
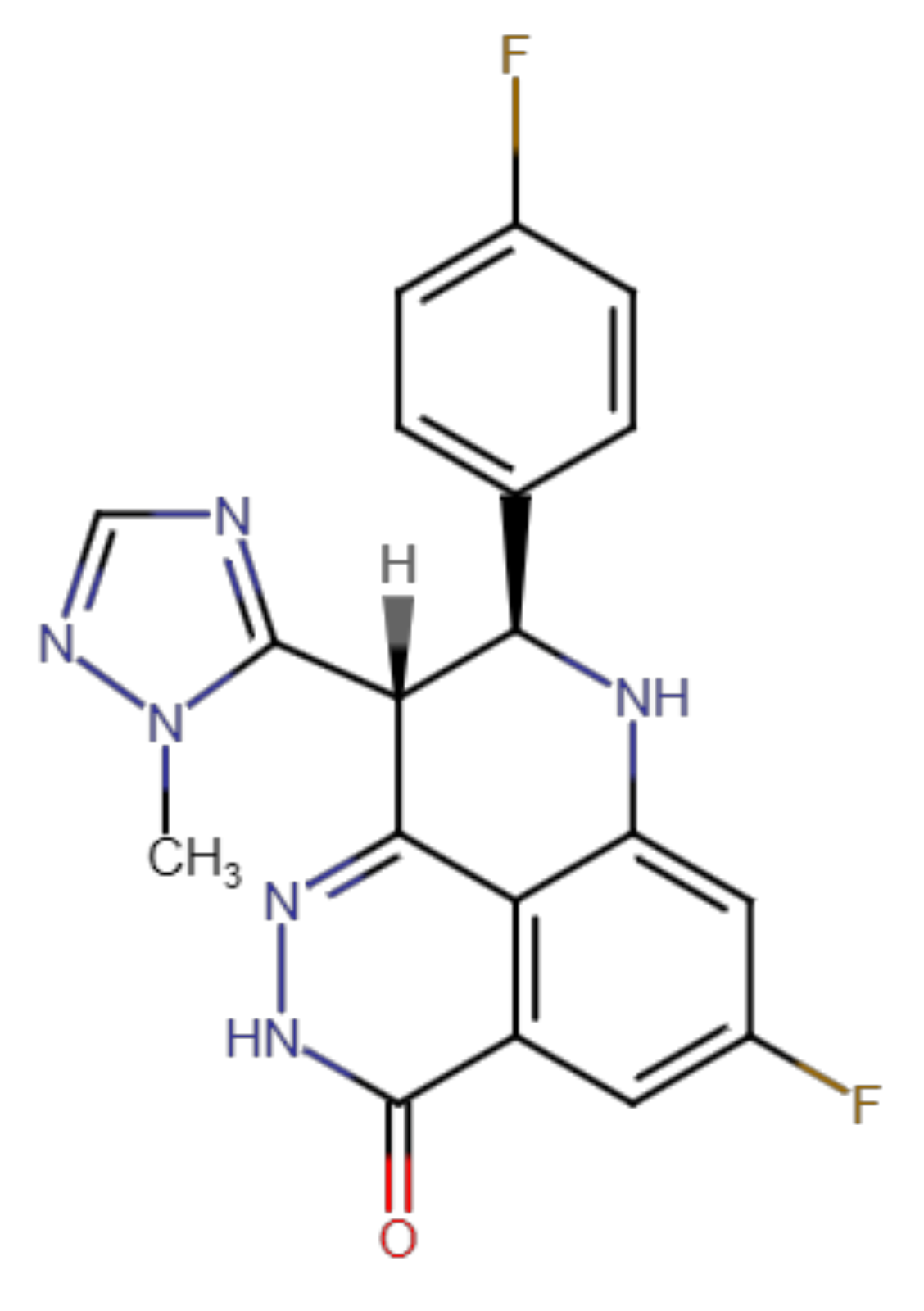
Talazoparib Does Not Interact with ABCB1 Transporter or Cytochrome P450s, but Modulates Multidrug Resistance Mediated by ABCC1 and ABCG2: An in Vitro and Ex Vivo Study

 , ,
, ,
Abstract
1. Introduction
2. Results
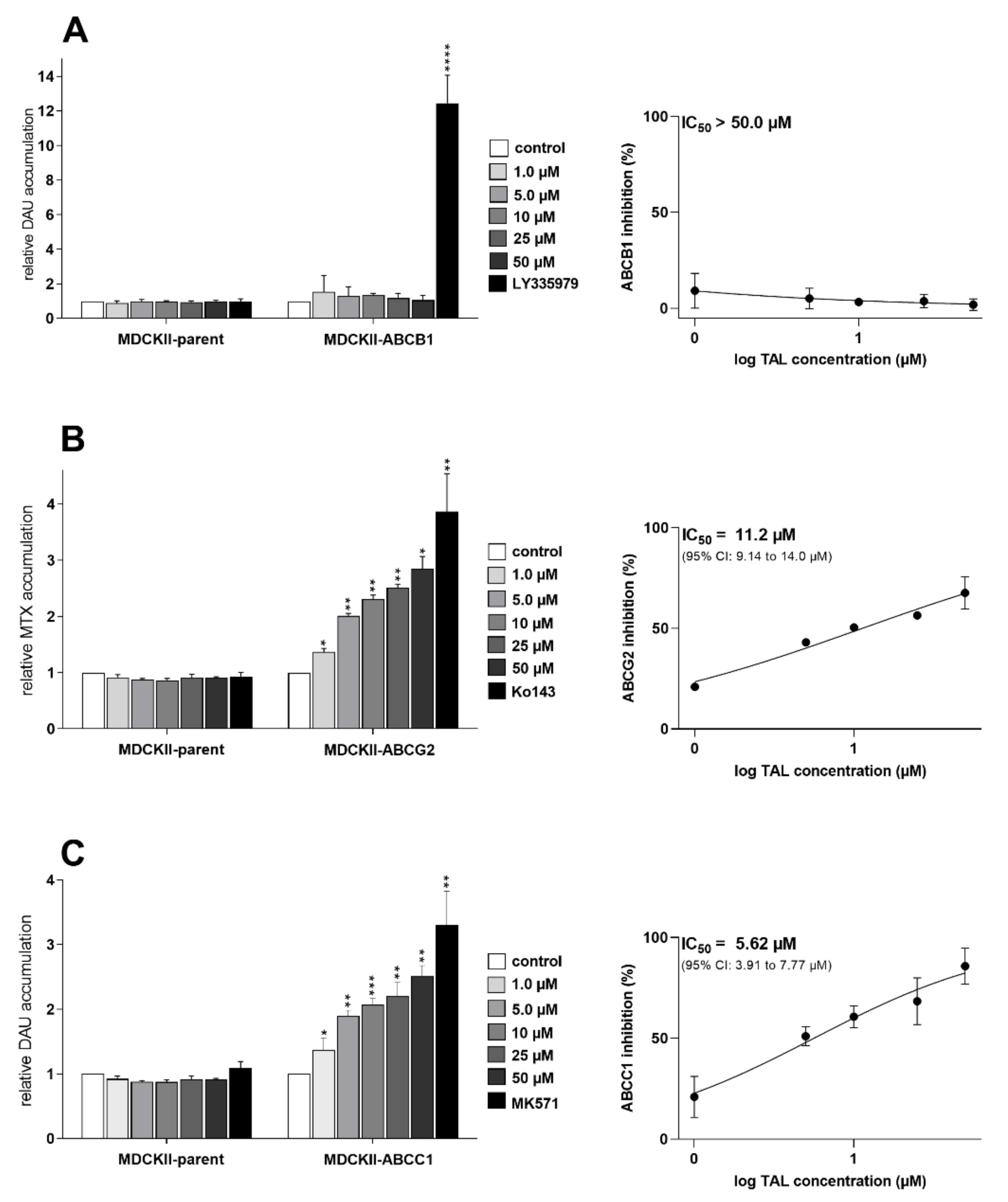
2.1. Talazoparib Significantly Inhibits ABCG2- and ABCC1-Mediated Transport of Probe Substrate Drugs
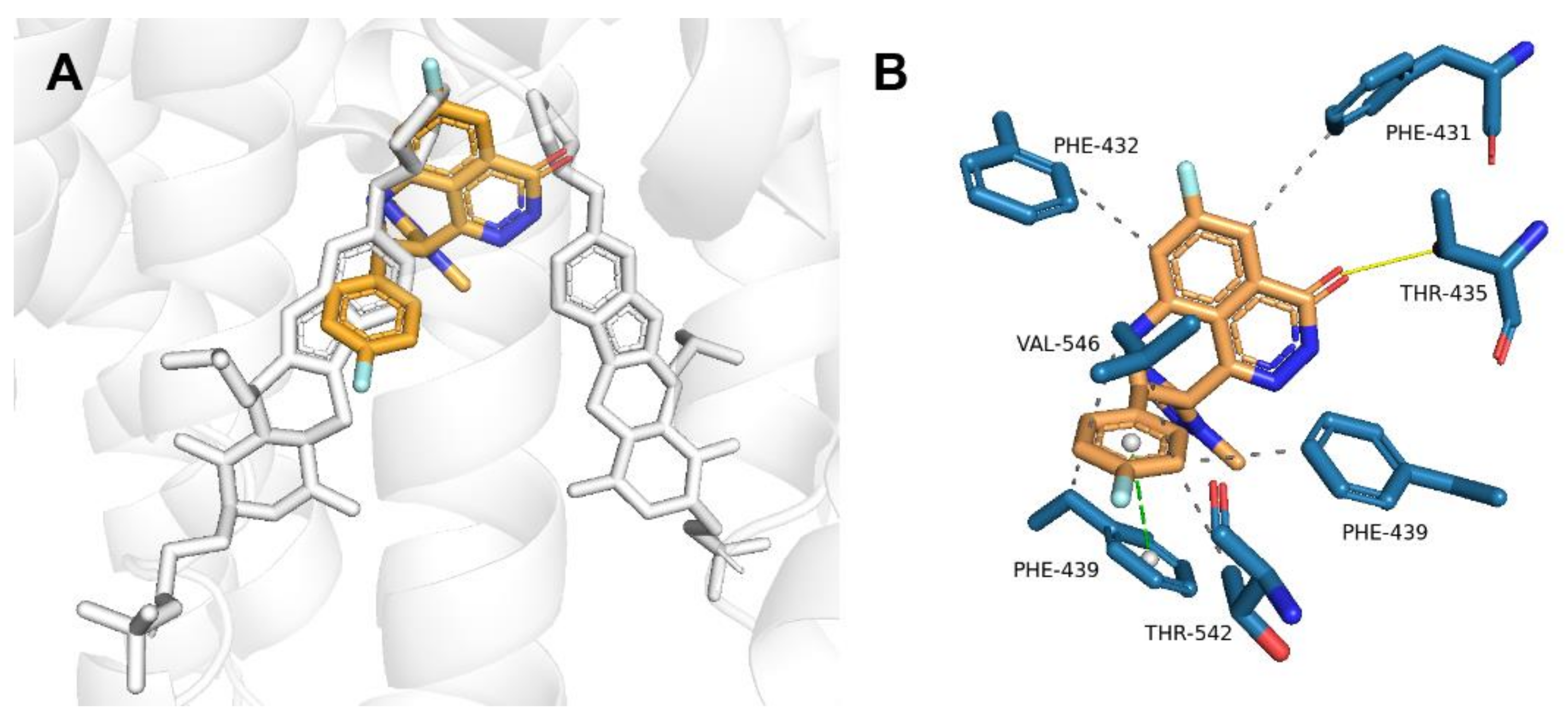
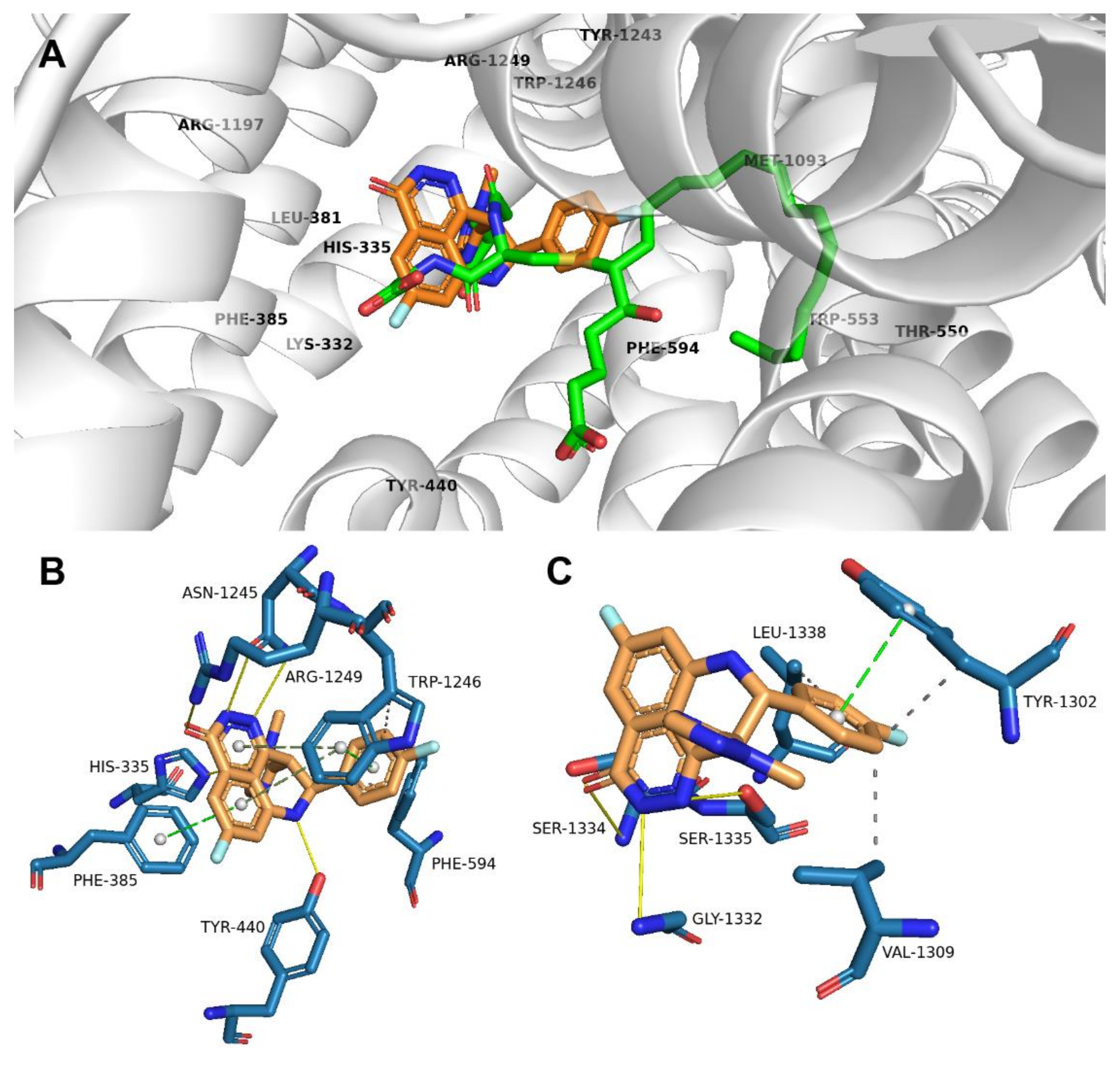
2.2. Talazoparib Is Predicted to Interact with the Ligand-Binding Sites of ABCG2 and ABCC1 and Nucleotide Binding Domain 2 of ABCC1
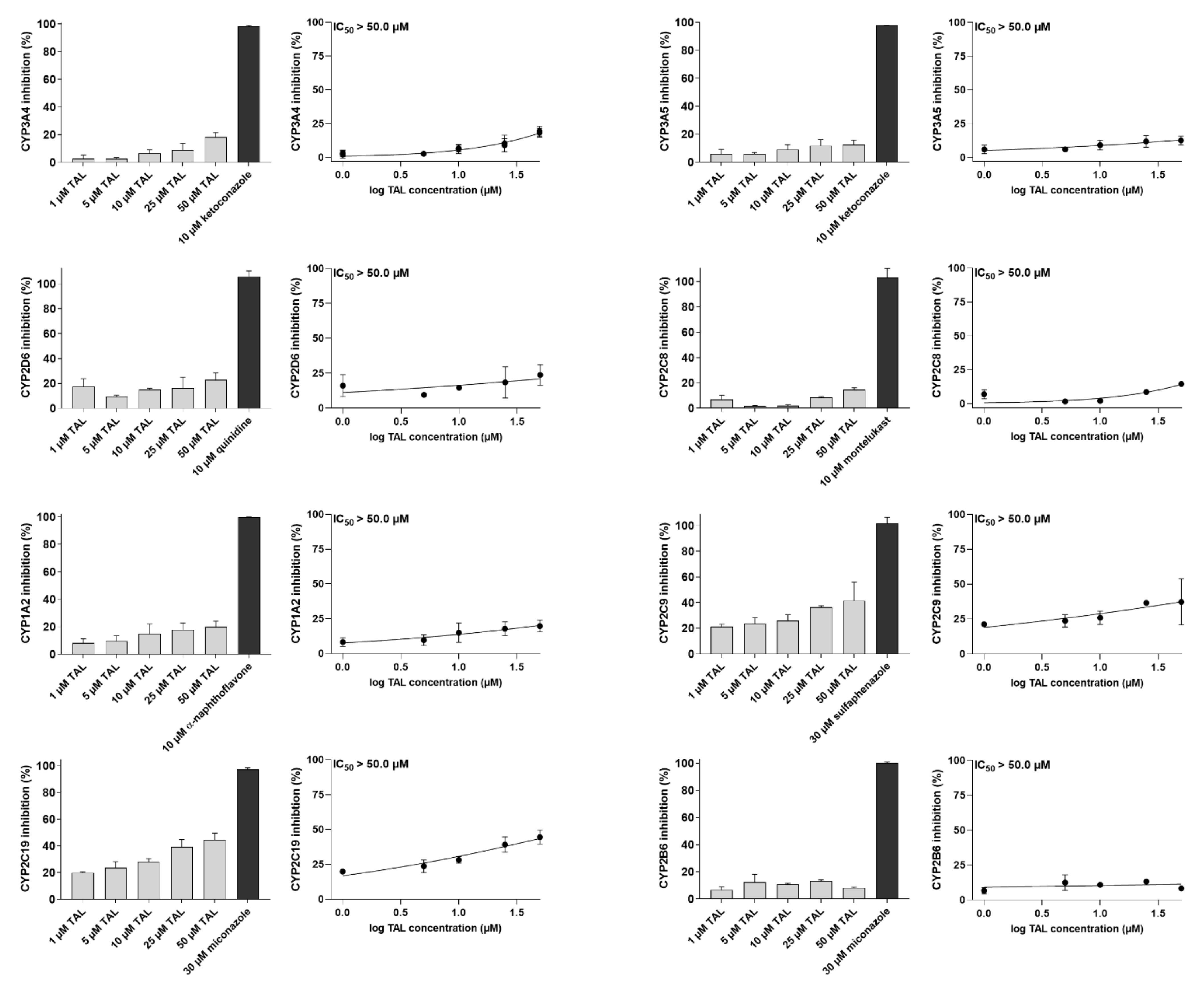
2.3. Talazoparib Does Not Interfere with the Metabolic Activities of Clinically Relevant CYP Isoforms
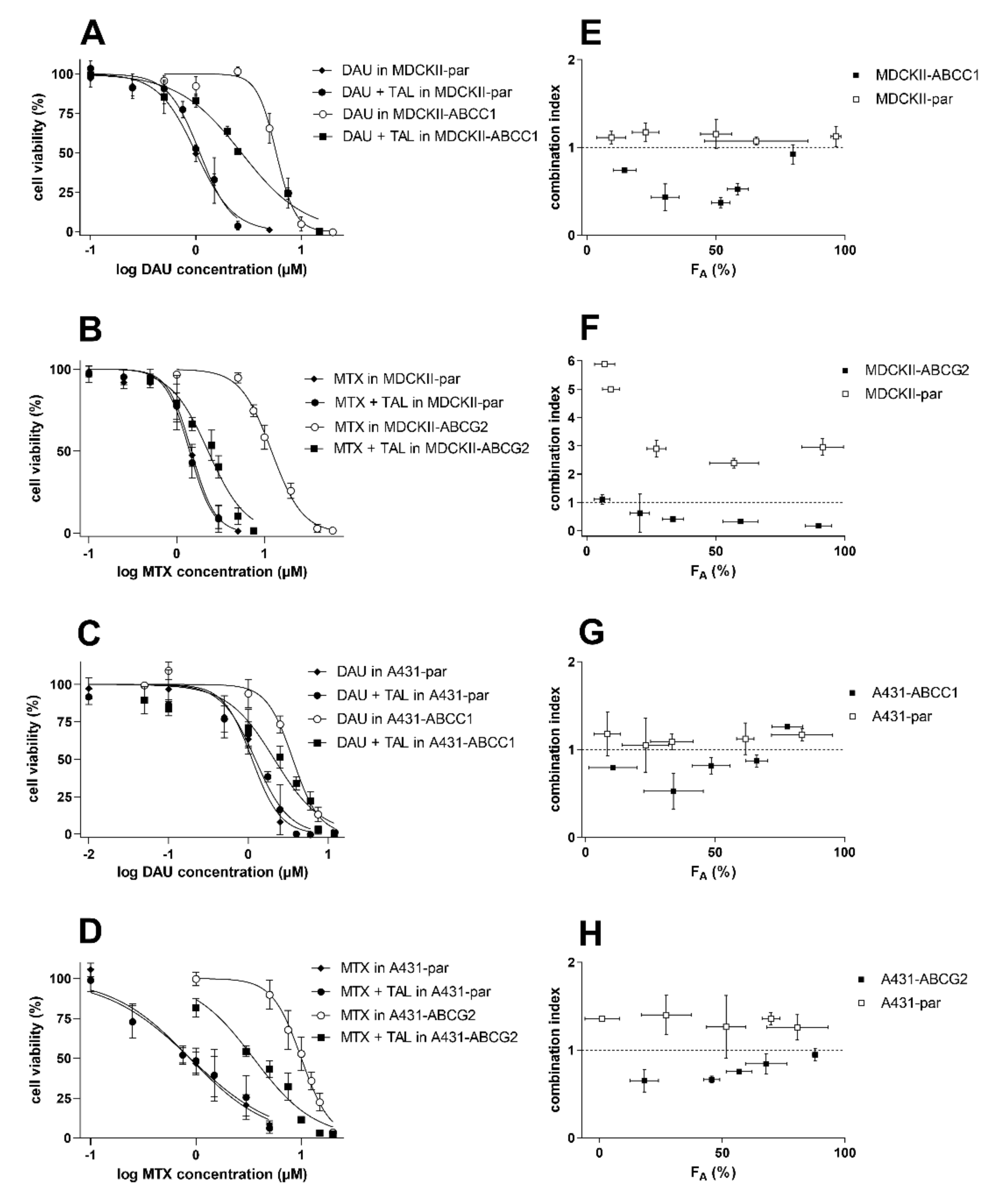
2.4. Talazoparib Combats ABCC1- and ABCG2-Mediated Chemotherapeutic Resistance In Vitro
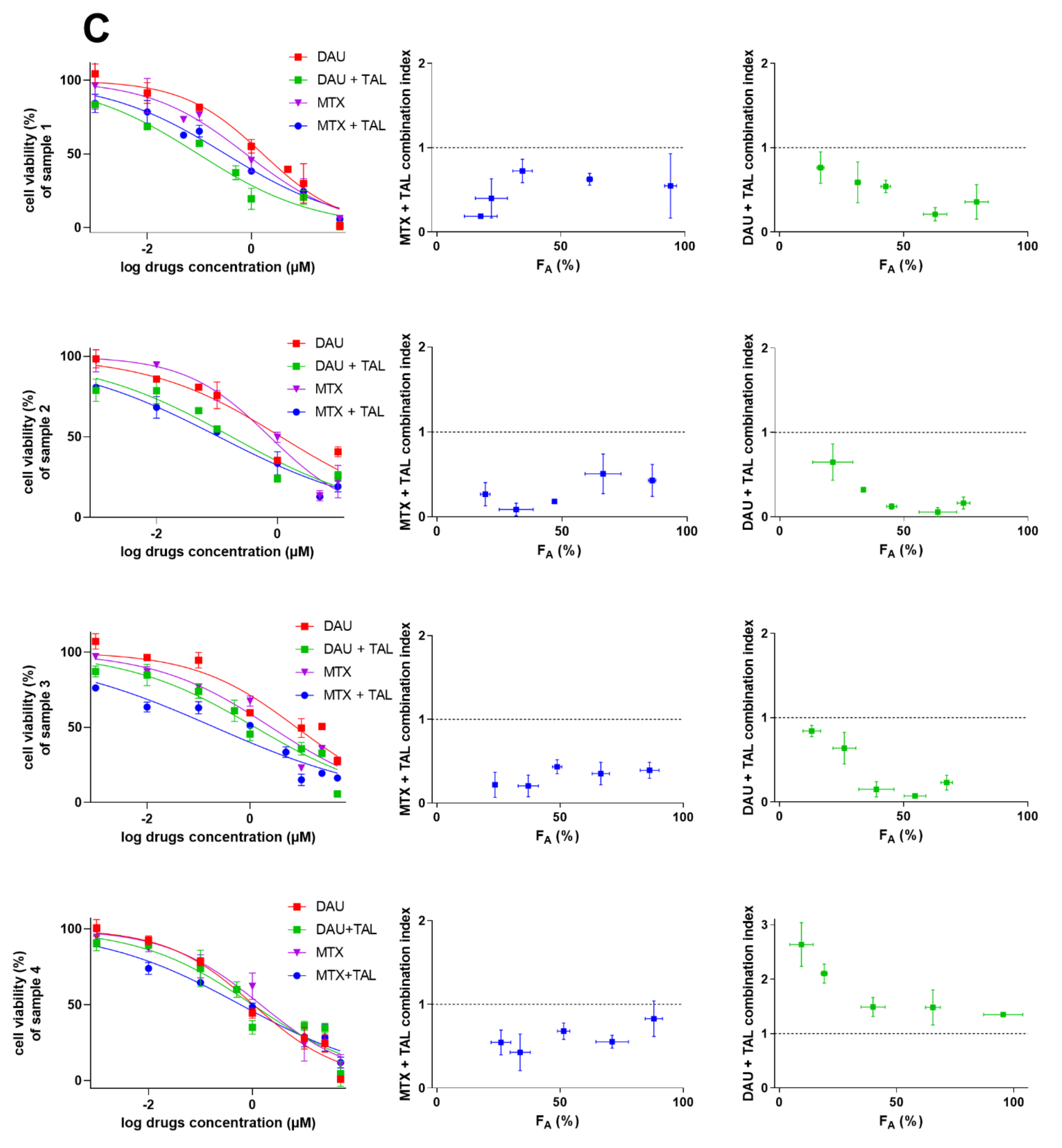
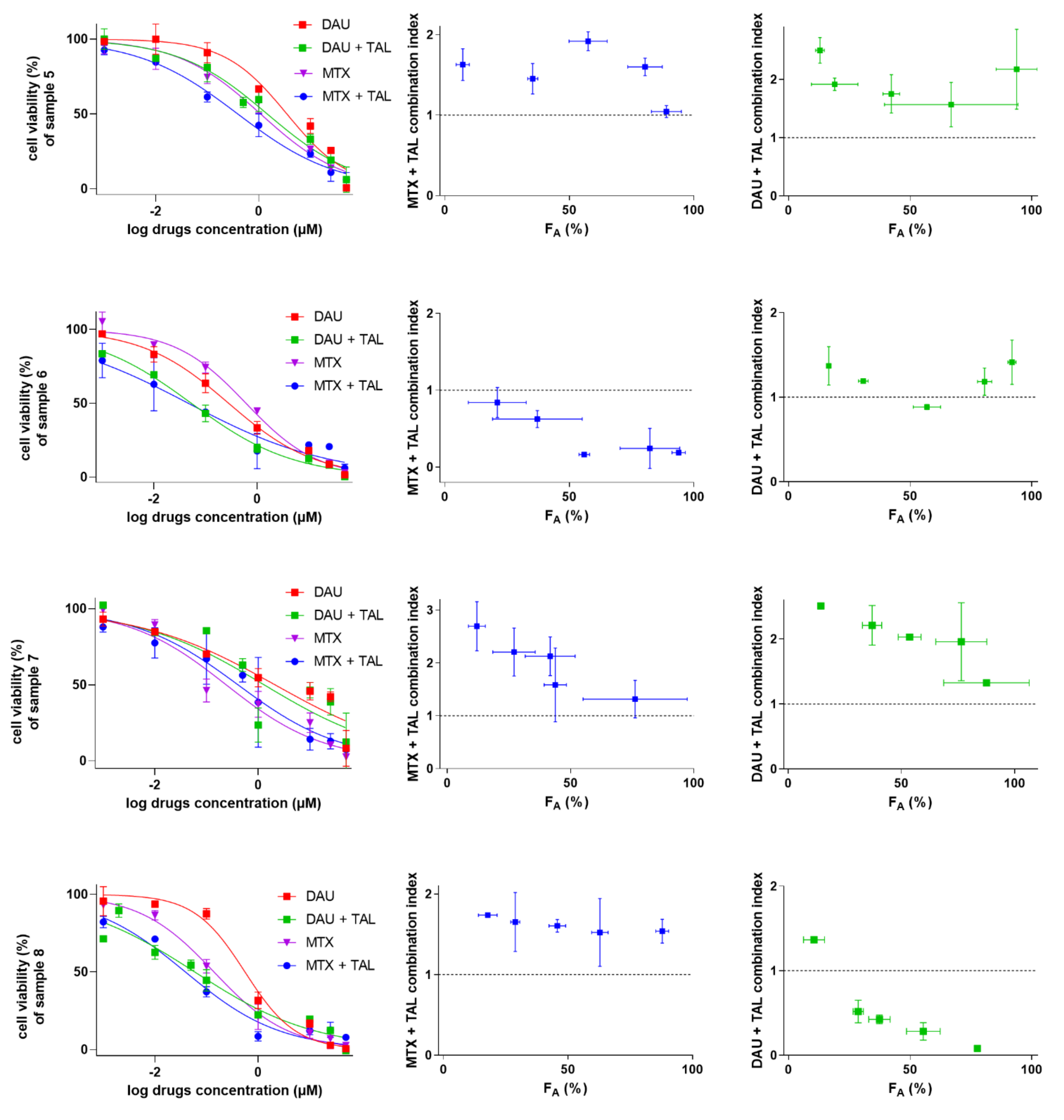
2.5. Talazoparib Targets Cytostatic MDR in Patient-Derived NSCLC Explants Ex Vivo
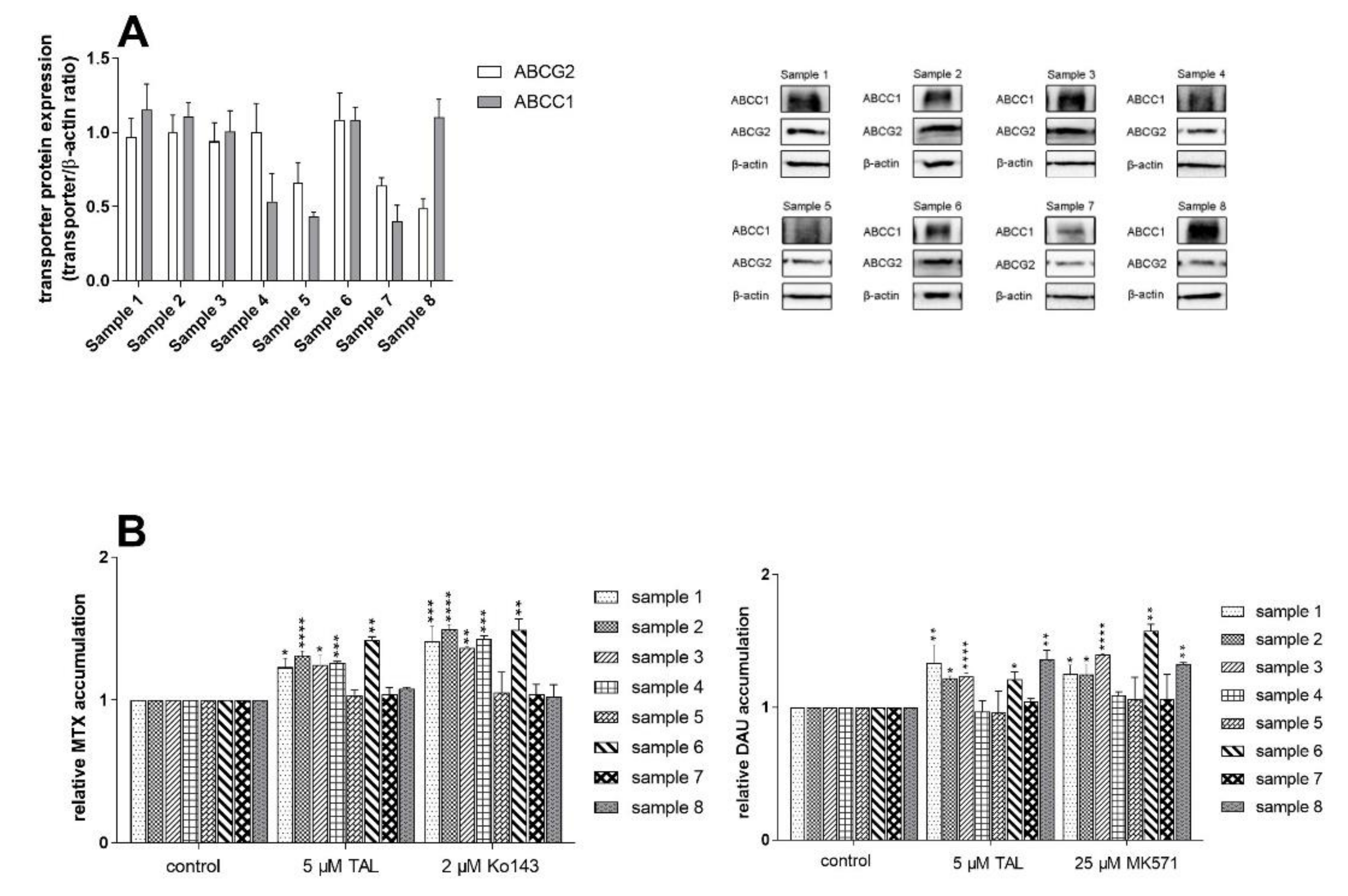
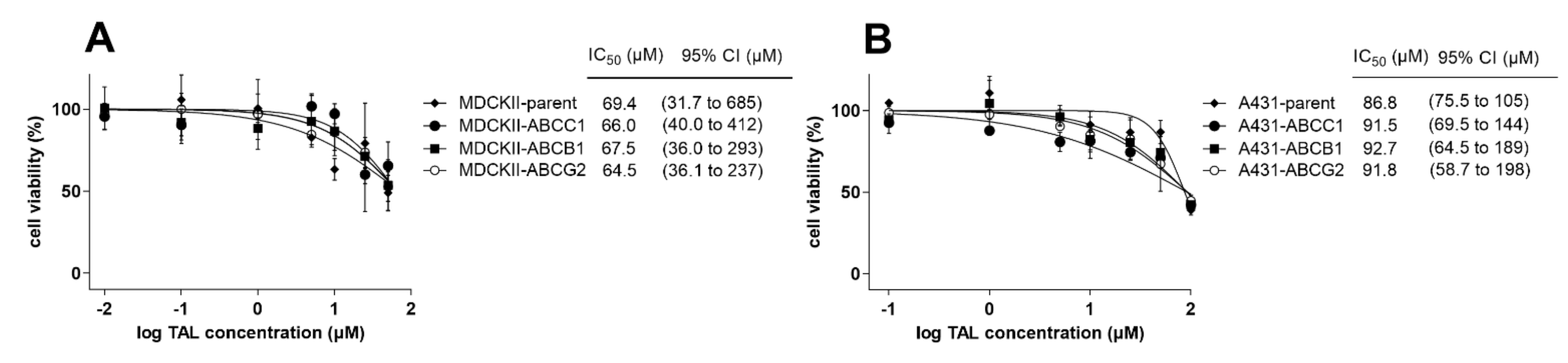
2.6. MDR-Related Transporters Do Not Establish Resistance to Talazoparib
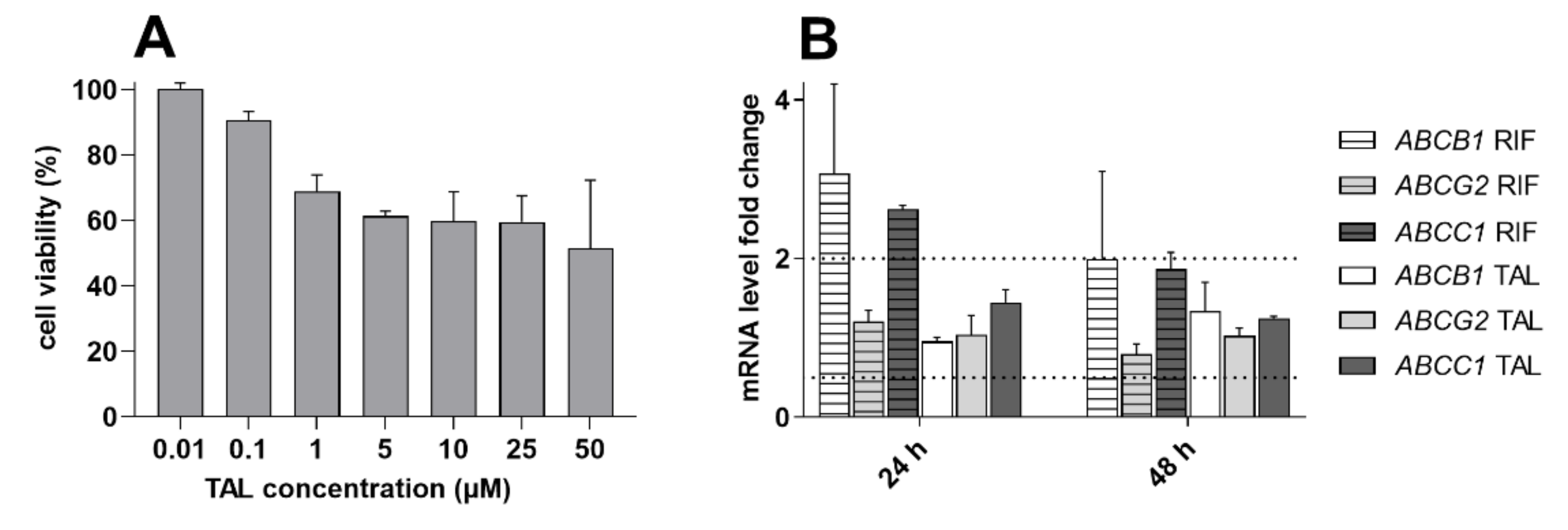
2.7. Talazoparib Does Not Change the Expressions of MDR-Related Transporters in Breast Cancer Model
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Cell Cultures
4.3. Generation of Primary Explants from Patients’ NSCLC Tissue Samples
4.4. Accumulation Assay with Fluorescent Cytostatic Substrates
4.5. Molecular Docking
4.6. Incubation Assay for Human Recombinant CYPs
4.7. MTT Proliferation Assay
4.8. Drug Combination Assays
4.9. Western Blotting Analysis
4.10. Gene Induction Studies
4.11. Statistical and Data Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shang, S.; Liu, J.; Verma, V.; Wu, M.; Welsh, J.; Yu, J.; Chen, D. Combined treatment of non-small cell lung cancer using radiotherapy and immunotherapy: Challenges and updates. Cancer Commun. 2021, 41, 1086–1099. [Google Scholar] [CrossRef] [PubMed]
- Thandra, K.C.; Barsouk, A.; Saginala, K.; Aluru, J.S.; Barsouk, A. Epidemiology of lung cancer. Contemp. Oncol. 2021, 25, 45. [Google Scholar] [CrossRef]
- Devesa, S.S.; Bray, F.; Vizcaino, A.P.; Parkin, D.M. International lung cancer trends by histologic type: Male: Female differences diminishing and adenocarcinoma rates rising. Int. J. Cancer 2005, 117, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-Y.; Ju, D.-T.; Chang, C.-F.; Reddy, P.M.; Velmurugan, B.K. A review on the effects of current chemotherapy drugs and natural agents in treating non–small cell lung cancer. Biomedicine 2017, 7. [Google Scholar] [CrossRef]
- Baudino, T.A. Targeted cancer therapy: The next generation of cancer treatment. Curr. Drug Discov. Technol. 2015, 12, 3–20. [Google Scholar] [CrossRef]
- Jemal, A.; Siegel, R.; Xu, J.; Ward, E. Cancer statistics, 2010. CA: A Cancer J. Clin. 2010, 60, 277–300. [Google Scholar] [CrossRef]
- Røsland, G.V.; Engelsen, A.S.T. Novel points of attack for targeted cancer therapy. Basic Clin. Pharmacol. Toxicol. 2015, 116, 9–18. [Google Scholar] [CrossRef]
- Morales, J.; Li, L.; Fattah, F.J.; Dong, Y.; Bey, E.A.; Patel, M.; Gao, J.; Boothman, D.A. Review of poly (ADP-ribose) polymerase (PARP) mechanisms of action and rationale for targeting in cancer and other diseases. Crit. Rev. ™ Eukaryot. Gene Expr. 2014, 24. [Google Scholar] [CrossRef]
- Curtin, N. PARP inhibitors for anticancer therapy. Biochem. Soc. Trans. 2014, 42, 82–88. [Google Scholar] [CrossRef]
- Hoy, S.M. Talazoparib: First global approval. Drugs 2018, 78, 1939–1946. [Google Scholar] [CrossRef]
- Exman, P.; Barroso-Sousa, R.; Tolaney, S.M. Evidence to date: Talazoparib in the treatment of breast cancer. OncoTargets Ther. 2019, 12, 5177. [Google Scholar] [CrossRef]
- Przybytkowski, E.; Davis, T.; Hosny, A.; Eismann, J.; Matulonis, U.A.; Wulf, G.M.; Nabavi, S. An immune-centric exploration of BRCA1 and BRCA2 germline mutation related breast and ovarian cancers. BMC Cancer 2020, 20, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, R. Hereditary breast and ovarian cancer (HBOC): Review of its molecular characteristics, screening, treatment, and prognosis. Breast Cancer 2021, 28, 1167–1180. [Google Scholar] [CrossRef] [PubMed]
- Seelig, A. P-Glycoprotein: One mechanism, many tasks and the consequences for pharmacotherapy of cancers. Front. Oncol. 2020, 1989. [Google Scholar] [CrossRef] [PubMed]
- Nikolaou, M.; Pavlopoulou, A.; Georgakilas, A.G.; Kyrodimos, E. The challenge of drug resistance in cancer treatment: A current overview. Clin. Exp. Metastasis 2018, 35, 309–318. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, H.; Chen, X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resist. (Alhambra Calif.) 2019, 2, 141. [Google Scholar] [CrossRef]
- Giddings, E.L.; Champagne, D.P.; Wu, M.-H.; Laffin, J.M.; Thornton, T.M.; Valenca-Pereira, F.; Culp-Hill, R.; Fortner, K.A.; Romero, N.; East, J. Mitochondrial ATP fuels ABC transporter-mediated drug efflux in cancer chemoresistance. Nat. Commun. 2021, 12, 1–19. [Google Scholar] [CrossRef]
- Kathawala, R.J.; Gupta, P.; Ashby Jr, C.R.; Chen, Z.-S. The modulation of ABC transporter-mediated multidrug resistance in cancer: A review of the past decade. Drug Resist. Updates 2015, 18, 1–17. [Google Scholar] [CrossRef]
- Thomas, C.; Aller, S.G.; Beis, K.; Carpenter, E.P.; Chang, G.; Chen, L.; Dassa, E.; Dean, M.; Duong Van Hoa, F.; Ekiert, D. Structural and functional diversity calls for a new classification of ABC transporters. FEBS Lett. 2020, 594, 3767–3775. [Google Scholar] [CrossRef]
- Crawford, R.R.; Potukuchi, P.K.; Schuetz, E.G.; Schuetz, J.D. Beyond competitive inhibition: Regulation of ABC transporters by kinases and protein-protein interactions as potential mechanisms of drug-drug interactions. Drug Metab. Dispos. 2018, 46, 567–580. [Google Scholar] [CrossRef]
- Hofman, J.; Vagiannis, D.; Chen, S.; Guo, L. Roles of CYP3A4, CYP3A5 and CYP2C8 drug-metabolizing enzymes in cellular cytostatic resistance. Chem. -Biol. Interact. 2021, 340, 109448. [Google Scholar] [CrossRef] [PubMed]
- van Schaik, R.H. CYP450 pharmacogenetics for personalizing cancer therapy. Drug Resist. Updates 2008, 11, 77–98. [Google Scholar] [CrossRef] [PubMed]
- Han, X.-M.; Zhou, H.-H. Polymorphism of CYP450 and cancer susceptibility. Acta Pharmacol. Sin. 2000, 21, 673–679. [Google Scholar] [PubMed]
- EMA. European Medicines Agency, Guideline on the Investigation of Drug Interactions. CPMP/EWP/560/95/Rev. 1 Corr. 2. 2012. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-drug-interactions-revision-1_en.pdf (accessed on 16 November 2022).
- FDA. Food and Drug Administration. In Vitro Drug Interaction Studies— Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions Guidance for Industry. Clin. Pharmacol. Silver Spring MD 2020. Available online: https://www.fda.gov/media/134582/download (accessed on 16 November 2022).
- Beretta, G.L.; Cassinelli, G.; Pennati, M.; Zuco, V.; Gatti, L. Overcoming ABC transporter-mediated multidrug resistance: The dual role of tyrosine kinase inhibitors as multitargeting agents. Eur. J. Med. Chem. 2017, 142, 271–289. [Google Scholar] [CrossRef]
- Wu, S.; Fu, L. Tyrosine kinase inhibitors enhanced the efficacy of conventional chemotherapeutic agent in multidrug resistant cancer cells. Mol. Cancer 2018, 17, 1–13. [Google Scholar] [CrossRef]
- Johnson, Z.L.; Chen, J. Structural basis of substrate recognition by the multidrug resistance protein MRP1. Cell 2017, 168, 1075–1085.e1079. [Google Scholar] [CrossRef]
- Aoyagi-Scharber, M.; Gardberg, A.S.; Yip, B.K.; Wang, B.; Shen, Y.; Fitzpatrick, P.A. Structural basis for the inhibition of poly (ADP-ribose) polymerases 1 and 2 by BMN 673, a potent inhibitor derived from dihydropyridophthalazinone. Acta Crystallogr. Sect. F: Struct. Biol. Commun. 2014, 70, 1143–1149. [Google Scholar] [CrossRef]
- Yu, Y.; Chung, C.H.; Plotka, A.; Quinn, K.; Shi, H.; Pápai, Z.; Nguyen, L.; Wang, D. A Phase 1 Mass Balance Study of 14C-Labeled Talazoparib in Patients With Advanced Solid Tumors. J. Clin. Pharmacol. 2019, 59, 1195–1203. [Google Scholar] [CrossRef]
- Du, W.; Elemento, O. Cancer systems biology: Embracing complexity to develop better anticancer therapeutic strategies. Oncogene 2015, 34, 3215–3225. [Google Scholar] [CrossRef]
- Omran, O.M. The prognostic value of breast cancer resistance protein (BCRB/ABCG2) expression in breast carcinomas. J. Environ. Pathol. Toxicol. Oncol. 2012, 31. [Google Scholar] [CrossRef] [PubMed]
- Wind, N.; Holen, I. Multidrug resistance in breast cancer: From in vitro models to clinical studies. Int. J. Breast Cancer 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Nooter, K.; Brutel De La Riviere, G.; Look, M.; Van Wingerden, K.; Henzen-Logmans, S.; Scheper, R.; Flens, M.; Klijn, J.; Stoter, G.; Foekens, J. The prognostic significance of expression of the multidrug resistance-associated protein (MRP) in primary breast cancer. Br. J. Cancer 1997, 76, 486–493. [Google Scholar] [CrossRef] [PubMed]
- Mo, W.; Liu, J.-Y.; Zhang, J.-T. Biochemistry and pharmacology of human ABCC1/MRP1 and its role in detoxification and in multidrug resistance of cancer chemotherapy. Recent Adv. Cancer Res. Ther. 2012, 371–404. [Google Scholar]
- Federico, S.M.; Stewart, E.; Coleman, J.L.; Bishop, M.W.; Santana, V.M.; Lam, C.; Hawkins, D.; Wu, J.; Mao, S.; Goshorn, D.R. Phase I study of talazoparib and irinotecan in children and young adults with recurrent/refractory solid tumors. J. Clin. Oncol. 2017, 35. [Google Scholar] [CrossRef]
- Kumar, A.; Jaitak, V. Natural products as multidrug resistance modulators in cancer. Eur. J. Med. Chem. 2019, 176, 268–291. [Google Scholar] [CrossRef]
- Li, M.; Seiser, E.L.; Baldwin, R.M.; Ramirez, J.; Ratain, M.J.; Innocenti, F.; Kroetz, D.L. ABC transporter polymorphisms are associated with irinotecan pharmacokinetics and neutropenia. Pharm. J. 2018, 18, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Xing, W.; Yu, H.; Zhang, W.; Si, T. ABCB1 and ABCG2 restricts the efficacy of gedatolisib (PF-05212384), a PI3K inhibitor in colorectal cancer cells. Cancer Cell Int. 2021, 21, 1–16. [Google Scholar] [CrossRef]
- Lok, B.H.; Gardner, E.E.; Schneeberger, V.E.; Ni, A.; Desmeules, P.; Rekhtman, N.; De Stanchina, E.; Teicher, B.A.; Riaz, N.; Powell, S.N. PARP inhibitor activity correlates with SLFN11 expression and demonstrates synergy with temozolomide in small cell lung cancer. Clin. Cancer Res. 2017, 23, 523–535. [Google Scholar] [CrossRef]
- Veringa, S.J.; Biesmans, D.; van Vuurden, D.G.; Jansen, M.H.; Wedekind, L.E.; Horsman, I.; Wesseling, P.; Vandertop, W.P.; Noske, D.P.; Kaspers, G.J. In vitro drug response and efflux transporters associated with drug resistance in pediatric high grade glioma and diffuse intrinsic pontine glioma. PLoS ONE 2013, 8, e61512. [Google Scholar] [CrossRef]
- Engert, F.; Kovac, M.; Baumhoer, D.; Nathrath, M.; Fulda, S. Osteosarcoma cells with genetic signatures of BRCAness are susceptible to the PARP inhibitor talazoparib alone or in combination with chemotherapeutics. Oncotarget 2017, 8, 48794. [Google Scholar] [CrossRef] [PubMed]
- Kizilbash, S.H.; Gupta, S.K.; Chang, K.; Kawashima, R.; Parrish, K.E.; Carlson, B.L.; Bakken, K.K.; Mladek, A.C.; Schroeder, M.A.; Decker, P.A. Restricted delivery of talazoparib across the blood–brain barrier limits the sensitizing effects of PARP inhibition on temozolomide therapy in glioblastoma. Mol. Cancer Ther. 2017, 16, 2735–2746. [Google Scholar] [CrossRef] [PubMed]
- Elmeliegy, M.; Láng, I.; Smolyarchuk, E.A.; Chung, C.H.; Plotka, A.; Shi, H.; Wang, D. Evaluation of the effect of P-glycoprotein inhibition and induction on talazoparib disposition in patients with advanced solid tumours. Br. J. Clin. Pharmacol. 2020, 86, 771–778. [Google Scholar] [CrossRef] [PubMed]
- Vagiannis, D.; Budagaga, Y.; Morell, A.; Zhang, Y.; Novotná, E.; Skarka, A.; Kammerer, S.; Küpper, J.-H.; Hanke, I.; Rozkoš, T. Tepotinib Inhibits Several Drug Efflux Transporters and Biotransformation Enzymes: The Role in Drug–Drug Interactions and Targeting Cytostatic Resistance In Vitro and Ex Vivo. Int. J. Mol. Sci. 2021, 22, 11936. [Google Scholar] [CrossRef]
- Vagiannis, D.; Novotna, E.; Skarka, A.; Kammerer, S.; Küpper, J.-H.; Chen, S.; Guo, L.; Staud, F.; Hofman, J. Ensartinib (X-396) effectively modulates pharmacokinetic resistance mediated by ABCB1 and ABCG2 drug efflux transporters and CYP3A4 biotransformation enzyme. Cancers 2020, 12, 813. [Google Scholar] [CrossRef]
- Vagiannis, D.; Zhang, Y.; Budagaga, Y.; Novotna, E.; Skarka, A.; Kammerer, S.; Küpper, J.-H.; Hofman, J. Alisertib shows negligible potential for perpetrating pharmacokinetic drug-drug interactions on ABCB1, ABCG2 and cytochromes P450, but acts as dual-activity resistance modulator through the inhibition of ABCC1 transporter. Toxicol. Appl. Pharmacol. 2022, 434, 115823. [Google Scholar] [CrossRef]
- Goos, J.A.; Verbeek, J.; Geldof, A.A.; Hiemstra, A.C.; van de Wiel, M.A.; Adamzek, K.A.; Delis-Van Diemen, P.M.; Stroud, S.G.; Bradley, D.P.; Meijer, G.A. Molecular imaging of aurora kinase A (AURKA) expression: Synthesis and preclinical evaluation of radiolabeled alisertib (MLN8237). Nucl. Med. Biol. 2016, 43, 63–72. [Google Scholar] [CrossRef]
- Michaelis, M.; Selt, F.; Rothweiler, F.; Löschmann, N.; Nüsse, B.; Dirks, W.G.; Zehner, R.; Cinatl Jr, J. Aurora kinases as targets in drug-resistant neuroblastoma cells. PLoS ONE 2014, 9, e108758. [Google Scholar] [CrossRef]
- Guney Eskiler, G.; Cecener, G.; Egeli, U.; Tunca, B. Talazoparib nanoparticles for overcoming multidrug resistance in triple-negative breast cancer. J. Cell. Physiol. 2020, 235, 6230–6245. [Google Scholar] [CrossRef]
- Wu, R. Growth of Human Lung Tumor Cells in Culture. In Culture of Human Tumor Cells; Pfragner, R., Freshney, R.I., Eds.; Wiley-Liss Inc: Hoboken, NJ, USA, 2004; pp. 1–21. [Google Scholar] [PubMed]
- Hofman, J.; Sorf, A.; Vagiannis, D.; Sucha, S.; Kammerer, S.; Küpper, J.-H.; Chen, S.; Guo, L.; Ceckova, M.; Staud, F. Brivanib Exhibits Potential for Pharmacokinetic Drug–Drug Interactions and the Modulation of Multidrug Resistance through the Inhibition of Human ABCG2 Drug Efflux Transporter and CYP450 Biotransformation Enzymes. Mol. Pharm. 2019, 16, 4436–4450. [Google Scholar] [CrossRef]
- Hofman, J.; Sorf, A.; Vagiannis, D.; Sucha, S.; Novotna, E.; Kammerer, S.; Küpper, J.-H.; Ceckova, M.; Staud, F. Interactions of alectinib with human ATP-binding cassette drug efflux transporters and cytochrome P450 biotransformation enzymes: Effect on pharmacokinetic multidrug resistance. Drug Metab. Dispos. 2019, 47, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Vagiannis, D.; Yu, Z.; Novotna, E.; Morell, A.; Hofman, J. Entrectinib reverses cytostatic resistance through the inhibition of ABCB1 efflux transporter, but not the CYP3A4 drug-metabolizing enzyme. Biochem. Pharmacol. 2020, 178, 114061. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A free tool to discover chemistry for biology. J. Chem. Inf. Modeling 2012, 52, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Schüttelkopf, A.W.; Van Aalten, D.M. PRODRG: A tool for high-throughput crystallography of protein–ligand complexes. Acta Crystallogr. Sect. D: Biol. Crystallogr. 2004, 60, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Jackson, S.M.; Manolaridis, I.; Kowal, J.; Zechner, M.; Taylor, N.M.; Bause, M.; Bauer, S.; Bartholomaeus, R.; Bernhardt, G.; Koenig, B. Structural basis of small-molecule inhibition of human multidrug transporter ABCG2. Nat. Struct. Mol. Biol. 2018, 25, 333–340. [Google Scholar] [CrossRef]
- Johnson, Z.L.; Chen, J. ATP binding enables substrate release from multidrug resistance protein 1. Cell 2018, 172, 81–89.e10. [Google Scholar] [CrossRef]
- Manolaridis, I.; Jackson, S.M.; Taylor, N.M.; Kowal, J.; Stahlberg, H.; Locher, K.P. Cryo-EM structures of a human ABCG2 mutant trapped in ATP-bound and substrate-bound states. Nature 2018, 563, 426–430. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Adasme, M.F.; Linnemann, K.L.; Bolz, S.N.; Kaiser, F.; Salentin, S.; Haupt, V.J.; Schroeder, M. PLIP 2021: Expanding the scope of the protein–ligand interaction profiler to DNA and RNA. Nucleic Acids Res. 2021, 49, W530–W534. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Vagiannis, D.; Budagaga, Y.; Sabet, Z.; Hanke, I.; Rozkoš, T.; Hofman, J. Sonidegib potentiates the cancer cells’ sensitivity to cytostatic agents by functional inhibition of ABCB1 and ABCG2 in vitro and ex vivo. Biochem. Pharmacol. 2022, 115009. [Google Scholar] [CrossRef]
- Chou, T.-C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Drug(s) | IC50 (µM) | 95% CI (µM) | RR |
|---|---|---|---|---|
| MDCKII-parent | ||||
| daunorubicin | 1.00 | (0.878–1.13) | ||
| mitoxantrone | 1.48 | (1.35–1.62) | ||
| daunorubicin + talazoparib | 1.09 ns | (1.01–1.18) | 0.917 | |
| mitoxantrone + talazoparib | 1.40 ns | (1.30–1.53) | 1.06 | |
| MDCKII-ABCC1 | ||||
| daunorubicin | 5.80 | (5.42–6.19) | ||
| daunorubicin + talazoparib | 2.71 ** | (2.36–3.12) | 2.14 | |
| MDCKII-ABCG2 | ||||
| mitoxantrone | 12.1 | (11.3–12.9) | ||
| mitoxantrone + talazoparib | 2.29 *** | (2.04–2.55) | 5.28 | |
| A431-parent | ||||
| daunorubicin | 1.11 | (0.966–1.29) | ||
| mitoxantrone | 0.88 | (0.714–1.06) | ||
| daunorubicin + talazoparib | 1.22 ns | (1.05–1.43) | 0.909 | |
| mitoxantrone + talazoparib | 0.88 ns | (0.692–1.11) | 1.00 | |
| A431-ABCC1 | ||||
| daunorubicin | 3.62 | (3.19–4.10) | ||
| daunorubicin + talazoparib | 2.13 * | (1.70–2.64) | 1.69 | |
| A431-ABCG2 | ||||
| mitoxantrone | 10.0 | (9.36–10.7) | ||
| mitoxantrone + talazoparib | 3.53 ** | (2.99–4.08) | 2.83 | |
| Sample No. | Gender | Age | Histopathological Diagnosis |
|---|---|---|---|
| 1 | male | 73 | adenocarcinoma |
| 2 | male | 66 | squamous carcinoma |
| 3 | female | 71 | adenocarcinoma |
| 4 | male | 73 | squamous carcinoma |
| 5 | male | 72 | adenocarcinoma |
| 6 | female | 69 | adenocarcinoma |
| 7 | male | 64 | combined neuroendocrine carcinoma (large cell + small cell) |
| 8 | male | 70 | squamous carcinoma |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sabet, Z.; Vagiannis, D.; Budagaga, Y.; Zhang, Y.; Novotná, E.; Hanke, I.; Rozkoš, T.; Hofman, J. Talazoparib Does Not Interact with ABCB1 Transporter or Cytochrome P450s, but Modulates Multidrug Resistance Mediated by ABCC1 and ABCG2: An in Vitro and Ex Vivo Study. Int. J. Mol. Sci. 2022, 23, 14338. https://doi.org/10.3390/ijms232214338
Sabet Z, Vagiannis D, Budagaga Y, Zhang Y, Novotná E, Hanke I, Rozkoš T, Hofman J. Talazoparib Does Not Interact with ABCB1 Transporter or Cytochrome P450s, but Modulates Multidrug Resistance Mediated by ABCC1 and ABCG2: An in Vitro and Ex Vivo Study. International Journal of Molecular Sciences. 2022; 23(22):14338. https://doi.org/10.3390/ijms232214338
Chicago/Turabian StyleSabet, Ziba, Dimitrios Vagiannis, Youssif Budagaga, Yu Zhang, Eva Novotná, Ivo Hanke, Tomáš Rozkoš, and Jakub Hofman. 2022. "Talazoparib Does Not Interact with ABCB1 Transporter or Cytochrome P450s, but Modulates Multidrug Resistance Mediated by ABCC1 and ABCG2: An in Vitro and Ex Vivo Study" International Journal of Molecular Sciences 23, no. 22: 14338. https://doi.org/10.3390/ijms232214338
APA StyleSabet, Z., Vagiannis, D., Budagaga, Y., Zhang, Y., Novotná, E., Hanke, I., Rozkoš, T., & Hofman, J. (2022). Talazoparib Does Not Interact with ABCB1 Transporter or Cytochrome P450s, but Modulates Multidrug Resistance Mediated by ABCC1 and ABCG2: An in Vitro and Ex Vivo Study. International Journal of Molecular Sciences, 23(22), 14338. https://doi.org/10.3390/ijms232214338