Scholar] [CrossRef] [PubMed]
- Hernanz-Schulman, M.; Vanholder, R.; Waterloos, M.A.; Hakim, R.; Schulman, G. Effect of Radiographic Contrast Agents on Leukocyte Metabolic Response. Pediatr. Radiol. 2000, 30, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Bartnikowski, M.; Dargaville, T.R.; Ivanovski, S.; Hutmacher, D.W. Degradation mechanisms of polycaprolactone in the context of chemistry, geometry and environment. Prog. Polym. Sci. 2019, 96, 1–20. [Google Scholar] [CrossRef]
- Jiang, L.; Shen, T.; Xu, P.; Zhao, X.; Li, X.; Dong, W.; Ma, P.; Chen, M. Crystallization modification of poly(lactide) by using nucleating agents and stereocomplexation. e-Polymers 2016, 16, 1–13. [Google Scholar] [CrossRef]
- Fang, H.; Zhang, Y.; Bai, J.; Wang, Z. Shear-Induced Nucleation and Morphological Evolution for Bimodal Long Chain Branched Polylactide. Macromolecules 2013, 46, 6555–6565. [Google Scholar] [CrossRef]
- Rapier, C.E.; Shea, K.J.; Lee, A.P. Investigating PLGA microparticle swelling behavior reveals an interplay of expansive intermolecular forces. Sci. Rep. 2021, 11, 14512. [Google Scholar] [CrossRef]
- Sato, H.; Miyada, M.; Yamamoto, S.; Reddy, K.R.; Ozaki, Y. C–H⋯O (ether) hydrogen bonding along the (110) direction in polyglycolic acid studied by infrared spectroscopy, wide-angle X-ray diffraction, quantum chemical calculations and natural bond orbital calculations. RSC Adv. 2016, 6, 16817–16823. [Google Scholar] [CrossRef]
- Funaki, C.; Yamamoto, S.; Hoshina, H.; Ozaki, Y.; Sato, H. Three different kinds of weak C-H⋯O=C inter- and intramolecular interactions in poly(ε-caprolactone) studied by using terahertz spectroscopy, infrared spectroscopy and quantum chemical calculations. Polymer 2018, 137, 245–254. [Google Scholar] [CrossRef]
- Nishimae, A.; Sato, H. Study of Co-crystallization and Intermolecular Hydrogen Bondings of Poly(glycolide-co-l-lactide) Copolymers by Terahertz and Low-Frequency Raman Spectroscopy. Macromolecules 2021, 54, 6440–6448. [Google Scholar] [CrossRef]
- Tatsuoka, S.; Sato, H. Stress-induced crystal transition of poly(butylene succinate) studied by terahertz and low-frequency Raman spectroscopy and quantum chemical calculation. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2018, 197, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Song, C.; Han, Y.; Xi, Z.; Zhao, L.; Cen, L.; Yang, Y. Regulation of inflammatory response to polyglycolic acid scaffolds through incorporation of sodium tripolyphosphate. Eur. Polym. J. 2020, 122, 109349. [Google Scholar] [CrossRef]
- Górecka, Ż.; Choińska, E.; Szlązak, K.; Święszkowski, W. Increase of Radiopacity of PCL Scaffolds for Their In Vivo Monitoring Using X-rays Imaging. Eur. Cell Mater. 2016, 31, P207. [Google Scholar]
- Pawlik, J.; Łukowicz, K.; Cholewa-Kowalska, K.; Osyczka, A.M. New Insights into the PLGA and PCL Blending: Physico-Mechanical Properties and Cell Response. Mater. Res. Express 2019, 6, 085344. [Google Scholar] [CrossRef]
- Li, Y.; Weng, W. Surface Modification of Hydroxyapatite by Stearic Acid: Characterization and In Vitro Behaviors. J. Mater. Sci. Mater. Med. 2007, 19, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Longlade, J.; Delaite, C.; Schuller, A.-S.; Longlade, J.; Delaite, C.; Schuller, A.-S. Surface Modification of Barium Sulfate Particles. Mater. Sci. Appl. 2021, 12, 106532. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | Form | Filler Content ± SD [%] | Mn [kDa] | Mw [kDa] | PDI | [°C] | [°C]/ [J/g] | [°C]/ [J/g] | [J/g] |
|---|---|---|---|---|---|---|---|---|---|
| PCL-1010-BH | P | 125.7 | 229.0 | 1.8 | −61.6 | - | 57.9/59.3 | 59.3 | |
| C | 19.9 ± 0.2 | 126.8 | 198.8 | 1.6 | −63.0 | - | 56.2/51.7 | 64.5 | |
| R | 19.8 ± 0.6 | 120.1 | 195.9 | 1.6 | −62.9 | - | 57.0/53.5 | 66.6 | |
| PLC70-1010-BH | P | 99.1 | 173.4 | 1.7 | 30.3 | 128.0/1.7 | 159.8/1.8 | 0.1 | |
| C | 19.7 ± 0.4 | 84.7 | 161.9 | 1.8 | 30.1 | 106.4/14.8 | 158.8/15.4 | 0.7 | |
| R | 20.1 ± 1.2 | 64.2 | 142.2 | 2.2 | 26.9 | 89.8/17.2 | 160.0/18.6 | 1.8 | |
| PLC85-1010-BH | P | 71.9 | 228.4 | 3.2 | 42.5 | - | - | 0.0 | |
| C | 19.3 ± 0.6 | 73.7 | 228.3 | 3.1 | 42.1 | - | - | 0.0 | |
| R | 19.9 ± 0.3 | 58.8 | 207.0 | 3.5 | 42.6 | - | - | 0.0 | |
| PLG82-1010-BH | P | 122.8 | 220.2 | 1.8 | 58.7 | - | - | 0.0 | |
| C | 19.8 ± 0.5 | 125.2 | 227.7 | 1.8 | 57.0 | - | - | 0.0 | |
| R | 19.8 ± 0.4 | 56.5 | 151.1 | 2.7 | 54.6 | - | - | 0.0 |
| Material | Matrix | Composition [%wt] | Temperature of Extrusion [°C] | ||
|---|---|---|---|---|---|
| Polymer | BaSO4 | HAp | |||
| PCL-1010-BH | PCL 80 kDa | 80 | 10 | 10 | 150 |
| PLC70-1010-BH | RESOMER LC 703 S | 80 | 10 | 10 | 190 |
| PLC85-1010-BH | PURASORB PLC 8516 | 80 | 10 | 10 | 165 |
| PLG82- 1010-BH | PURASORB PLG 8218 | 80 | 10 | 10 | 200 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Górecka, Ż.; Choińska, E.; Heljak, M.; Święszkowski, W. Long-Term In Vitro Assessment of Biodegradable Radiopaque Composites for Fiducial Marker Fabrication. Int. J. Mol. Sci. 2022, 23, 14363. https://doi.org/10.3390/ijms232214363
Górecka Ż, Choińska E, Heljak M, Święszkowski W. Long-Term In Vitro Assessment of Biodegradable Radiopaque Composites for Fiducial Marker Fabrication. International Journal of Molecular Sciences. 2022; 23(22):14363. https://doi.org/10.3390/ijms232214363
Chicago/Turabian StyleGórecka, Żaneta, Emilia Choińska, Marcin Heljak, and Wojciech Święszkowski. 2022. "Long-Term In Vitro Assessment of Biodegradable Radiopaque Composites for Fiducial Marker Fabrication" International Journal of Molecular Sciences 23, no. 22: 14363. https://doi.org/10.3390/ijms232214363
APA StyleGórecka, Ż., Choińska, E., Heljak, M., & Święszkowski, W. (2022). Long-Term In Vitro Assessment of Biodegradable Radiopaque Composites for Fiducial Marker Fabrication. International Journal of Molecular Sciences, 23(22), 14363. https://doi.org/10.3390/ijms232214363