
Genetic Dissection of Phosphorus Use Efficiency and Genotype-by-Environment Interaction in Maize

 , ,
, ,  , , , , and
, , , , and 
Abstract
1. Introduction
2. Results
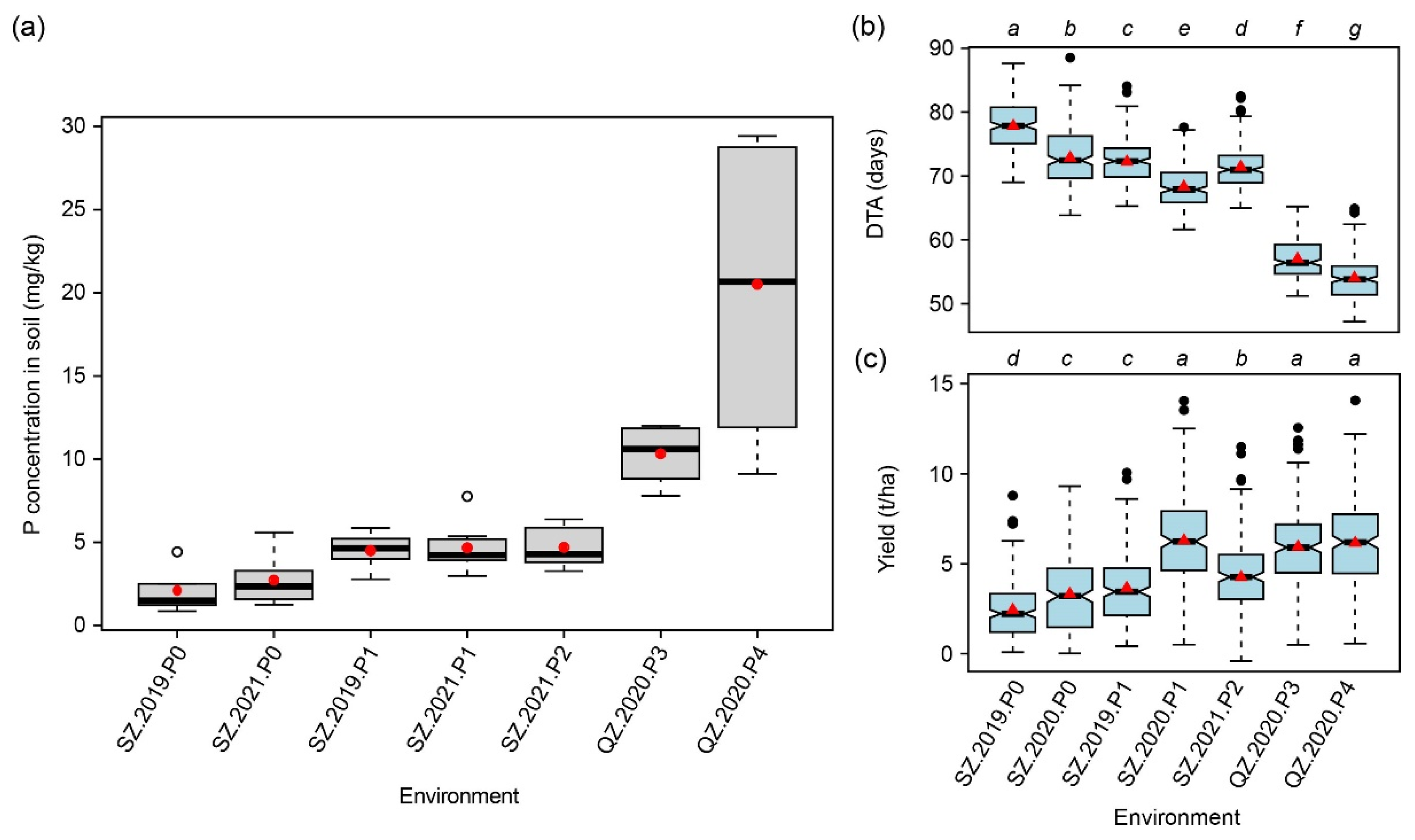
2.1. Analysis of Olsen-P in the Soil
2.2. Population Analysis
2.3. Summary Statistics for 15 Traits Evaluated at Seven Macro-Environments
2.4. Finlay–Wilkinson Regression with Seven Macro-Environments
2.5. Contribution of Different Gene Regions to the Linear and Non-Linear Plasticity
2.6. Genome-Wide Association Mapping for Genotypic Performance, Linear Plasticity, and Non-Linear Plasticity
2.7. Genome-Wide Association Mapping for Genotype-by-Environment Interactions
2.8. Genomic Prediction for Genotype-by-Environment Interaction
3. Discussion
3.1. The Macro-Environment in Plant Breeding
3.2. How to Interpret and Utilize Genotype-by-Environment Interactions
3.3. Contribution of Different Gene Regions to Genotypic Performance and Plasticity
3.4. Mine Gene with Genotype-by-P Treatment
4. Materials and Methods
4.1. Materials
4.2. Phenotypic Analysis
4.3. Genotyping and Quality Control
4.4. Population Analysis
4.5. Finlay–Wilkinson Regression Analysis
4.6. Genome-Wide Association Analysis
4.7. Confidence Intervals of QTL and Candidate Gene Identification
4.8. Variance Component Estimation for SNPs in Different Regions
4.9. Genomic Prediction of Genotype-by-Environment Interaction
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hickey, L.T.; Hafeez, A.N.; Robinson, H.; Jackson, S.A.; Leal-Bertioli, S.C.M.; Tester, M.; Gao, C.; Godwin, I.D.; Hayes, B.J.; Wulff, B.B.H. Breeding Crops to Feed 10 Billion. Nat. Biotechnol. 2019, 37, 744–754. [Google Scholar] [CrossRef] [PubMed]
- Schnable, P.S.; Ware, D.; Fulton, R.S.; Stein, J.C.; Wei, F.; Pasternak, S.; Liang, C.; Zhang, J.; Fulton, L.; Graves, T.A.; et al. The B73 Maize Genome: Complexity, Diversity, and Dynamics. Science 2009, 326, 1112–1115. [Google Scholar] [CrossRef] [PubMed]
- Malosetti, M.; Ribaut, J.M.; van Eeuwijk, F.A. The Statistical Analysis of Multi-Environment Data: Modeling Genotype-by-Environment Interaction and Its Genetic Basis. Front. Physiol. 2013, 4, 44. [Google Scholar] [CrossRef] [PubMed]
- Finlay, K.W.; Wilkinson, G.N. The Analysis of Adaptation in a Plant-Breeding Programme. Aust. J. Agric. Res. 1963, 14, 742–754. [Google Scholar] [CrossRef]
- Kusmec, A.; Srinivasan, S.; Nettleton, D.; Schnable, P.S. Distinct Genetic Architectures for Phenotype Means and Plasticities in Zea Mays. Nat. Plants 2017, 3, 715–723. [Google Scholar] [CrossRef]
- Gage, J.L.; Jarquin, D.; Romay, C.; Lorenz, A.; Buckler, E.S.; Kaeppler, S.; Alkhalifah, N.; Bohn, M.; Campbell, D.A.; Edwards, J.; et al. The Effect of Artificial Selection on Phenotypic Plasticity in Maize. Nat. Commun. 2017, 8, 1348. [Google Scholar] [CrossRef]
- Desmidt, E.; Ghyselbrecht, K.; Zhang, Y.; Pinoy, L.; Van Der Bruggen, B.; Verstraete, W.; Rabaey, K.; Meesschaert, B. Global Phosphorus Scarcity and Full-Scale P-Recovery Techniques: A Review. Crit. Rev. Environ. Sci. Technol. 2015, 45, 336–384. [Google Scholar] [CrossRef]
- Guo, Z.; Cao, H.; Zhao, J.; Bai, S.; Peng, W.; Li, J.; Sun, L.; Chen, L.; Lin, Z.; Shi, C.; et al. A Natural UORF Variant Confers Phosphorus Acquisition Diversity in Soybean. Nat. Commun. 2022, 13, 3796. [Google Scholar] [CrossRef]
- Li, X.; Guo, T.; Mu, Q.; Li, X.; Yu, J. Genomic and Environmental Determinants and Their Interplay Underlying Phenotypic Plasticity. Proc. Natl. Acad. Sci. USA 2018, 115, 6679–6684. [Google Scholar] [CrossRef]
- Li, X.; Guo, T.; Wang, J.; Bekele, W.A.; Sukumaran, S.; Vanous, A.E.; McNellie, J.P.; Cortes, L.T.; Lopes, M.S.; Lamkey, K.R.; et al. An Integrated Framework Reinstating the Environmental Dimension for GWAS and Genomic Selection in Crops. Mol. Plant 2021, 14, 874–887. [Google Scholar] [CrossRef]
- Mu, Q.; Guo, T.; Li, X.; Yu, J. Phenotypic Plasticity in Plant Height Shaped by Interaction between Genetic Loci and Diurnal Temperature Range. New Phytol. 2022, 233, 1768–1779. [Google Scholar] [CrossRef] [PubMed]
- Grotewold, E.; Drummond, B.J.; Bowen, B.; Peterson, T. The Myb-Homologous P Gene Controls Phlobaphene Pigmentation in Maize Floral Organs by Directly Activating a Flavonoid Biosynthetic Gene Subset. Cell 1994, 76, 543–553. [Google Scholar] [CrossRef]
- Farfan, I.D.B.; De La Fuente, G.N.; Murray, S.C.; Isakeit, T.; Huang, P.C.; Warburton, M.; Williams, P.; Windham, G.L.; Kolomiets, M. Genome Wide Association Study for Drought, Aflatoxin Resistance, and Important Agronomic Traits of Maize Hybrids in the Sub-Tropics. PLoS ONE 2015, 10, e0117737. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Galindo, J.C.; Malvar, R.A.; Butrón, A.; Santiago, R.; Samayoa, L.F.; Caicedo, M.; Ordás, B. Mapping of Resistance to Corn Borers in a MAGIC Population of Maize. BMC Plant Biol. 2019, 19, 431. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yuan, Y.; Liao, Z.; Jiang, Y.; Wang, Q.; Zhang, L.; Gao, S.; Wu, F.; Li, M.; Xie, W.; et al. Genome-Wide Association Study of 13 Traits in Maize Seedlings under Low Phosphorus Stress. Plant Genome 2019, 12, 190039. [Google Scholar] [CrossRef]
- Salazar-Vidal, M.N.; Acosta-Segovia, E.; Sanchez-León, N.; Ahern, K.R.; Brutnell, T.P.; Sawers, R.J.H. Characterization and Transposon Mutagenesis of the Maize (Zea mays) Pho1 Gene Family. PLoS ONE 2016, 11, e0161882. [Google Scholar] [CrossRef]
- Li, D.; Wang, H.; Wang, M.; Li, G.; Chen, Z.; Leiser, W.L.; Weiß, T.M.; Lu, X.; Wang, M.; Chen, S.; et al. Genetic Dissection of Phosphorus Use Efficiency in a Maize Association Population under Two P Levels in the Field. Int. J. Mol. Sci. 2021, 22, 9311. [Google Scholar] [CrossRef]
- Miura, K.; Rus, A.; Sharkhuu, A.; Yokoi, S.; Karthikeyan, A.S.; Raghothama, K.G.; Baek, D.; Duck Koo, Y.; Bo Jin, J.; Bressan, R.A.; et al. The Arabidopsis SUMO E3 Ligase SIZ1 Controls Phosphate Deficiency Responses. Proc. Natl. Acad. Sci. USA 2005, 102, 7760–7765. [Google Scholar] [CrossRef]
- Li, Z.; Gao, Q.; Liu, Y.; He, C.; Zhang, X.; Zhang, J. Overexpression of Transcription Factor ZmPTF1 Improves Low Phosphate Tolerance of Maize by Regulating Carbon Metabolism and Root Growth. Planta 2011, 233, 1129–1143. [Google Scholar] [CrossRef]
- Szakiel, A.; Pączkowski, C.; Henry, M. Influence of Environmental Abiotic Factors on the Content of Saponins in Plants. Phytochem. Rev. 2011, 10, 471–491. [Google Scholar] [CrossRef]
- Bos, I.; Kleikamp, A. Reduction of Micro-Envrionmental Variation in a Selection Field of Rye. Euphytica 1985, 34, 1–6. [Google Scholar] [CrossRef]
- Xu, Y. Envirotyping for Deciphering Environmental Impacts on Crop Plants. Theor. Appl. Genet. 2016, 129, 653–673. [Google Scholar] [CrossRef] [PubMed]
- Moore, R.; Casale, F.P.; Jan Bonder, M.; Horta, D.; Heijmans, B.T.; Peter, P.A.; van Meurs, J.; Isaacs, A.; Jansen, R.; Franke, L.; et al. A Linear Mixed-Model Approach to Study Multivariate Gene–Environment Interactions. Nat. Genet. 2019, 51, 180–186. [Google Scholar] [CrossRef]
- Li, D.; Zhou, Z.; Lu, X.; Jiang, Y.; Li, G.; Li, J.; Wang, H.; Chen, S.; Li, X.; Würschum, T.; et al. Genetic Dissection of Hybrid Performance and Heterosis for Yield-Related Traits in Maize. Front. Plant Sci. 2021, 12, 774478. [Google Scholar] [CrossRef]
- Mei, W.; Stetter, M.G.; Gates, D.J.; Stitzer, M.C.; Ross-Ibarra, J. Adaptation in Plant Genomes: Bigger Is Different. Am. J. Bot. 2018, 105, 16–19. [Google Scholar] [CrossRef]
- Liu, J.; Jung, C.; Xu, J.; Wang, H.; Deng, S.; Bernad, L.; Arenas-Huertero, C.; Chua, N.H. Genome-Wide Analysis Uncovers Regulation of Long Intergenic Noncoding RNAs in Arabidopsis. Plant Cell 2012, 24, 4333–4345. [Google Scholar] [CrossRef] [PubMed]
- Olsen, S.R.; Cole, C.V.; Watanabe, F.S.; Daen, L.A. Estimation of Available Phosphorus in Soils by Extraction with Sodium Bicarbonate; Department of Agriculture: Washington, DC, USA, 1954. [Google Scholar]
- Li, D.; Chen, Z.; Wang, M.; Leiser, W.L.; Weiß, T.M.; Zhao, Z.; Cheng, S.; Chen, S.; Chen, F.; Yuan, L.; et al. Dissecting the Phenotypic Response of Maize to Low Phosphorus Soils by Field Screening of a Large Diversity Panel. Euphytica 2021, 217, 12. [Google Scholar] [CrossRef]
- Xu, C.; Zhang, H.W.; Sun, J.H.; Guo, Z.F.; Zou, C.; Li, W.-X.; Xie, C.X.; Huang, C.L.; Xu, R.N.; Liao, H.; et al. Genome-Wide Association Study Dissects Yield Components Associated with Low-Phosphorus Stress Tolerance in Maize. Theor. Appl. Genet. 2018, 131, 1699–1714. [Google Scholar] [CrossRef]
- Yang, N.; Lu, Y.; Yang, X.; Huang, J.; Zhou, Y.; Ali, F.; Wen, W.; Liu, J.; Li, J.; Yan, J. Genome Wide Association Studies Using a New Nonparametric Model Reveal the Genetic Architecture of 17 Agronomic Traits in an Enlarged Maize Association Panel. PLoS Genet. 2014, 10, e1004573. [Google Scholar] [CrossRef]
- Bernal-Vasquez, A.M.; Utz, H.F.; Piepho, H.P. Outlier Detection Methods for Generalized Lattices: A Case Study on the Transition from ANOVA to REML. Theor. Appl. Genet. 2016, 129, 787–804. [Google Scholar] [CrossRef]
- Cullis, B.R.; Smith, A.B.; Coombes, N.E. On the Design of Early Generation Variety Trials with Correlated Data. J. Agric. Biol. Environ. Stat. 2006, 11, 381–393. [Google Scholar] [CrossRef]
- Butler, D.G.; Cullis, B.R.; Gilmour, A.R.; Gogel, B.J.; Thompson, R. ASReml-R Reference Manual Version 4; VSN International Ltd.: Hemel Hempstead, UK, 2017. [Google Scholar]
- Doyle, J.J.; Doyle, J.L. A Rapid DNA Isolation Procedure for Small Quantities of Fresh Leaf Tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The Variant Call Format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for Association Mapping of Complex Traits in Diverse Samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Browning, B.L.; Zhou, Y.; Browning, S.R. A One-Penny Imputed Genome from Next-Generation Reference Panels. Am. J. Hum. Genet. 2018, 103, 338–348. [Google Scholar] [CrossRef]
- Huang, X.; Feng, Q.; Qian, Q.; Zhao, Q.; Wang, L.; Wang, A.; Guan, J.; Fan, D.; Weng, Q.; Huang, T.; et al. High-Throughput Genotyping by Whole-Genome Resequencing. Genome Res. 2009, 19, 1068–1076. [Google Scholar] [CrossRef]
- Xu, S. Genetic Mapping and Genomic Selection Using Recombination Breakpoint Data. Genetics 2013, 195, 1103–1115. [Google Scholar] [CrossRef]
- Xiao, Y.; Tong, H.; Yang, X.; Xu, S.; Pan, Q.; Qiao, F.; Raihan, M.S.; Luo, Y.; Liu, H.; Zhang, X.; et al. Genome-Wide Dissection of the Maize Ear Genetic Architecture Using Multiple Populations. New Phytol. 2016, 210, 1095–1106. [Google Scholar] [CrossRef]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.Y. Ggtree: An r Package for Visualization and Annotation of Phylogenetic Trees with Their Covariates and Other Associated Data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Zhang, C.; Dong, S.S.; Xu, J.Y.; He, W.M.; Yang, T.L. PopLDdecay: A Fast and Effective Tool for Linkage Disequilibrium Decay Analysis Based on Variant Call Format Files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef] [PubMed]
- Xu, S. Mapping Quantitative Trait Loci by Controlling Polygenic Background Effects. Genetics 2013, 195, 1209–1222. [Google Scholar] [CrossRef]
- Lian, L.; De Los Campos, G. FW: An R Package for Finlay-Wilkinson Regression That Incorporates Genomic/Pedigree Information and Covariance Structures between Environments. G3 Genes Genomes Genet. 2016, 6, 589–597. [Google Scholar] [CrossRef]
- Yu, J.; Pressoir, G.; Briggs, W.H.; Bi, I.V.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S.; Nielsen, D.M.; Holland, J.B.; et al. A Unified Mixed-Model Method for Association Mapping That Accounts for Multiple Levels of Relatedness. Nat. Genet. 2006, 38, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, Z. GAPIT Version 3: Boosting Power and Accuracy for Genomic Association and Prediction. Genom. Proteom. Bioinform. 2021, 19, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, E.; Matsunaga, H. Exploring Efficient Linear Mixed Models to Detect Quantitative Trait Locus-by-Environment Interactions. G3 Genes Genomes Genet. 2021, 11, jkab119. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Storey, J.D.; Tibshirani, R.; Green, P.P. Statistical Significance for Genomewide Studies. Proc. Natl. Acad. Sci. USA 2003, 100, 9440–9445. [Google Scholar] [CrossRef]
- Zheng, X.; Levine, D.; Shen, J.; Gogarten, S.M.; Laurie, C.; Weir, B.S. A High-Performance Computing Toolset for Relatedness and Principal Component Analysis of SNP Data. Bioinformatics 2012, 28, 3326–3328. [Google Scholar] [CrossRef]
- Lawrence, M.; Huber, W.; Pagès, H.; Aboyoun, P.; Carlson, M.; Gentleman, R.; Morgan, M.T.; Carey, V.J. Software for Computing and Annotating Genomic Ranges. PLoS Comput. Biol. 2013, 9, e1003118. [Google Scholar] [CrossRef] [PubMed]
- Tian, D.; Wang, P.; Tang, B.; Teng, X.; Li, C.; Liu, X.; Zou, D.; Song, S.; Zhang, Z. GWAS Atlas: A Curated Resource of Genome-Wide Variant-Trait Associations in Plants and Animals. Nucleic Acids Res. 2020, 48, D927–D932. [Google Scholar] [CrossRef] [PubMed]
- Durinck, S.; Moreau, Y.; Kasprzyk, A.; Davis, S.; De Moor, B.; Brazma, A.; Huber, W. BioMart and Bioconductor: A Powerful Link between Biological Databases and Microarray Data Analysis. Bioinformatics 2005, 21, 3439–3440. [Google Scholar] [CrossRef] [PubMed]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping Identifiers for the Integration of Genomic Datasets with the R/Bioconductor Package BiomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef] [PubMed]
- Pérez, P.; Campos, G. de los Genome-Wide Regression and Prediction with the BGLR Statistical Package. Genetics 2014, 198, 483–495. [Google Scholar] [CrossRef]
- Lopez-Cruz, M.; Crossa, J.; Bonnett, D.; Dreisigacker, S.; Poland, J.; Jannink, J.L.; Singh, R.P.; Autrique, E.; de los Campos, G. Increased Prediction Accuracy in Wheat Breeding Trials Using a Marker × Environment Interaction Genomic Selection Model. G3 Genes Genomes Genet. 2015, 5, 569–582. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Min | Max | Mean | Ratio | H2 | ||
|---|---|---|---|---|---|---|---|
| DTS | 63.83 | 78.67 | 69.99 | 7.58 ** | 1.61 ** | 0.21 | 0.91 |
| DTH | 59.45 | 73.98 | 65.09 | 6.68 ** | 1.32 ** | 0.20 | 0.92 |
| DTA | 62.12 | 76.51 | 67.76 | 6.54 ** | 1.51 ** | 0.23 | 0.91 |
| ASI | 0.19 | 6.56 | 2.14 | 1.13 ** | 0.45 ** | 0.40 | 0.82 |
| PH | 129.25 | 238.54 | 184.88 | 369.22 ** | 56.22 ** | 0.15 | 0.95 |
| EH | 30.84 | 103.68 | 65.35 | 145.78 ** | 24.58 ** | 0.17 | 0.95 |
| ELL | 55.54 | 84.90 | 69.85 | 29.78 ** | 6.42 ** | 0.22 | 0.93 |
| ELW | 5.75 | 9.39 | 7.72 | 0.51 ** | 0.11 ** | 0.21 | 0.93 |
| ELO | 5.01 | 7.65 | 6.29 | 0.24 ** | 0.05 ** | 0.21 | 0.93 |
| EL | 9.21 | 15.81 | 12.42 | 2.08 ** | 0.65 ** | 0.32 | 0.88 |
| ED | 30.37 | 43.55 | 36.92 | 7.31 ** | 3.08 ** | 0.42 | 0.85 |
| RNPE | 10.48 | 18.04 | 13.64 | 1.66 ** | 0.41 ** | 0.25 | 0.90 |
| KNPR | 12.13 | 30.13 | 21.14 | 11.17 ** | 5.13 ** | 0.46 | 0.85 |
| HGW | 16.99 | 34.22 | 24.75 | 9.35 ** | 2.52 ** | 0.27 | 0.90 |
| Yield | 1.46 | 8.15 | 4.61 | 1.78 ** | 0.75 ** | 0.42 | 0.83 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, D.; Li, G.; Wang, H.; Guo, Y.; Wang, M.; Lu, X.; Luo, Z.; Zhu, X.; Weiß, T.M.; Roller, S.; et al. Genetic Dissection of Phosphorus Use Efficiency and Genotype-by-Environment Interaction in Maize. Int. J. Mol. Sci. 2022, 23, 13943. https://doi.org/10.3390/ijms232213943
Li D, Li G, Wang H, Guo Y, Wang M, Lu X, Luo Z, Zhu X, Weiß TM, Roller S, et al. Genetic Dissection of Phosphorus Use Efficiency and Genotype-by-Environment Interaction in Maize. International Journal of Molecular Sciences. 2022; 23(22):13943. https://doi.org/10.3390/ijms232213943
Chicago/Turabian StyleLi, Dongdong, Guoliang Li, Haoying Wang, Yuhang Guo, Meng Wang, Xiaohuan Lu, Zhiheng Luo, Xintian Zhu, Thea Mi Weiß, Sandra Roller, and et al. 2022. "Genetic Dissection of Phosphorus Use Efficiency and Genotype-by-Environment Interaction in Maize" International Journal of Molecular Sciences 23, no. 22: 13943. https://doi.org/10.3390/ijms232213943
APA StyleLi, D., Li, G., Wang, H., Guo, Y., Wang, M., Lu, X., Luo, Z., Zhu, X., Weiß, T. M., Roller, S., Chen, S., Yuan, L., Würschum, T., & Liu, W. (2022). Genetic Dissection of Phosphorus Use Efficiency and Genotype-by-Environment Interaction in Maize. International Journal of Molecular Sciences, 23(22), 13943. https://doi.org/10.3390/ijms232213943