
Bilirubin Prevents the TH+ Dopaminergic Neuron Loss in a Parkinson’s Disease Model by Acting on TNF-α



Abstract
1. Introduction
2. Results
2.1. Low Levels of UCB Protect from the TH+ Positive Neurons Loss in the OBCs-SN Model of PD
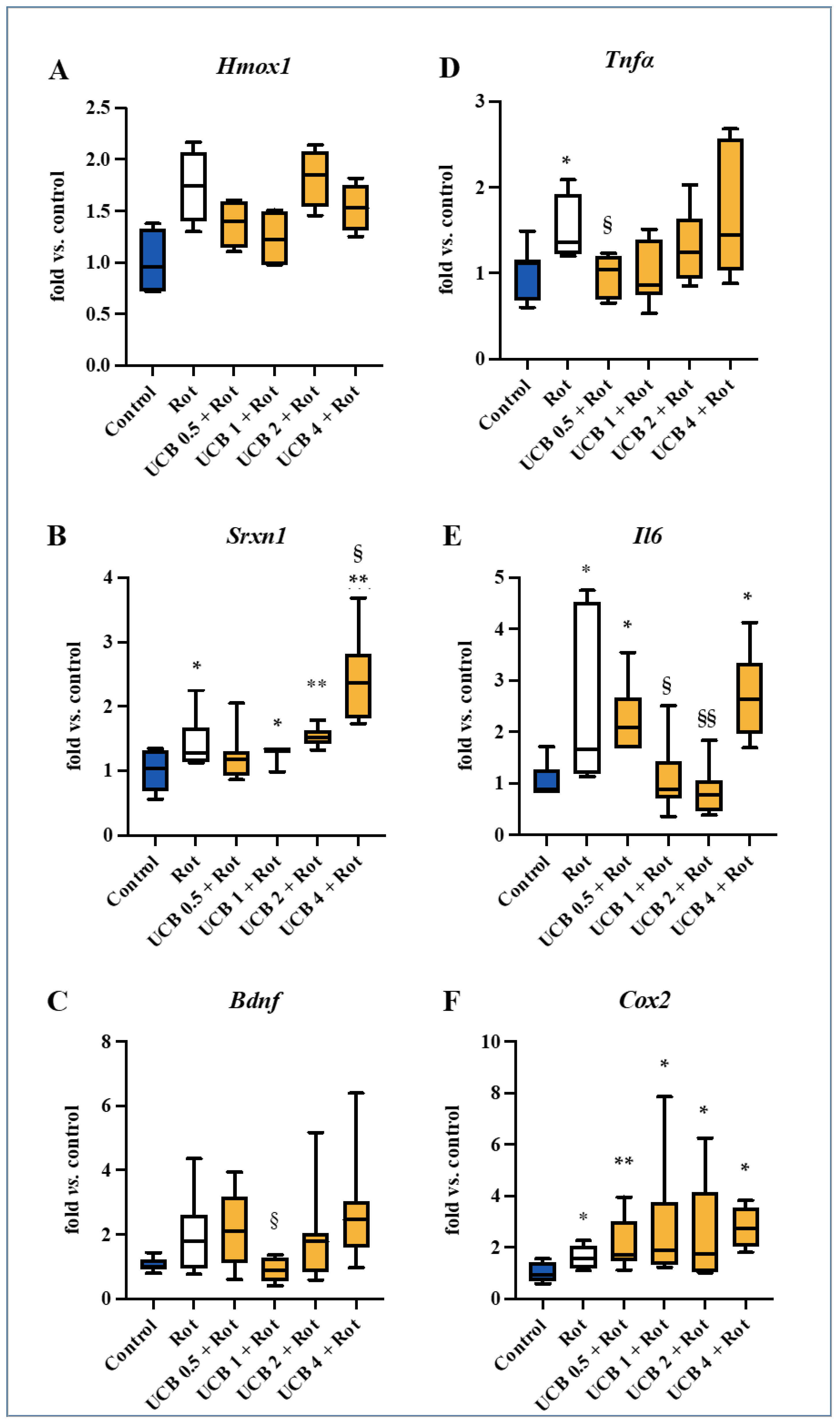
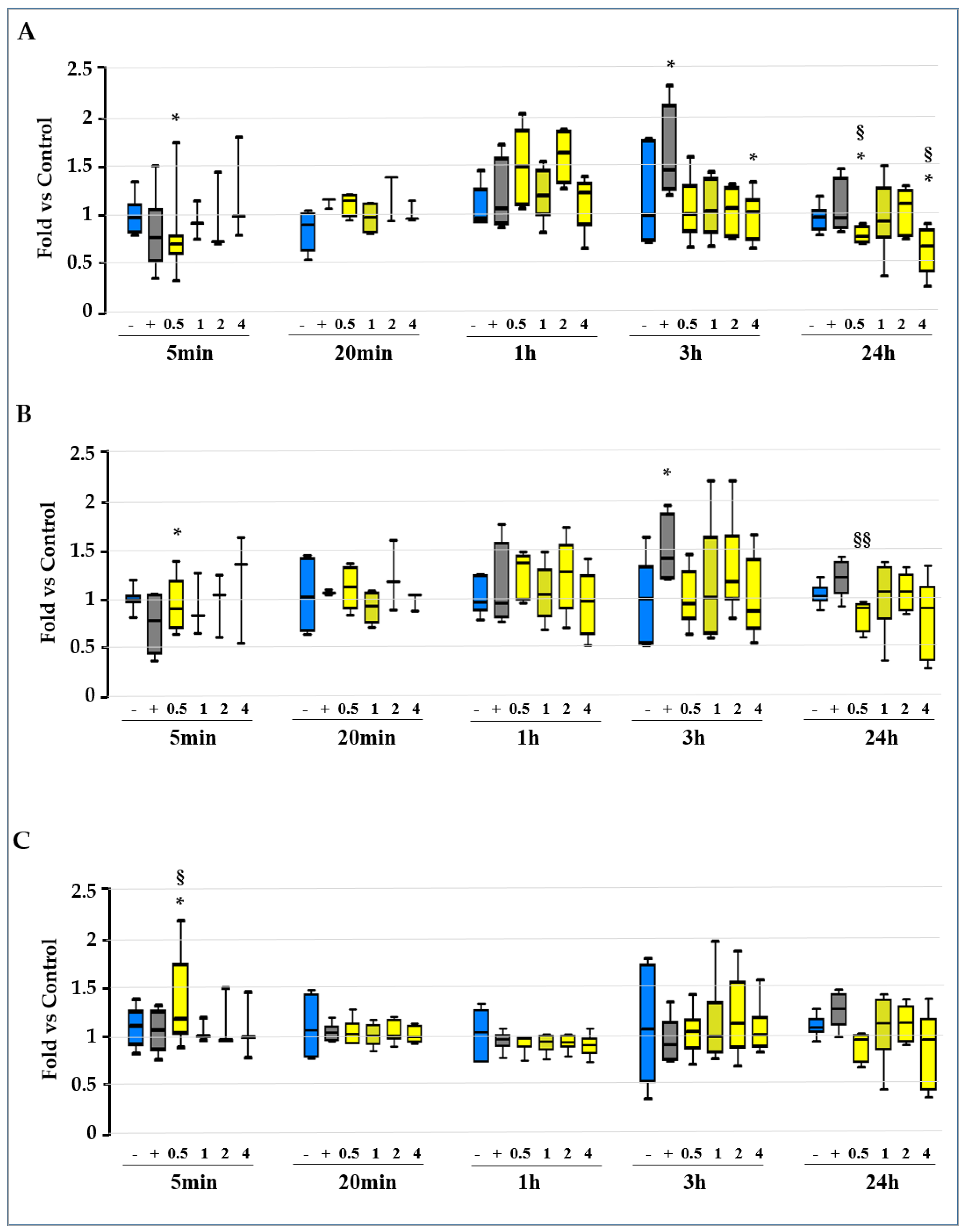
2.2. Screening for the Molecular Mechanisms of Protection
2.3. Unravelling the Role of Redox Imbalance in the Model
2.3.1. Evaluation of the GSH/GSSG Ratio, GSH, and GSSG Level along the Experimental Time
2.3.2. NAC Protection against the Rot-Induced Redox Imbalance
2.4. TNF-α Role in DOPAn Demise
3. Discussion
4. Materials and Methods
4.1. Organotypic Brain Culture Preparation
4.2. Treatments
- A PD model of rotenone (Rot, R8875, Sigma-Aldrich, St. Louis, MO, USA), a pesticide known to trigger PD in humans and animal models [8,67], was used in this study. Rot was dissolved in DMSO (Sigma-Aldrich, St. Louis, MO, USA) and was used in a final concentration of 10 nM in the culture medium [8]. The final concentration has been experimentally identified for recreating a slow progressive demise of the TH+ DOPAn, mimicking the clinical scenario corresponding to the diagnosis in patients (40% TH+ DOPAn loss), after 24 h of challenge. At that time, the early mechanisms acting on DOPAn demise were maximally modulated by Rot [8]. For this reason, we chose 24 h as the time period to study their role and the potential protection conferred by bilirubin in this work.
- For the evaluation of UCB protection, UCB (B4126, Sigma-Aldrich, St. Louis, MO, USA. PubChem CID 5280352) was purified as previously described [68] and dissolved in DMSO to reach the desired concentrations. As preliminary data for this work, we carefully selected the concentrations of the potential protection conferred by UCB. Doses of 10 μM and 20 μM UCB were toxic to heathy slices, inducing TH+ DOPAn loss (60% and 70%, respectively); thus, they were excluded. Doses of 2 μM and 4 μM concentrations were not so high as to be toxic (adding toxicity to Rot challenged slices), but not so low as to be protective toward Rot-induced damage. The highest concentration of 4 μM UCB in the culture medium was determined using the spectrophotometer (absorbance of 0.190, λ = 440 nm). The final concentrations of 2 μM, 1 μM, and 0.5 μM UCB were reached by dilution; then, Rot was added to UCB-containing medium to perform co-treatments. The control medium was prepared by dissolving an equal amount of DMSO, the vehicle of both UCB and Rot (control: ctrl). Because FBS contains albumin, a binder of UCB, we quantified the amount of UCB not bound to albumin (namely, free bilirubin—Bf; the moiety of UCB entering the tissue) in the OBCs-SN medium, as described by Roca et al. [69]. All the selected UCB concentrations corresponded to a Bf < 4 nM, considered to be physiological concentrations, or Gilbert-like conditions [18,70].
- For the evaluation of redox imbalance to TH+ DOPAn loss, N-acetyl-cysteine (NAC, Sigma-Aldrich, St. Louis, MO, USA) treatment was used. We applied a broad range of NAC concentrations, based on the methods in the literature [71,72]. NAC was dissolved in medical water to a 125 mM concentration, then diluted it to the final concentrations ranging from 10 μM to 15,000 μM in the OBCs-SN medium, and Rot was added to perform co-treatments.
- For DOPAn loss by TNF-α challenging, TNF-α (Peprotech, London, UK) was diluted directly in the OBC-SN medium, starting with the TNF-α concentration suggested by the literature [38], and was increased at three concentrations (20 ng/mL, 40 ng/mL, and 60 ng/mL) until replication of the TH+ DOPAn loss observed in the Rot treatment was reached.
- For the infliximab protection in PD model, infliximab (Flixabi®, Biogen, Hillerod, Denmark), a TNF-α neutralizing antibody, was diluted in the OBC-SN medium at the final concentration of 41 µg/mL [26]; then, Rot was added to perform the co-treatments.
4.3. Immunofluorescent Staining of Dopaminergic Neuron
4.4. RNA Isolation, cDNA Synthesis, and Real-Time PCR Analysis
4.5. Glutathione Measurement
4.6. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Tysnes, O.-B.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef] [PubMed]
- Balestrino, R.; Schapira, A.H.V. Parkinson disease. Eur. J. Neurol. 2020, 27, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Dorsey, E.R.; Bloem, B.R. The Parkinson Pandemic—A Call to Action. JAMA Neurol. 2018, 75, 9–10. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, J.; Tan, E.K. Parkinson’s disease: Etiopathogenesis and treatment. J. Neurol. Neurosurg. Psychiatry 2020, 91, 795–808. [Google Scholar] [CrossRef]
- Postuma, R.B.; Berg, D.; Stern, M.; Poewe, W.; Olanow, C.W.; Oertel, W.; Obeso, J.; Marek, K.; Litvan, I.; Lang, A.E.; et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 2015, 30, 1591–1601. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.; Steur, E.N.J.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Rocha, E.M.; De Miranda, B.; Sanders, L.H. Alpha-synuclein: Pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol. Dis. 2018, 109, 249–257. [Google Scholar] [CrossRef]
- Ben, M.D.; Bongiovanni, R.; Tuniz, S.; Fioriti, E.; Tiribelli, C.; Moretti, R.; Gazzin, S. Earliest Mechanisms of Dopaminergic Neurons Sufferance in a Novel Slow Progressing Ex Vivo Model of Parkinson Disease in Rat Organotypic Cultures of Substantia Nigra. Int. J. Mol. Sci. 2019, 20, 2224. [Google Scholar] [CrossRef]
- Johnson, M.E.; Bobrovskaya, L. An update on the rotenone models of Parkinson’s disease: Their ability to reproduce the features of clinical disease and model gene–environment interactions. NeuroToxicology 2015, 46, 101–116. [Google Scholar] [CrossRef]
- Gazzin, S.; Vitek, L.; Watchko, J.; Shapiro, S.M.; Tiribelli, C. A Novel Perspective on the Biology of Bilirubin in Health and Disease. Trends Mol. Med. 2016, 22, 758–768. [Google Scholar] [CrossRef]
- Maruhashi, T.; Soga, J.; Fujimura, N.; Idei, N.; Mikami, S.; Iwamoto, Y.; Kajikawa, M.; Matsumoto, T.; Kihara, Y.; Chayama, K.; et al. Hyperbilirubinemia, Augmentation of Endothelial Function, and Decrease in Oxidative Stress in Gilbert Syndrome. Circulation 2012, 126, 598–603. [Google Scholar] [CrossRef]
- Sedlak, T.W.; Snyder, S.H. Bilirubin Benefits: Cellular Protection by a Biliverdin Reductase Antioxidant Cycle. Pediatrics 2004, 113, 1776–1782. [Google Scholar] [CrossRef]
- Jayanti, S.; Vítek, L.; Tiribelli, C.; Gazzin, S. The Role of Bilirubin and the Other “Yellow Players” in Neurodegenerative Diseases. Antioxidants 2020, 9, 900. [Google Scholar] [CrossRef]
- Barañano, D.E.; Rao, M.; Ferris, C.D.; Snyder, S.H. Biliverdin reductase: A major physiologic cytoprotectant. Proc. Natl. Acad. Sci. USA 2002, 99, 16093–16098. [Google Scholar] [CrossRef]
- Jayanti, S.; Moretti, R.; Tiribelli, C.; Gazzin, S. Bilirubin and inflammation in neurodegenerative and other neurological diseases. Neuroimmunol. Neuroinflammation 2020, 7, 92–108. [Google Scholar] [CrossRef]
- Liu, Y.; Li, P.; Lu, J.; Xiong, W.; Oger, J.; Tetzlaff, W.; Cynader, M. Bilirubin Possesses Powerful Immunomodulatory Activity and Suppresses Experimental Autoimmune Encephalomyelitis. J. Immunol. 2008, 181, 1887–1897. [Google Scholar] [CrossRef] [PubMed]
- Vítek, L.; Tiribelli, C. Bilirubin: The yellow hormone? J. Hepatol. 2021, 75, 1485–1490. [Google Scholar] [CrossRef]
- Wagner, K.-H.; Wallner, M.; Mölzer, C.; Gazzin, S.; Bulmer, A.C.; Tiribelli, C.; Vitek, L. Looking to the horizon: The role of bilirubin in the development and prevention of age-related chronic diseases. Clin. Sci. 2015, 129, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Vítek, L. The Role of Bilirubin in Diabetes, Metabolic Syndrome, and Cardiovascular Diseases. Front. Pharmacol. 2012, 3, 55. [Google Scholar] [CrossRef]
- Yu, M.; Su, D.; Yang, Y.; Qin, L.; Hu, C.; Liu, R.; Zhou, Y.; Yang, C.; Yang, X.; Wang, G.; et al. D-T7 Peptide-Modified PEGylated Bilirubin Nanoparticles Loaded with Cediranib and Paclitaxel for Antiangiogenesis and Chemotherapy of Glioma. ACS Appl. Mater. Interfaces 2019, 11, 176–186. [Google Scholar] [CrossRef]
- Liu, Y.; Zhu, B.; Wang, X.; Luo, L.; Li, P.; Paty, D.W.; Cynader, M.S. Bilirubin as a potent antioxidant suppresses experimental autoimmune encephalomyelitis: Implications for the role of oxidative stress in the development of multiple sclerosis. J. Neuroimmunol. 2003, 139, 27–35. [Google Scholar] [CrossRef]
- Giguère, N.; Nanni, S.B.; Trudeau, L.-E. On Cell Loss and Selective Vulnerability of Neuronal Populations in Parkinson’s Disease. Front. Neurol. 2018, 9, 455. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Duan, S.; Zhou, Y.; Yu, S.; Wu, J.; Wu, X.; Zhao, J.; Zhao, Y. Sulfiredoxin-1 Attenuates Oxidative Stress via Nrf2/ARE Pathway and 2-Cys Prdxs After Oxygen-Glucose Deprivation in Astrocytes. J. Mol. Neurosci. 2014, 55, 941–950. [Google Scholar] [CrossRef] [PubMed]
- Zagoura, D.; Canovas-Jorda, D.; Pistollato, F.; Bremer-Hoffmann, S.; Bal-Price, A. Evaluation of the rotenone-induced activation of the Nrf2 pathway in a neuronal model derived from human induced pluripotent stem cells. Neurochem. Int. 2017, 106, 62–73. [Google Scholar] [CrossRef]
- Monti, D.A.; Zabrecky, G.; Kremens, D.; Liang, T.-W.; Wintering, N.A.; Bazzan, A.J.; Zhong, L.; Bowens, B.K.; Chervoneva, I.; Intenzo, C.; et al. N-Acetyl Cysteine Is Associated With Dopaminergic Improvement in Parkinson’s Disease. Clin. Pharmacol. Ther. 2019, 106, 884–890. [Google Scholar] [CrossRef]
- Adedokun, O.J.; Sandborn, W.J.; Feagan, B.G.; Rutgeerts, P.; Xu, Z.; Marano, C.W.; Johanns, J.; Zhou, H.; Davis, H.M.; Cornillie, F.; et al. Association Between Serum Concentration of Infliximab and Efficacy in Adult Patients With Ulcerative Colitis. Gastroenterology 2014, 147, 1296–1307.e5. [Google Scholar] [CrossRef]
- McGuire, S.O.; Ling, Z.D.; Lipton, J.W.; Sortwell, C.E.; Collier, T.J.; Carvey, P.M. Tumor Necrosis Factor α Is Toxic to Embryonic Mesencephalic Dopamine Neurons. Exp. Neurol. 2001, 169, 219–230. [Google Scholar] [CrossRef]
- Tansey, M.G.; McCoy, M.K.; Frank-Cannon, T.C. Neuroinflammatory mechanisms in Parkinson’s disease: Potential environmental triggers, pathways, and targets for early therapeutic intervention. Exp. Neurol. 2007, 208, 1–25. [Google Scholar] [CrossRef]
- Yuyun, X.; Jinjun, Q.; Minfang, X.; Jing, Q.; Juan, X.; Rui, M.; Li, Z.; Jing, G. Effects of Low Concentrations of Rotenone Upon Mitohormesis in SH-SY5Y Cells. Dose-Response 2012, 11, 270–280. [Google Scholar] [CrossRef]
- Testa, C.M.; Sherer, T.B.; Greenamyre, J.T. Rotenone induces oxidative stress and dopaminergic neuron damage in organotypic substantia nigra cultures. Mol. Brain Res. 2005, 134, 109–118. [Google Scholar] [CrossRef]
- Pagida, M.A.; Konstantinidou, A.E.; Tsekoura, E.; Mangoura, D.; Patsouris, E.; Panayotacopoulou, M.T. Vulnerability of the Mesencephalic Dopaminergic Neurons of the Human Neonate to Prolonged Perinatal Hypoxia: An Immunohistochemical Study of Tyrosine Hydroxylase Expression in Autopsy Material. J. Neuropathol. Exp. Neurol. 2013, 72, 337–350. [Google Scholar] [CrossRef] [PubMed]
- Qaisiya, M.; Zabetta, C.D.C.; Bellarosa, C.; Tiribelli, C. Bilirubin mediated oxidative stress involves antioxidant response activation via Nrf2 pathway. Cell. Signal. 2014, 26, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Mateo, I.; Infante, J.; Sánchez-Juan, P.; García-Gorostiaga, I.; Rodríguez-Rodríguez, E.; Vázquez-Higuera, J.L.; Berciano, J.; Combarros, O. Serum heme oxygenase-1 levels are increased in Parkinson’s disease but not in Alzheimer’s disease. Acta Neurol. Scand. 2010, 121, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Schipperab, H.M.; Liberman, A.; Stopa, E. Neural Heme Oxygenase-1 Expression in Idiopathic Parkinson’s Disease. Exp. Neurol. 1998, 150, 60–68. [Google Scholar] [CrossRef]
- Moccia, M.; Picillo, M.; Erro, R.; Longo, K.; Amboni, M.; Santangelo, G.; Palladino, R.; Allocca, R.; Caporale, O.; Triassi, M.; et al. Increased bilirubin levels in de novo Parkinson’s disease. Eur. J. Neurol. 2015, 22, 954–959. [Google Scholar] [CrossRef]
- Berman, A.E.; Chan, W.Y.; Brennan, A.M.; Reyes, R.C.; Adler, B.L.; Suh, S.W.; Kauppinen, T.M.; Edling, Y.; Swanson, R.A. N-acetylcysteine prevents loss of dopaminergic neurons in the EAAC1−/− mouse. Ann. Neurol. 2010, 69, 509–520. [Google Scholar] [CrossRef]
- Kouchaki, E.; Kakhaki, R.D.; Tamtaji, O.R.; Dadgostar, E.; Behnam, M.; Nikoueinejad, H.; Akbari, H. Increased serum levels of TNF-α and decreased serum levels of IL-27 in patients with Parkinson disease and their correlation with disease severity. Clin. Neurol. Neurosurg. 2018, 166, 76–79. [Google Scholar] [CrossRef]
- McCoy, M.K.; Martinez, T.N.; Ruhn, K.A.; Szymkowski, D.; Smith, C.G.; Botterman, B.R.; Tansey, K.E.; Tansey, M.G. Blocking Soluble Tumor Necrosis Factor Signaling with Dominant-Negative Tumor Necrosis Factor Inhibitor Attenuates Loss of Dopaminergic Neurons in Models of Parkinson’s Disease. J. Neurosci. 2006, 26, 9365–9375. [Google Scholar] [CrossRef]
- Hung, S.-Y.; Liou, H.-C.; Kang, K.-H.; Wu, R.-M.; Wen, C.-C.; Fu, W.-M. Overexpression of Heme Oxygenase-1 Protects Dopaminergic Neurons against 1-Methyl-4-Phenylpyridinium-Induced Neurotoxicity. Mol. Pharmacol. 2008, 74, 1564–1575. [Google Scholar] [CrossRef]
- Zhu, G.; Li, J.; He, L.; Wang, X.; Hong, X. MPTP-induced changes in hippocampal synaptic plasticity and memory are prevented by memantine through the BDNF-TrkB pathway. J. Cereb. Blood Flow Metab. 2015, 172, 2354–2368. [Google Scholar] [CrossRef]
- Scalzo, P.; Kummer, A.; Bretas, T.; Cardoso, F.; Teixeira, A.L. Serum levels of brain-derived neurotrophic factor correlate with motor impairment in Parkinson’s disease. J. Neurol. 2009, 257, 540–545. [Google Scholar] [CrossRef] [PubMed]
- Khalil, H.; Alomari, M.A.; Khabour, O.F.; Al-Hieshan, A.; Bajwa, J.A. Relationship of circulatory BDNF with cognitive deficits in people with Parkinson’s disease. J. Neurol. Sci. 2016, 362, 217–220. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Chan, N.G.; Bannon, M.J.; Orozco-Barrios, C.E.; Escobedo, L.; Zamudio, S.; De la Cruz, F.; Gongora-Alfaro, J.L.; Armendáriz-Borunda, J.; Reyes-Corona, D.; Espadas-Alvarez, A.J.; et al. Neurotensin-polyplex-mediated brain-derived neurotrophic factor gene delivery into nigral dopamine neurons prevents nigrostriatal degeneration in a rat model of early Parkinson’s disease. J. Biomed. Sci. 2015, 22, 59. [Google Scholar] [CrossRef] [PubMed]
- Lucidi-Phillipi, C.A.; Gage, F.H.; Shults, C.W.; Jones, K.; Reichardt, L.F.; Kang, U. Brain-derived neurotrophic factor-transduced fibroblasts: Production of BDNF and effects of grafting to the adult rat brain. J. Comp. Neurol. 1995, 354, 361–376. [Google Scholar] [CrossRef] [PubMed]
- Samii, A.; Etminan, M.; Wiens, M.O.; Jafari, S.; Samii, A. NSAID Use and the Risk of Parkinson’s Disease. Drugs Aging 2009, 26, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Yi, J.; Yang, J.; Li, P.; Cheng, X.; Mao, P. Nonsteroidal anti-inflammatory drugs use and risk of Parkinson disease. Medicine 2018, 97, e12172. [Google Scholar] [CrossRef]
- Lin, Q.-S.; Chen, P.; Wang, W.-X.; Lin, C.-C.; Zhou, Y.; Yu, L.-H.; Lin, Y.-X.; Xu, Y.-F.; Kang, D.-Z. RIP1/RIP3/MLKL mediates dopaminergic neuron necroptosis in a mouse model of Parkinson disease. Lab. Investig. 2019, 100, 503–511. [Google Scholar] [CrossRef]
- Dufek, M.; Rektorova, I.; Thon, V.; Lokaj, J.; Rektor, I. Interleukin-6 May Contribute to Mortality in Parkinson’s Disease Patients: A 4-Year Prospective Study. Park. Dis. 2015, 2015, 898192. [Google Scholar] [CrossRef]
- Pereira, J.R.; dos Santos, L.V.; Santos, R.M.S.; Campos, A.L.F.; Pimenta, A.L.; de Oliveira, M.S.; Bacheti, G.G.; Rocha, N.P.; Teixeira, A.L.; Christo, P.P.; et al. IL-6 serum levels are elevated in Parkinson’s disease patients with fatigue compared to patients without fatigue. J. Neurol. Sci. 2016, 370, 153–156. [Google Scholar] [CrossRef]
- Teismann, P.; Vila, M.; Choi, D.-K.; Tieu, K.; Wu, D.C.; Jackson-Lewis, V.; Przedborski, S. COX-2 and neurodegeneration in Parkinson’s disease. Ann. N.Y. Acad. Sci. 2006, 991, 272–277. [Google Scholar] [CrossRef]
- Song, S.-Y.; Kim, I.-S.; Koppula, S.; Park, J.-Y.; Kim, B.-W.; Yoon, S.-H.; Choi, D.-K. 2-Hydroxy-4-Methylbenzoic Anhydride Inhibits Neuroinflammation in Cellular and Experimental Animal Models of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 8195. [Google Scholar] [CrossRef] [PubMed]
- Håkansson, A.; Westberg, L.; Nilsson, S.; Buervenich, S.; Carmine, A.; Holmberg, B.; Sydow, O.; Olson, L.; Johnels, B.; Eriksson, E.; et al. Interaction of polymorphisms in the genes encoding interleukin-6 and estrogen receptor beta on the susceptibility to Parkinson’s disease. Am. J. Med Genet. Part B Neuropsychiatr. Genet. 2005, 133, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.-W.; Wu, Y.-R.; Chen, Y.-C.; Fung, H.-C.; Chen, C.-M. Polymorphisms of Interleukin-6 and Interleukin-8 Are Not Associated with Parkinson’s Disease in Taiwan. Brain Sci. 2021, 11, 768. [Google Scholar] [CrossRef]
- Gómez-Santos, C.; Francisco, R.; Giménez-Xavier, P.; Ambrosio, S. Dopamine induces TNFα and TNF-R1 expression in SH-SY5Y human neuroblastoma cells. NeuroReport 2007, 18, 1725–1728. [Google Scholar] [CrossRef] [PubMed]
- De Lella Ezcurra, A.L.; Chertoff, M.; Ferrari, C.; Graciarena, M.; Pitossi, F. Chronic expression of low levels of tumor necrosis factor-α in the substantia nigra elicits progressive neurodegeneration, delayed motor symptoms and microglia/macrophage activation. Neurobiol. Dis. 2010, 37, 630–640. [Google Scholar] [CrossRef] [PubMed]
- Adin, C.A. Bilirubin as a Therapeutic Molecule: Challenges and Opportunities. Antioxidants 2021, 10, 1536. [Google Scholar] [CrossRef]
- Williams-Gray, C.H.; Wijeyekoon, R.; Yarnall, A.J.; Lawson, R.A.; Breen, D.P.; Evans, J.R.; Cummins, G.A.; Duncan, G.W.; Khoo, T.K.; Burn, D.J.; et al. Serum immune markers and disease progression in an incident P arkinson’s disease cohort (ICICLE-PD). Mov. Disord. 2016, 31, 995–1003. [Google Scholar] [CrossRef]
- Majbour, N.K.; Aasly, J.O.; Hustad, E.; Thomas, M.A.; Vaikath, N.N.; Elkum, N.; van de Berg, W.; Tokuda, T.; Mollenhauer, B.; Berendse, H.W.; et al. CSF total and oligomeric α-Synuclein along with TNF-α as risk biomarkers for Parkinson’s disease: A study in LRRK2 mutation carriers. Transl. Neurodegener. 2020, 9, 15. [Google Scholar] [CrossRef]
- Park, K.M.; Bowers, W.J. Tumor necrosis factor-alpha mediated signaling in neuronal homeostasis and dysfunction. Cell. Signal. 2010, 22, 977–983. [Google Scholar] [CrossRef]
- Mogi, M.; Togari, A.; Kondo, T.; Mizuno, Y.; Komure, O.; Kuno, S.; Ichinose, H.; Nagatsu, T. Caspase activities and tumor necrosis factor receptor R1 (p55) level are elevated in the substantia nigra from Parkinsonian brain. J. Neural Transm. 2000, 107, 335–341. [Google Scholar] [CrossRef]
- Peter, I.; Dubinsky, M.; Bressman, S.; Park, A.; Lu, C.; Chen, N.; Wang, A. Anti–Tumor Necrosis Factor Therapy and Incidence of Parkinson Disease Among Patients With Inflammatory Bowel Disease. JAMA Neurol. 2018, 75, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Olsen, A.L.; Riise, T.; Scherzer, C.R. Discovering New Benefits From Old Drugs With Big Data—Promise for Parkinson Disease. JAMA Neurol. 2018, 75, 917–920. [Google Scholar] [CrossRef] [PubMed]
- Brito, M.A.; Palmela, I.; Cardoso, F.L.; Sá-Pereira, I.; Brites, D. Blood–Brain Barrier and Bilirubin: Clinical Aspects and Experimental Data. Arch. Med. Res. 2014, 45, 660–676. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Hossieny, P.; Wu, B.J.; Qawasmeh, A.; Beck, K.; Stocker, R. Pharmacologic Induction of Heme Oxygenase-1. Antioxid. Redox Signal. 2007, 9, 2227–2240. [Google Scholar] [CrossRef]
- Jayanti, S.; Moretti, R.; Tiribelli, C.; Gazzin, S. Bilirubin: A Promising Therapy for Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 6223. [Google Scholar] [CrossRef] [PubMed]
- Kamothi, D.J.; Kant, V.; Jangir, B.L.; Joshi, V.G.; Ahuja, M.; Kumar, V. Novel preparation of bilirubin-encapsulated pluronic F-127 nanoparticles as a potential biomaterial for wound healing. Eur. J. Pharmacol. 2022, 919, 174809. [Google Scholar] [CrossRef] [PubMed]
- Tanner, C.M.; Kamel, F.; Ross, G.W.; Hoppin, J.; Goldman, S.; Korell, M.; Marras, C.; Bhudhikanok, G.S.; Kasten, M.; Chade, A.R.; et al. Rotenone, Paraquat, and Parkinson’s Disease. Environ. Health Perspect. 2011, 119, 866–872. [Google Scholar] [CrossRef]
- McDonagh, A.F.; Assisi, F. The ready isomerization of bilirubin IX-α in aqueous solution. Biochem. J. 1972, 129, 797–800. [Google Scholar] [CrossRef]
- Roca, L.; Calligaris, S.; Wennberg, R.P.; Ahlfors, C.E.; Malik, S.G.; Ostrow, J.D.; Tiribelli, C. Factors Affecting the Binding of Bilirubin to Serum Albumins: Validation and Application of the Peroxidase Method. Pediatr. Res. 2006, 60, 724–728. [Google Scholar] [CrossRef][Green Version]
- Ostrow, D.J.; Pascolo, L.; Tiribelli, C. Reassessment of the Unbound Concentrations of Unconjugated Bilirubin in Relation to Neurotoxicity In Vitro. Pediatr. Res. 2003, 54, 98–104. [Google Scholar] [CrossRef][Green Version]
- Monti, D.A.; Zabrecky, G.; Kremens, D.; Liang, T.-W.; Wintering, N.A.; Cai, J.; Wei, X.; Bazzan, A.J.; Zhong, L.; Bowen, B.; et al. N-Acetyl Cysteine May Support Dopamine Neurons in Parkinson’s Disease: Preliminary Clinical and Cell Line Data. PLoS ONE 2016, 11, e0157602. [Google Scholar] [CrossRef] [PubMed]
- Gleixner, A.M.; Hutchison, D.F.; Sannino, S.; Bhatia, T.N.; Leak, L.C.; Flaherty, P.T.; Wipf, P.; Brodsky, J.L.; Leak, R.K. N-Acetyl-l-Cysteine Protects Astrocytes against Proteotoxicity without Recourse to Glutathione. Mol. Pharmacol. 2017, 92, 564–575. [Google Scholar] [CrossRef] [PubMed]
- Vianello, E.; Zampieri, S.; Marcuzzo, T.; Tordini, F.; Bottin, C.; Dardis, A.; Zanconati, F.; Tiribelli, C.; Gazzin, S. Histone acetylation as a new mechanism for bilirubin-induced encephalopathy in the Gunn rat. Sci. Rep. 2018, 8, 13690. [Google Scholar] [CrossRef] [PubMed]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, research0034.1. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef]
- Mokrasch, L.C.; Teschke, E.J. Glutathione content of cultured cells and rodent brain regions: A specific fluorometric assay. Anal. Biochem. 1984, 140, 506–509. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hmox1 | Srnx1 | Bdnf | Tnf-α | Il6 | Cox2 | |
|---|---|---|---|---|---|---|
| r | −0.8286 | −0.7714 | −0.6571 | −0.7143 | −0.6377 | −0.2571 |
| p Value (two-tailed) | 0.0583 | 0.1028 | 0.175 | 0.1361 | 0.175 | 0.6583 |
| Significant | no | no | no | no | no | no |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jayanti, S.; Moretti, R.; Tiribelli, C.; Gazzin, S. Bilirubin Prevents the TH+ Dopaminergic Neuron Loss in a Parkinson’s Disease Model by Acting on TNF-α. Int. J. Mol. Sci. 2022, 23, 14276. https://doi.org/10.3390/ijms232214276
Jayanti S, Moretti R, Tiribelli C, Gazzin S. Bilirubin Prevents the TH+ Dopaminergic Neuron Loss in a Parkinson’s Disease Model by Acting on TNF-α. International Journal of Molecular Sciences. 2022; 23(22):14276. https://doi.org/10.3390/ijms232214276
Chicago/Turabian StyleJayanti, Sri, Rita Moretti, Claudio Tiribelli, and Silvia Gazzin. 2022. "Bilirubin Prevents the TH+ Dopaminergic Neuron Loss in a Parkinson’s Disease Model by Acting on TNF-α" International Journal of Molecular Sciences 23, no. 22: 14276. https://doi.org/10.3390/ijms232214276
APA StyleJayanti, S., Moretti, R., Tiribelli, C., & Gazzin, S. (2022). Bilirubin Prevents the TH+ Dopaminergic Neuron Loss in a Parkinson’s Disease Model by Acting on TNF-α. International Journal of Molecular Sciences, 23(22), 14276. https://doi.org/10.3390/ijms232214276