
_Putnam.png)
Synthesis of Radioiodinated Compounds. Classical Approaches and Achievements of Recent Years
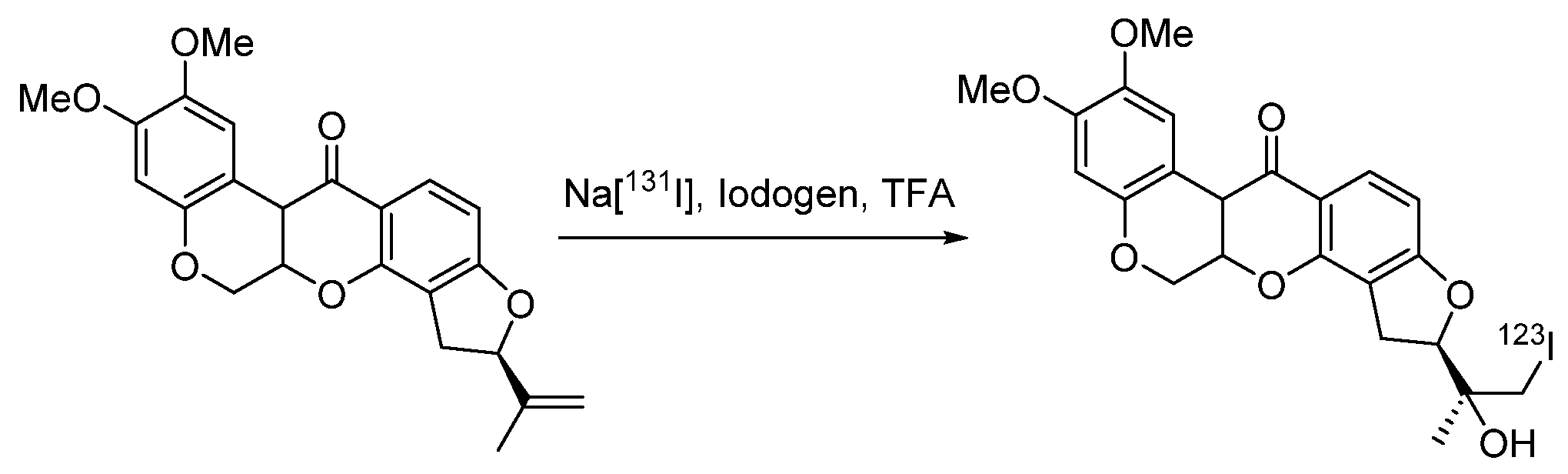
 , ,
, ,  and
and 
Abstract

1. Introduction
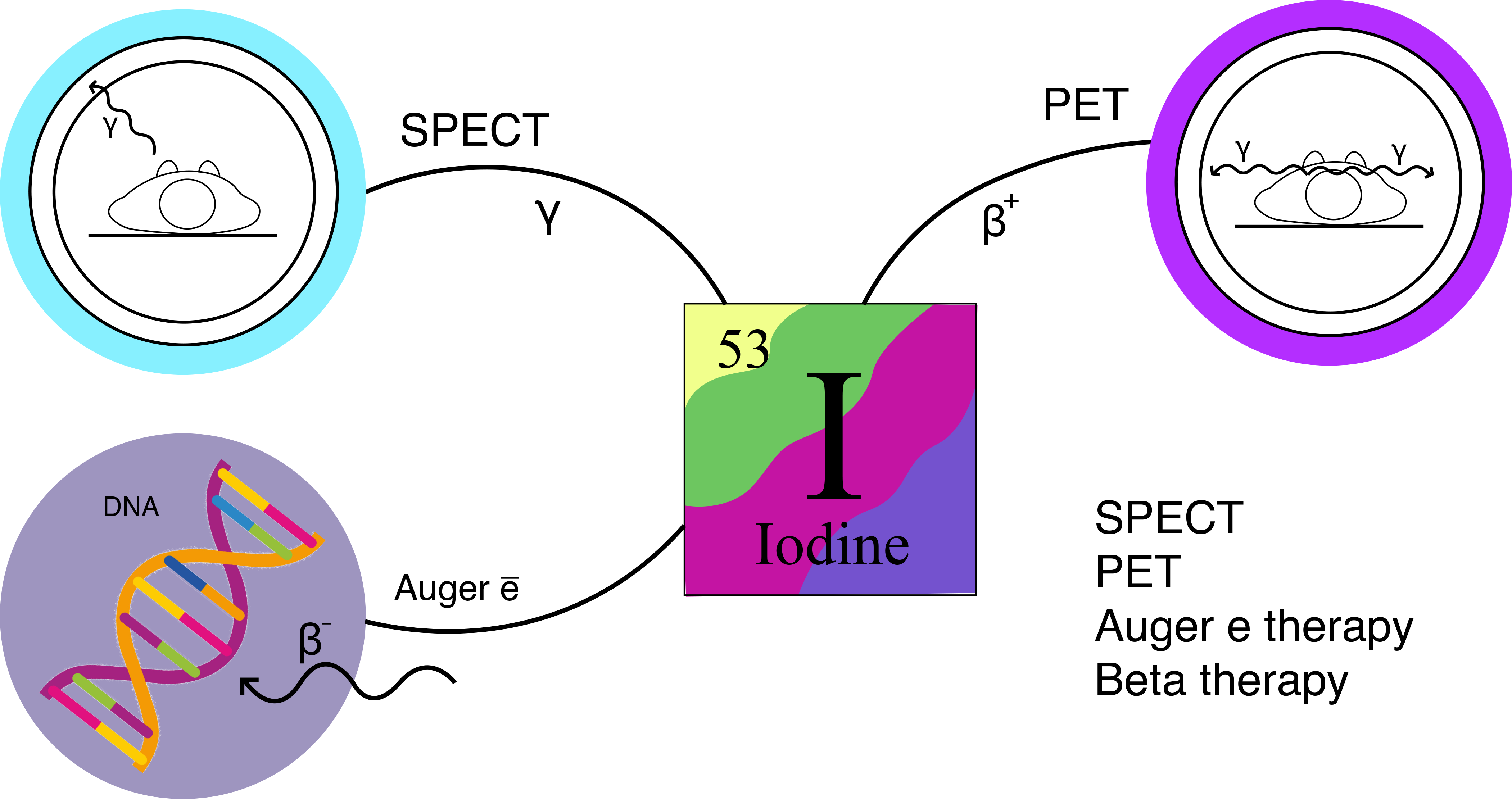
2. Iodine Isotopes Used in Radiopharmaceuticals
- iobenguane 131I, a form of 131I-MIBG, for the treatment of paragangliomas and pheochromocytomas
- 131I-labeled HSA for the determination of total blood and plasma volume, cardiac output, cardiac and pulmonary blood volumes and circulation times; for the study of protein metabolism and borders of the heart and large vessels; and for localization of the placenta and cerebral neoplasms
- [131I]NaI for diagnostics and therapeutic applications
3. Synthetic Methods for Incorporation of Radioactive Iodine into Molecules
3.1. The Problem of Iodine-Containing Drugs’ Stability and the Optimal Position of Radioactive Iodine in a Molecule
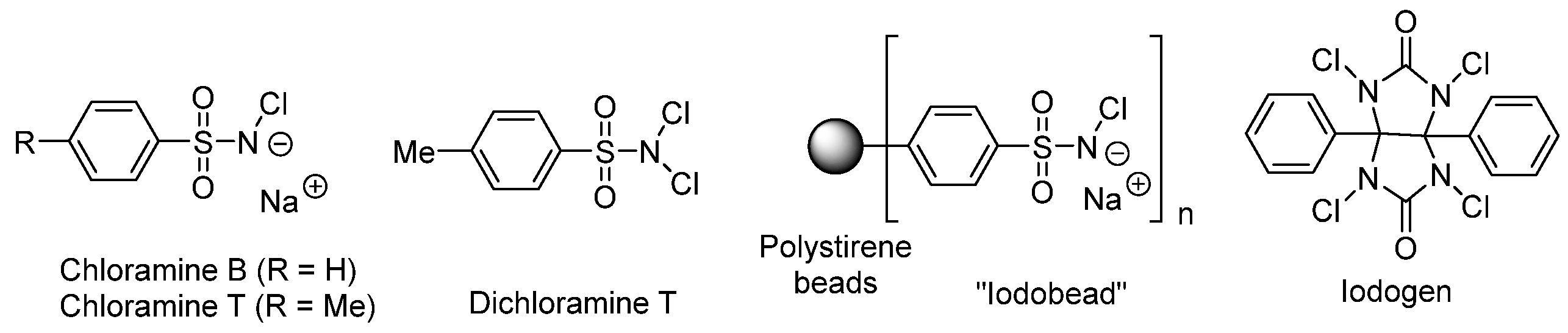
3.2. General Methods for Incorporation of Radioactive Iodine into a Molecule
3.2.1. Introduction of Radioactive Iodine into Small Molecules
Substrate Choice
Oxidizing Agent Choice
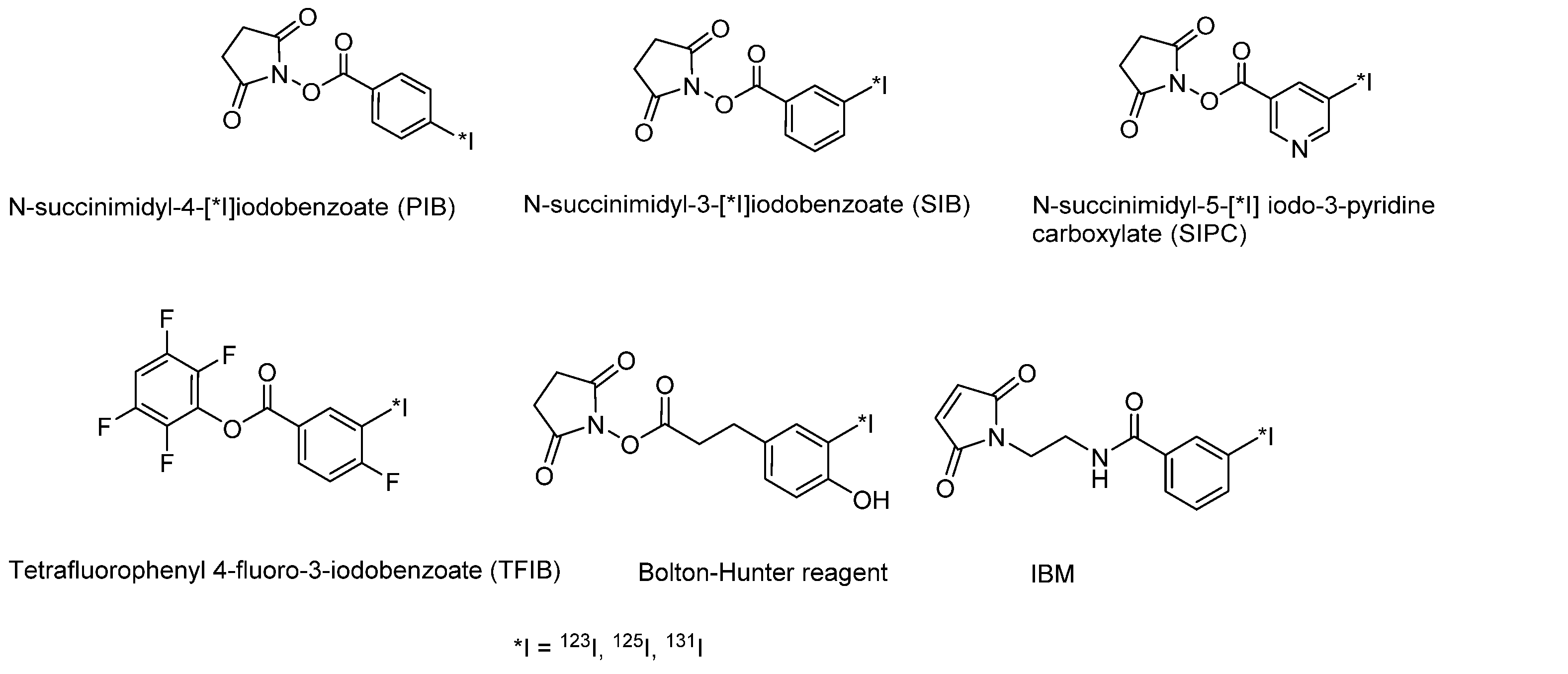
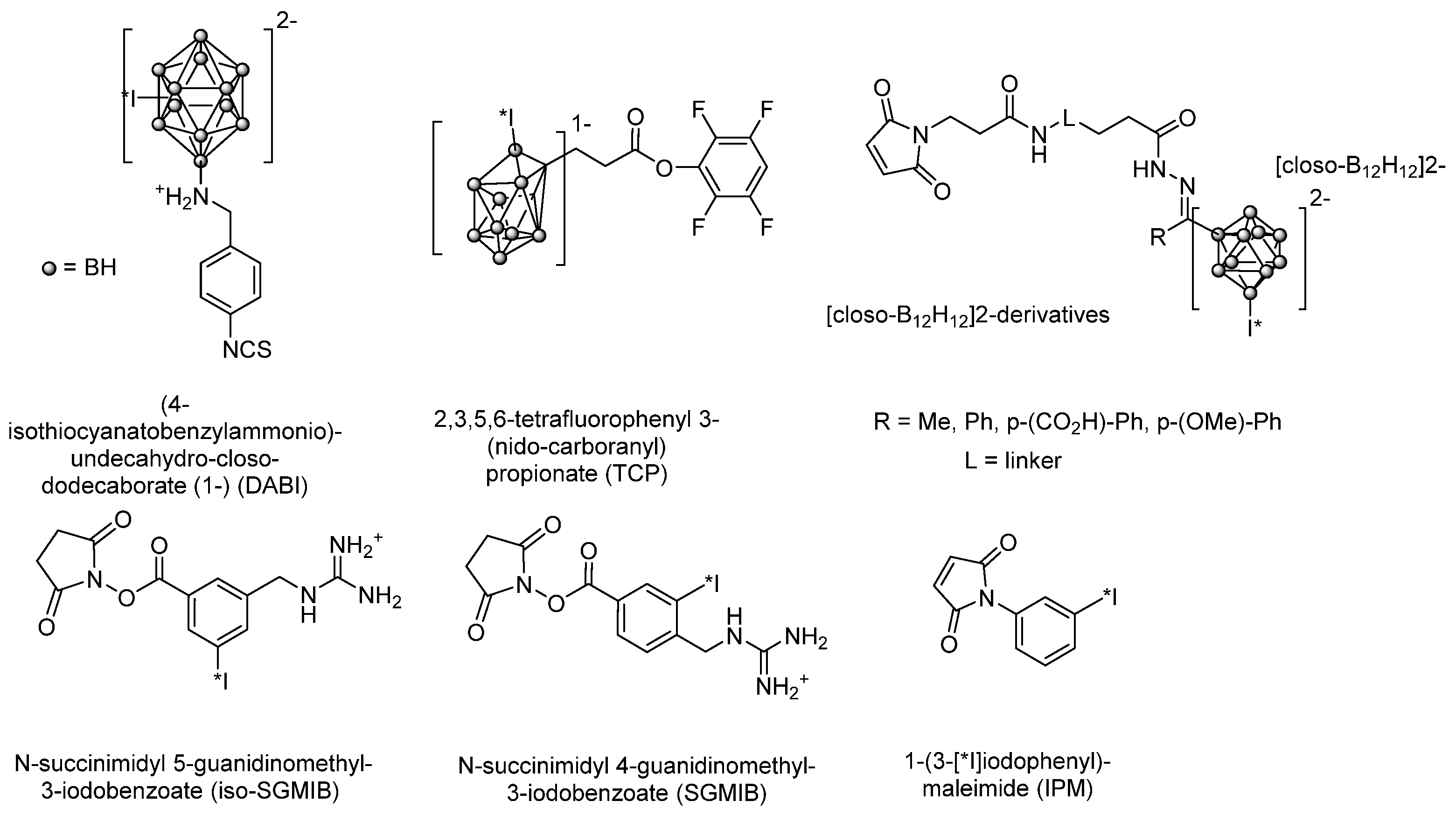
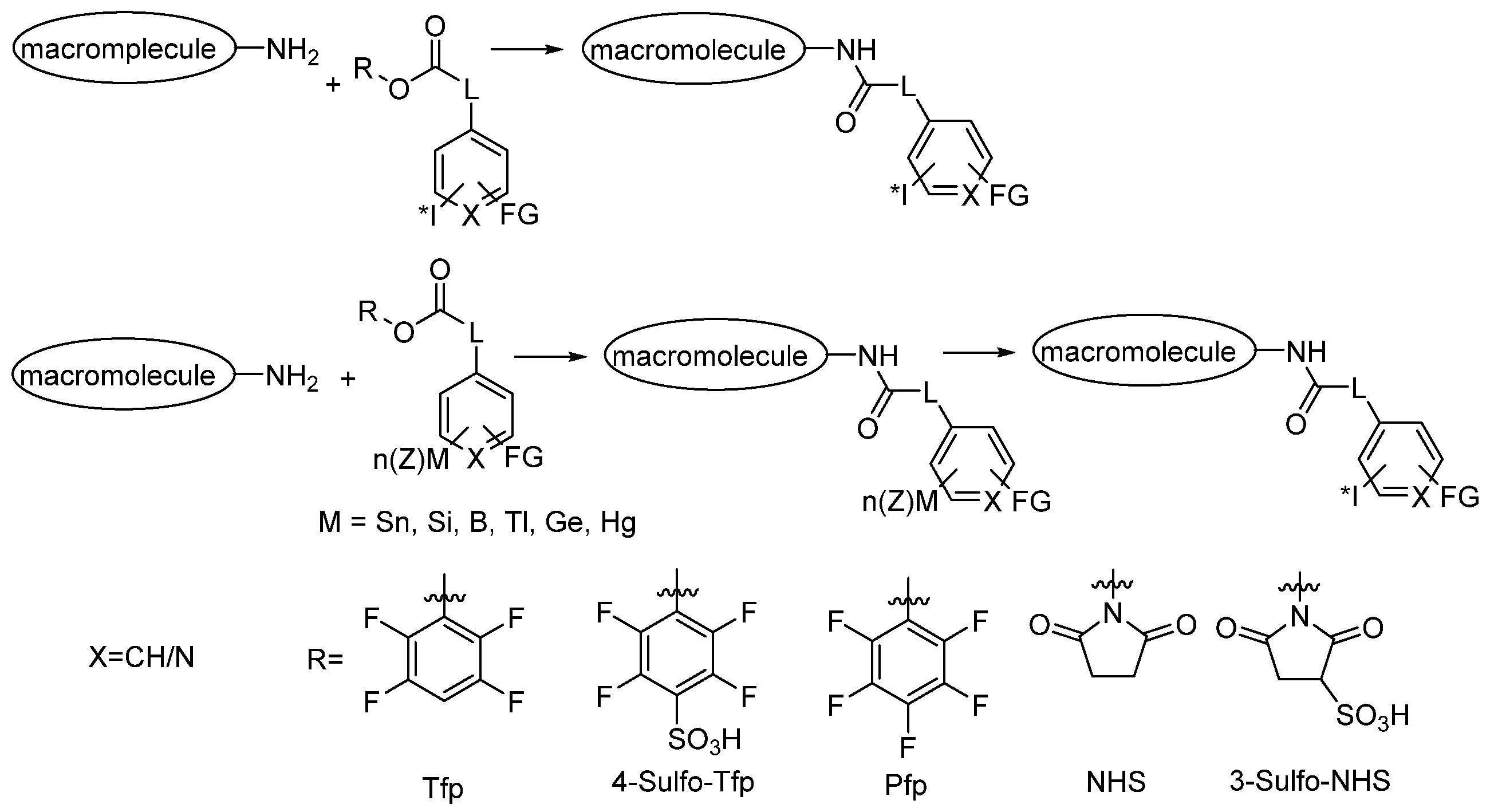
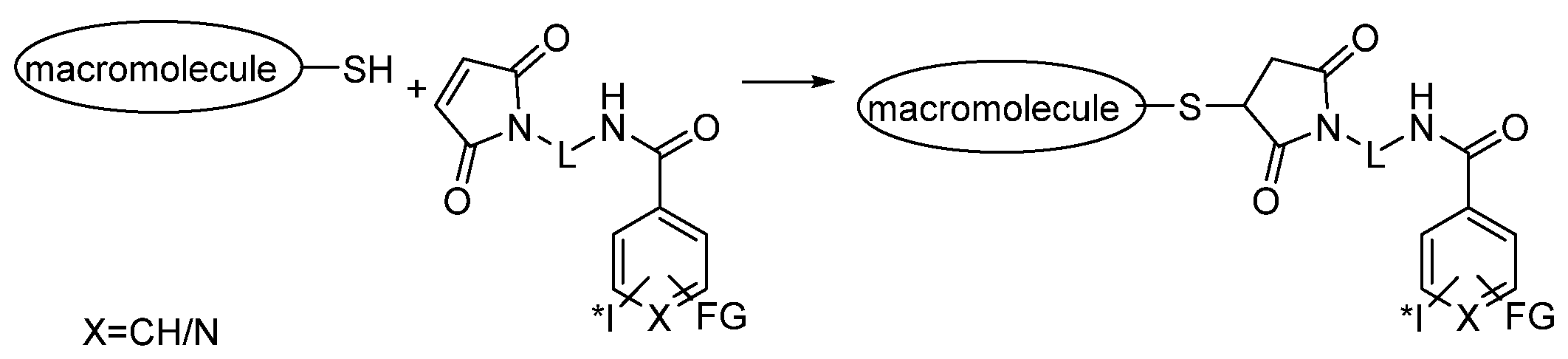
3.2.2. Radioiodination of Macromolecules Using Prosthetic Groups
Direct Radioiodination
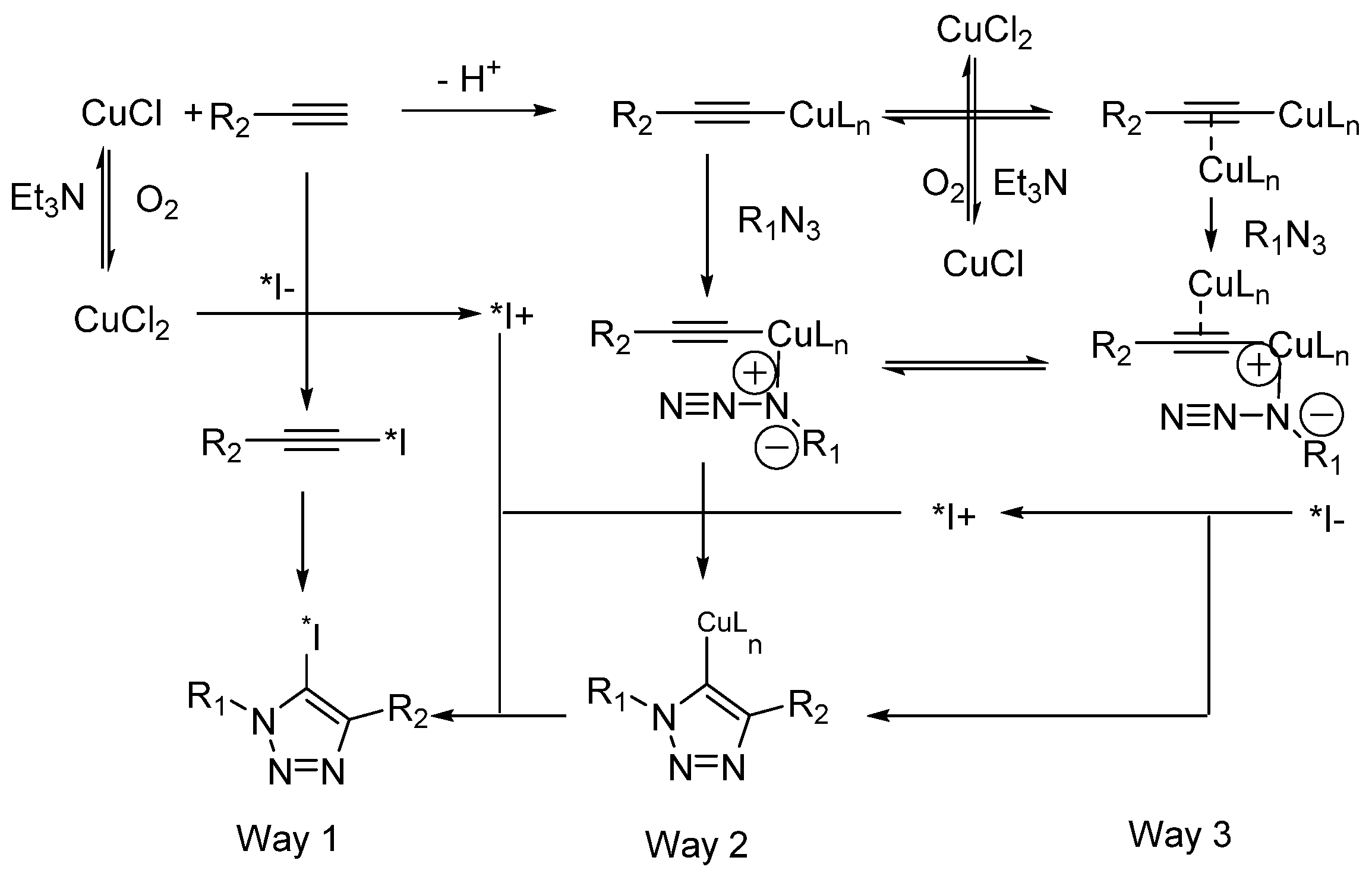
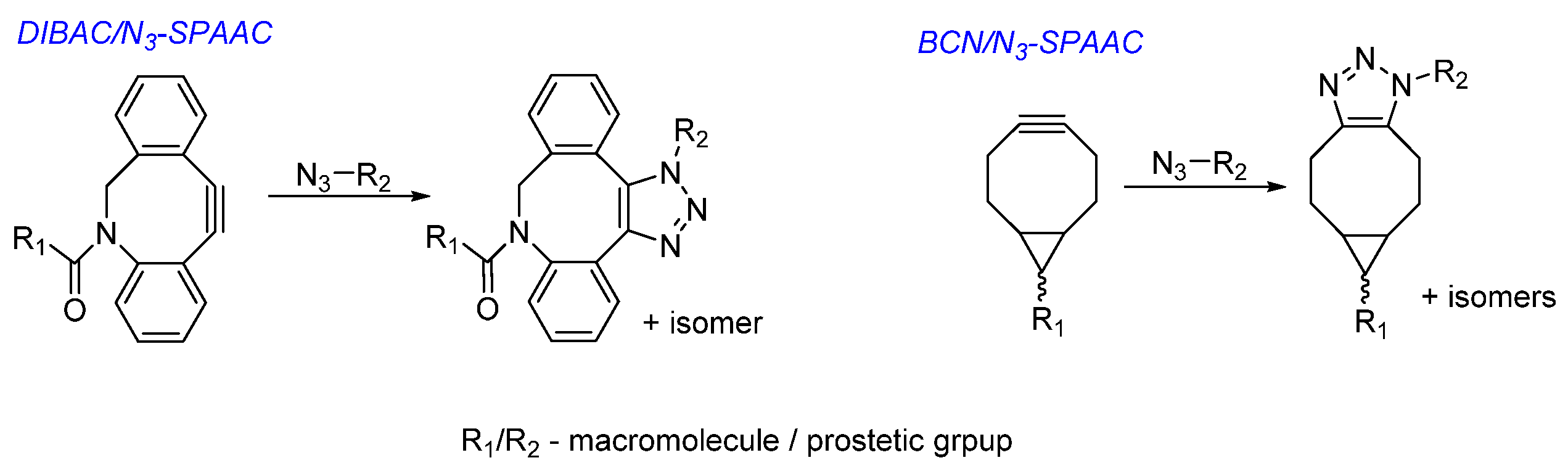
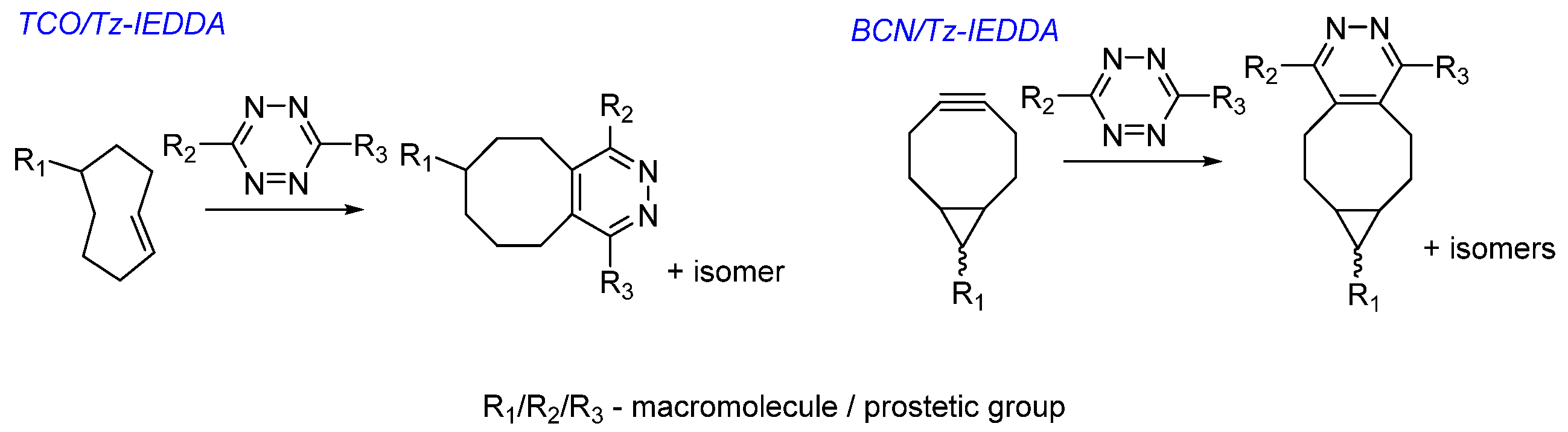
Choice of Bioconjugation Reaction
3.3. Synthesis of Radioiodinated Derivatives: Conclusion
4. Iodine-Containing Radiopharmaceuticals at the Stage of Clinical Trials
4.1. Low-Molecular-Weight Iodine-Containing Radiopharmaceuticals
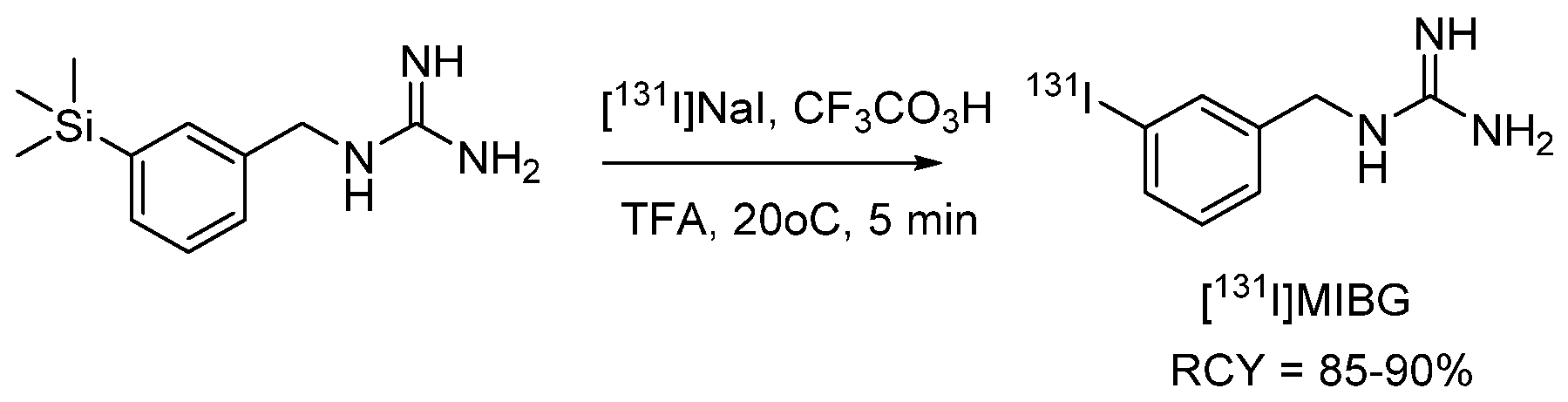
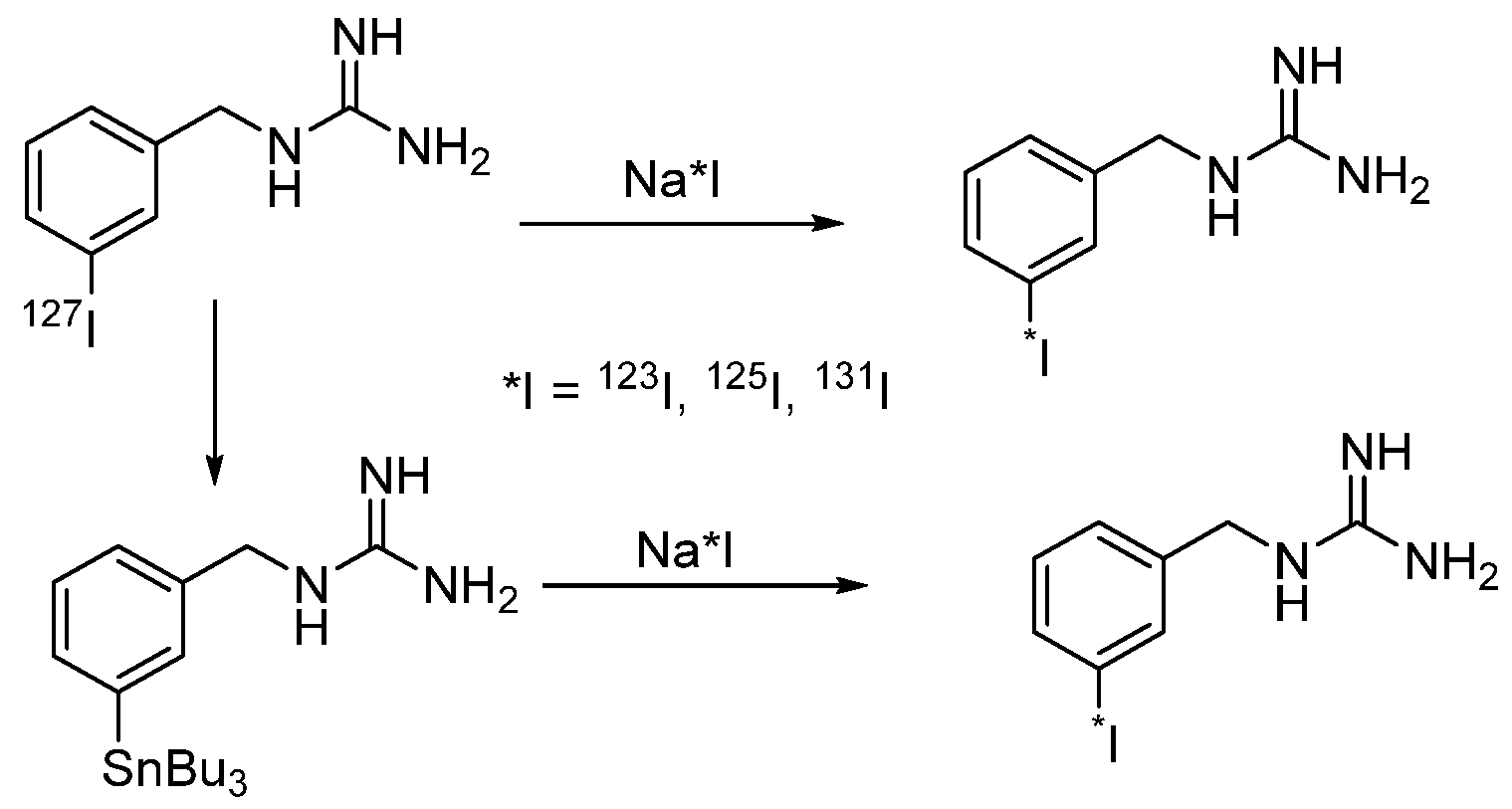
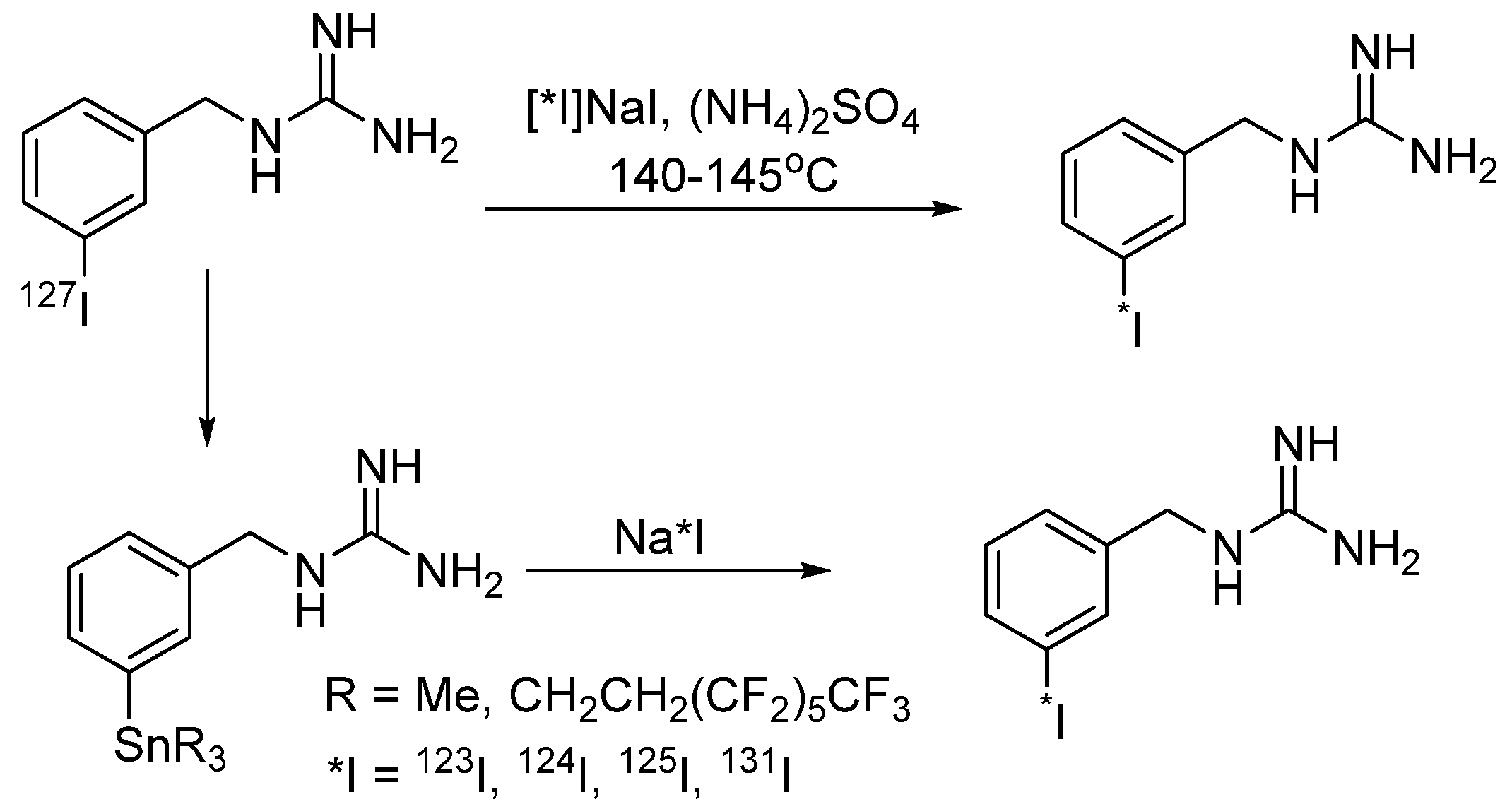
4.1.1. meta-Iodobenzylguanidine (MIBG)
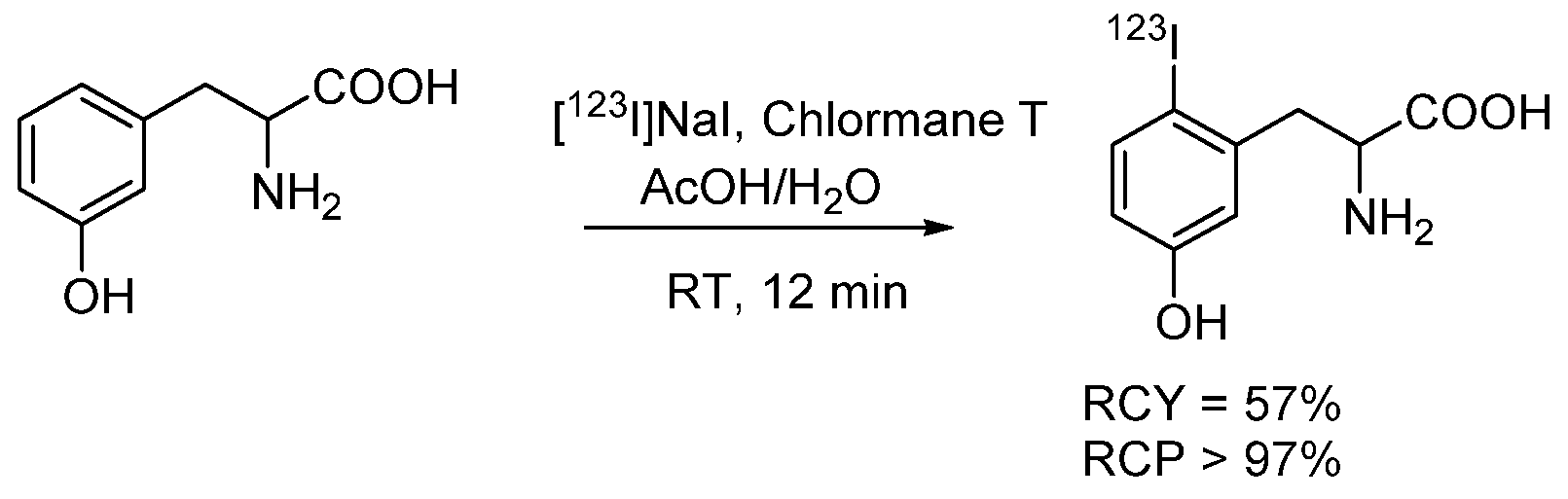
4.1.2. Radioiodinated Amino Acids
4.1.3. Radioiodinated Lipids
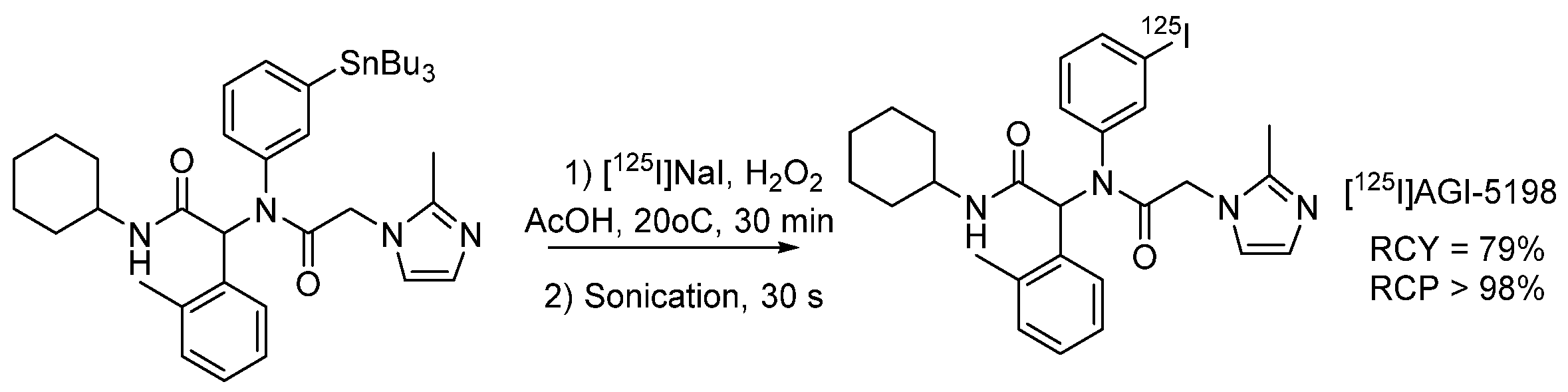
4.1.4. Radioiodinated Heterocyclic Compounds
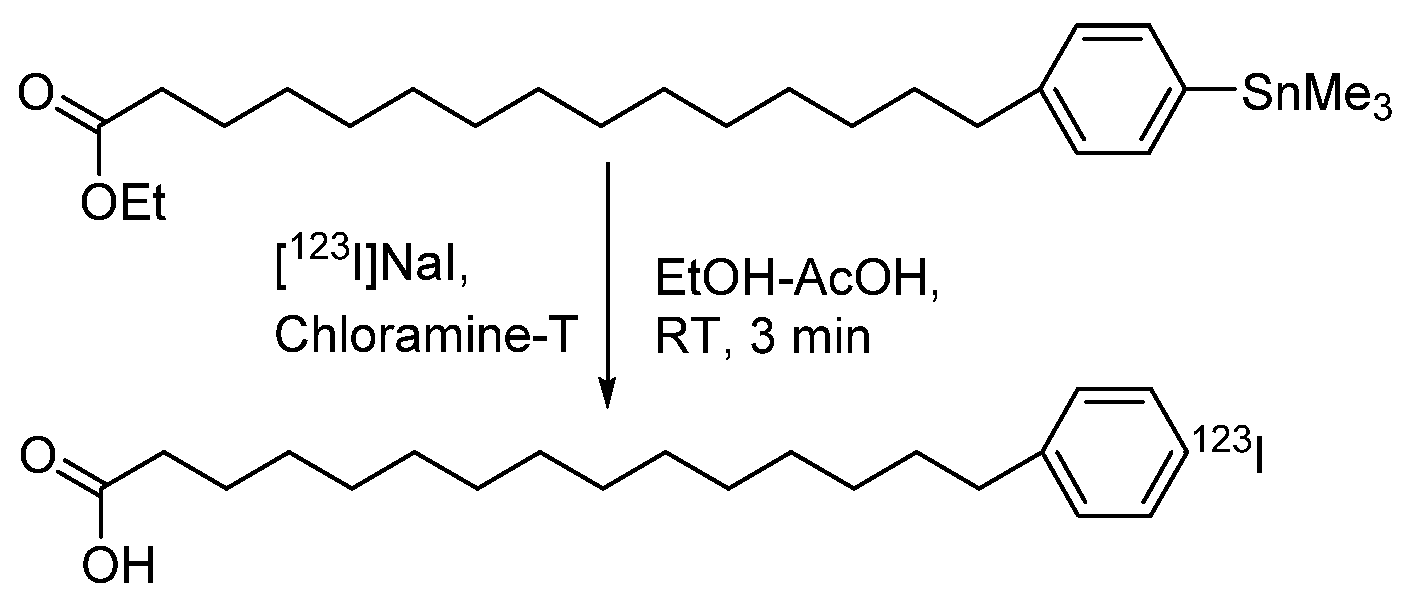
4.1.5. para-Iodophenylpentadecanoic Acid (IPPA) and β-Methyliodophenylpentadecanoic Acid (BMIPP)
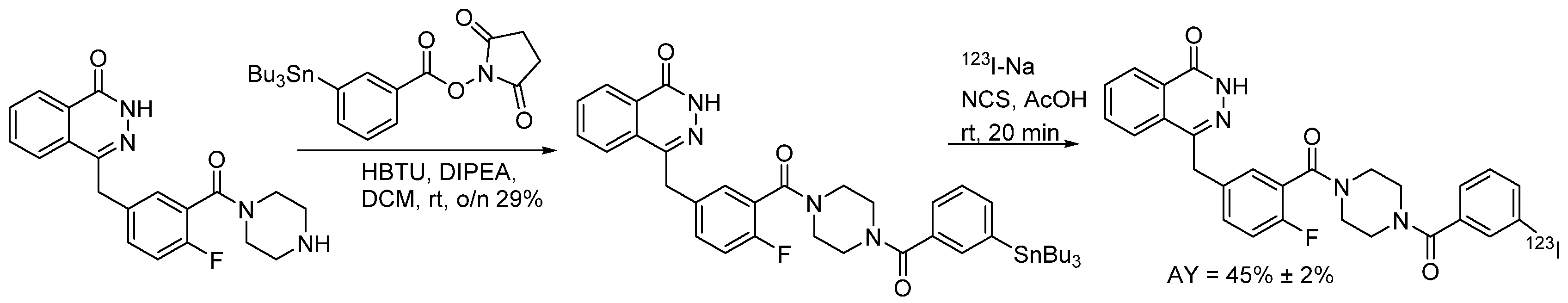
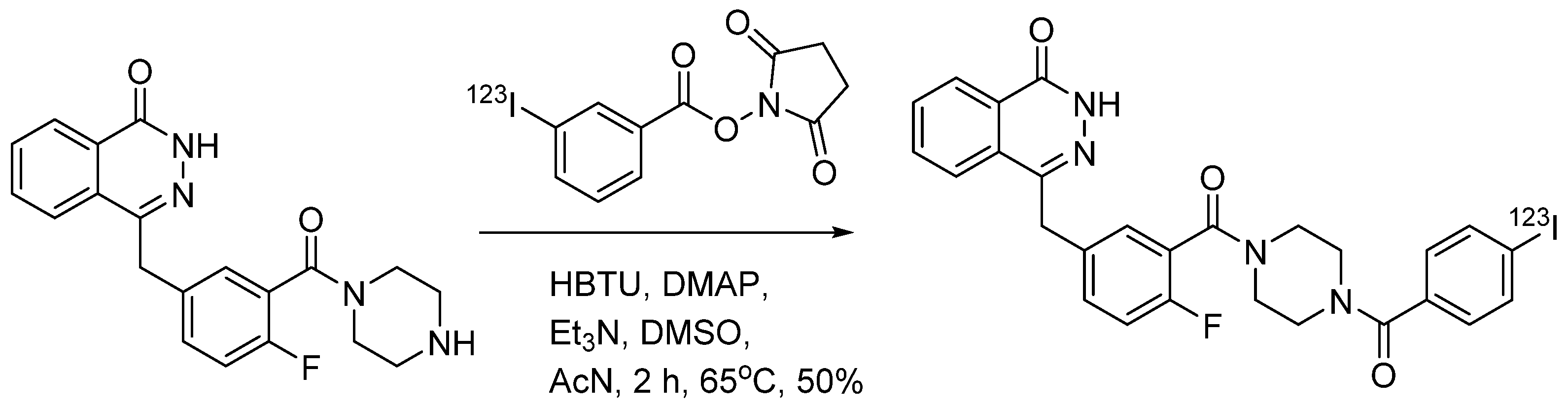
4.1.6. 4-(4-Fluoro-3-(4-(3-iodobenzoyl)piperazine-1-carbonyl)benzyl)phthalazin-1(2H)-one (MAPi)
4.1.7. 4-(6-Iodimidazo [1,2-a]pyridin-2-yl)-N,N-dimethylaniline (IMPY, TZDM)
4.1.8. Iodobenzovesamicol (IBVM)
4.1.9. 4-(2-(Bis(4-fluorophenyl)methoxy)ethyl)-1-(4-iodobenzyl)piperidine (β-CIT)
4.1.10. Ioflupan (FP-CIT)
4.1.11. (2R)-2-(2-Hydroxy-1-iodopropan-2-yl)-8,9-dimethoxy-1,2,12,12a-tetrahydrochromeno [3,4-b]fluoro [2,3-h] chromen-6(6aH)-one (CMICE-013)
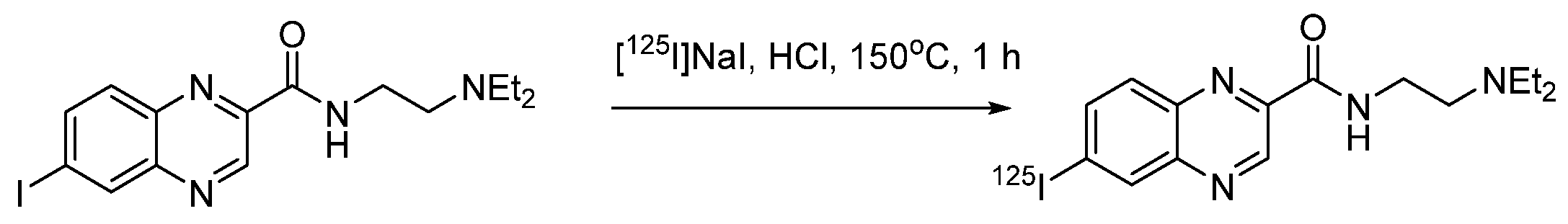
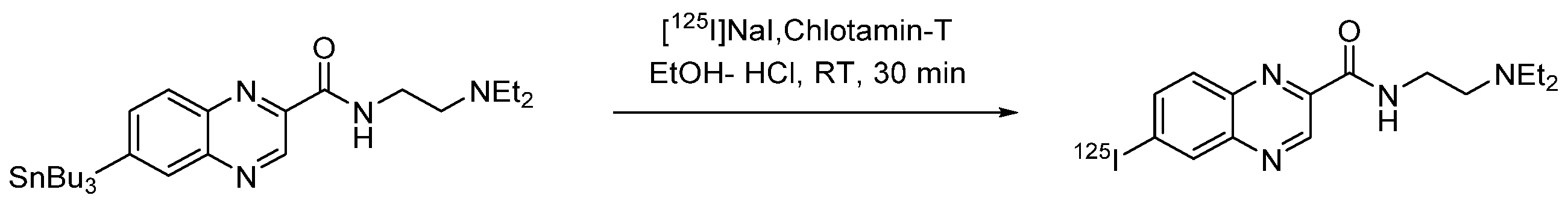
4.1.12. N-(2-(Diethylamino)ethyl)-6-iodoquinoxaline-2-carboxamide (ICF01012)
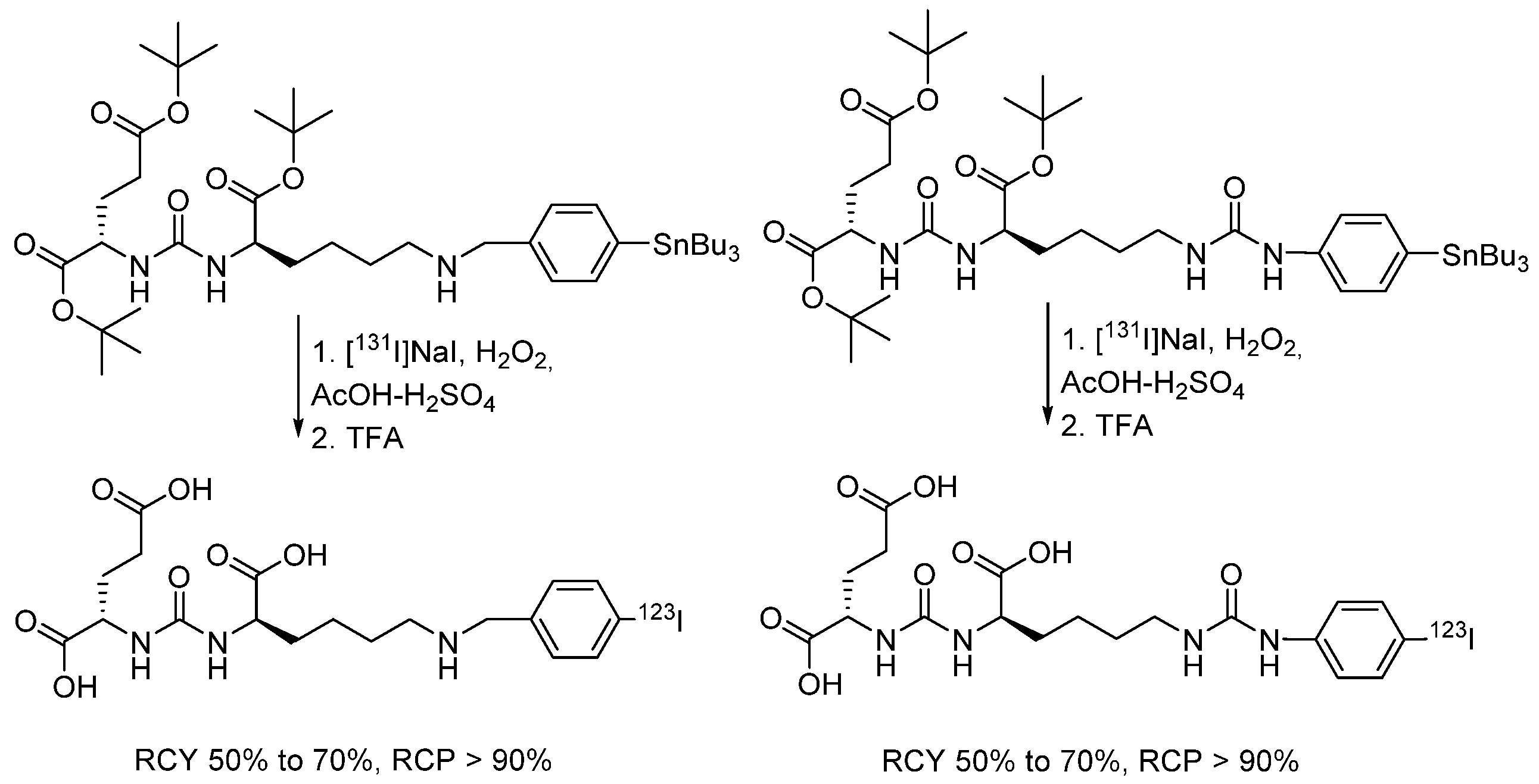
4.1.13. Radioiodinated Drugs Based on Peptide Conjugates
4.2. Radioiodinated Drugs Based on Macromolecular Compounds
5. Conclusions
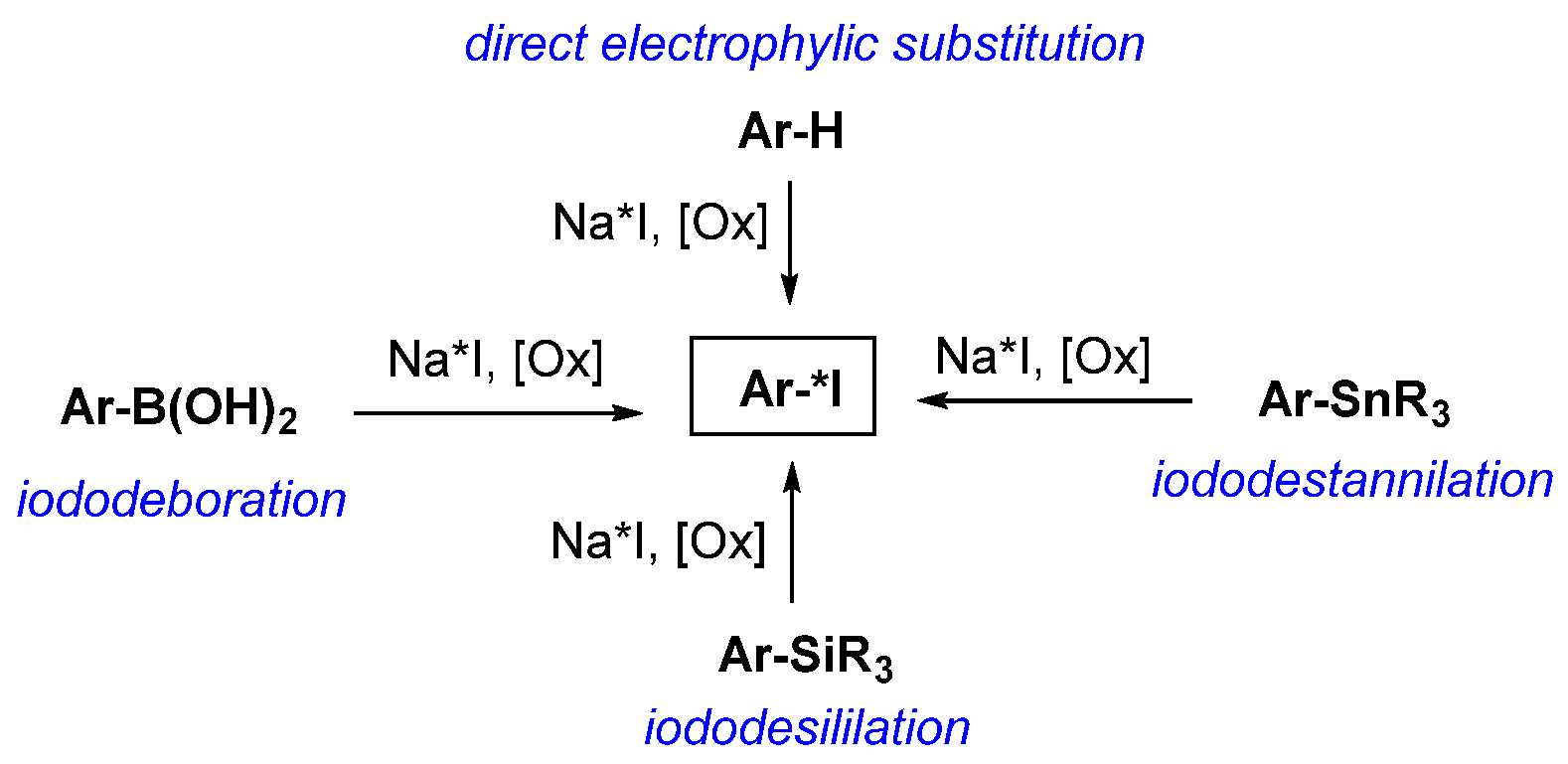
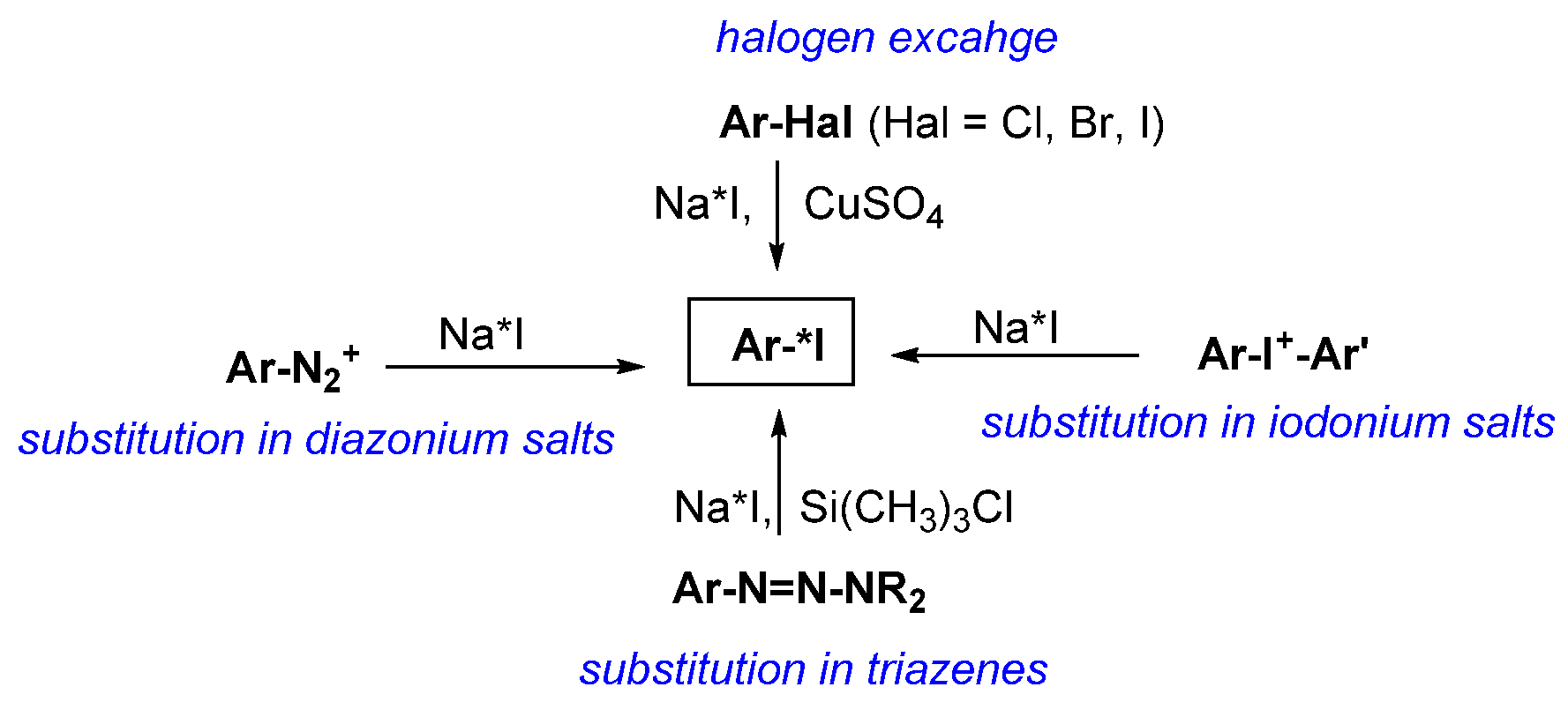
- Electrophilic and nucleophilic substitution reactions, especially the former, are the predominant synthetic approaches used in radioiodination.
- Iododestantnylation is the best and most widely used method for electrophilic radioiodination of low-molecular-weight compounds, provided that the product can be purified from the unreacted tin-containing precursor.
- Fixing tin precursors to insoluble resins or using ionic liquids may overcome the difficulties of separation from toxic tin derivatives.
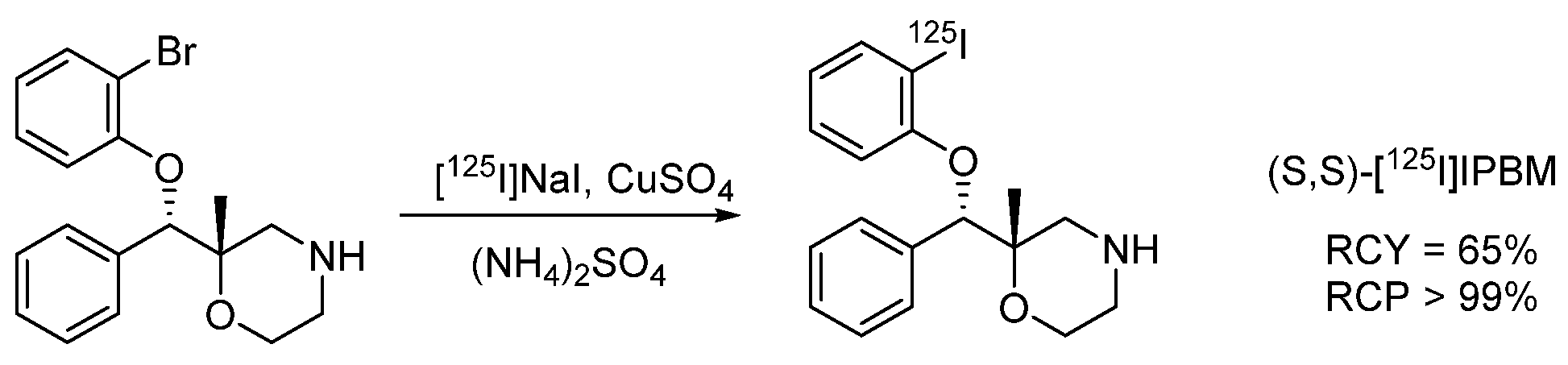
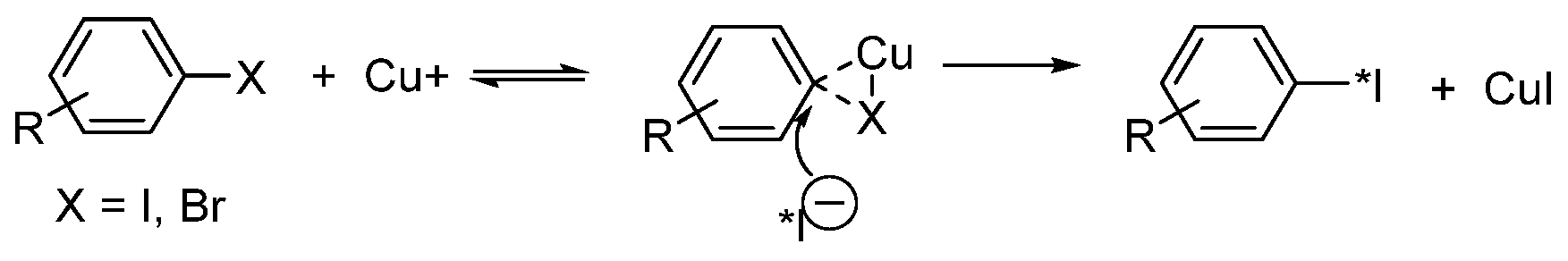
- Isotopic exchange of iodine-127 is the most commonly used method of nucleophilic radioiodination of low-molecular-weight compounds.
- Direct radioactive iodination of peptides and proteins is very easy, and it cannot be recommended as a method for the synthesis of radiopharmaceuticals, because the products are rapidly deiodinated in vivo.
- The problem of deiodination of proteins and peptides is generally solved by the use of prosthetic groups.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| A85380 | 5-Iodo-3-(2(S)-azetidinylmethoxy)pyridine |
| AGI-5198 | N-Cyclohexyl-2-(N-(3-iodophenyl)-2-(2-methyl-1H-imidazol-1- yl)acetamido)-2-(o-tolyl)acetamide |
| BMIPP | β-Methyliodophenylpentadecanoic acid |
| β-CIT | 2β-Carbomethoxy-3β-(4-iodophenyl)tropane |
| CLR-131 | Iopofosin I-131, 12-[4-(131I)iodophenyl]dodecylphosphocholine |
| CMICE-013 | (2R)-2-(2-Hydroxy-1-iodopropan-2-yl)-8,9-dimethoxy-1,2,12,12a- tetrahydrochromeno [3,4-b]fluoro [2,3-h] chromen-6(6aH)-one |
| CNS1261 | N-(1-Naphthyl)-N’-(3-iodophenyl)-N-methylguanidine |
| (1-)-DABI | (4-isothiocyanatobenzylammonio)-undecahydro-closo-dodecaborate |
| FDA | Food and Drug Administration (Food and Drug Administration, USA) |
| FP-CIT | Ioflupane, N-ω-fluoropropyl-2β-carbomethoxy-3β-(4- iodophenyl)northropan |
| HAS | Human serum albumin |
| IAZA | 1-(5-Iodo-5-deoxy-β-D-arabinofuranosyl)-2-nitroimidazole |
| IBOX | 2-(4’-Dimethylaminophenyl)-6-iodobenzoxazole |
| IBM | N-(2-Aminoethyl)maleimide) |
| IBVM | Iodobenzovemicol |
| ICF01012 | N-(2-(Diethylamino)ethyl)-6-iodoquinoxaline-2-carboxyamide () |
| IMPY, TZDM | 4-(6-Iodimidazo [1,2-a]pyridin-2-yl)-N,N-dimethylaniline |
| IMTO | Iodomethomidate |
| IMT | L-3-Iodine-α-methyltyrosine |
| IPA | para-I-Iodo-L-Phenylalanine |
| IPBM | 2-((2-Iodophenyloxy)(phenyl)methyl)-2-methylmorpholine |
| IPM | 1-(3-Iodophenyl)maleimide |
| IPPA | para-Iodophenylpentadecanoic acid |
| mAb | Monoclonal antibodies |
| MAPi | 4-(4-Fluoro-3-(4-(3-iodobenzoyl)piperazine-1-carbonyl)benzyl) phthalazin-1(2H)-one |
| MIBG, MIBG | meta-Iodobenzylguanidine |
| MIP-1072 | (S)-2-(3-((S)-1-carboxy-5-(4-iodobenzylamino)pentyl)ureido) pentanedioic acid |
| MNI-187 | 4-(5-Iodo-2H-benzo[d][1,2,3]triazol-2-yl)-N,N-dimethylaniline |
| MZINT | 2β-Carbomethoxy-3β-(4′-((Z)-2-iodoethenyl)phenyl)northropan |
| NBS | N-Bromosuccinimide |
| NCS | N-Chlorosuccinimide |
| NCTFS | N-Chloro-tetra-fluorosuccinimide |
| PET | Positron emission tomography |
| PIB | N-Succinimidyl-4-iodobenzoate |
| RCC | Radiochemical Conversion |
| RCP | Radiochemical purity |
| RCY | Radiochemical yield |
| SIB | N-Succinimidyl-3-iodobenzoate |
| SIPC | N-Succinimidyl-5-iodine-3-pyridinecarboxylate |
| SPECT | Single photon emission tomography |
| TCP | 2,3,5,6-Tetrafluorophenyl-3-(nidocarboranyl)propionate |
| TFIB | Tetrafluorophenyl-4-fluoro-3-iodobenzoate. |
References
- Weissleder, R. Molecular Imaging in Cancer. Science 2006, 312, 1168–1171. [Google Scholar] [CrossRef] [PubMed]
- Weissleder, R.; Mahmood, U. Molecular Imaging. Radiology 2001, 219, 316–333. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, A. Radiohalogenation of Organic Compounds: Practical Considerations and Challenges for Molecular Imaging. Synthesis 2019, 51, 4368–4373. [Google Scholar] [CrossRef]
- Dubost, E.; McErlain, H.; Babin, V.; Sutherland, A.; Cailly, T. Recent Advances in Synthetic Methods for Radioiodination. J. Org. Chem. 2020, 85, 8300–8310. [Google Scholar] [CrossRef]
- Tolmachev, V.; Orlova, A.; Lundqvist, H. Approaches to Improve Cellular Retention of Radiohalogen Labels Delivered by Internalising Tumour-Targeting Proteins and Peptides. Curr. Med. Chem. 2003, 10, 2447–2460. [Google Scholar] [CrossRef] [PubMed]
- Coenen, H.H.; Mertens, J.; Mazieère, B.; Bläuenstein, P.; Emond, P.; Frangin, Y.; Guilloteau, D.; Gysemans, M.; Holschbach, M.; Loc’H, C.; et al. Radioionidation Reactions for Radio Pharmaceuticals; Coenen, H.H., Mertens, J., Mazieère, B., Eds.; Springer: Dordrecht, The Netherlands, 2006; Volume 128, ISBN 978-1-4020-4560-8. [Google Scholar]
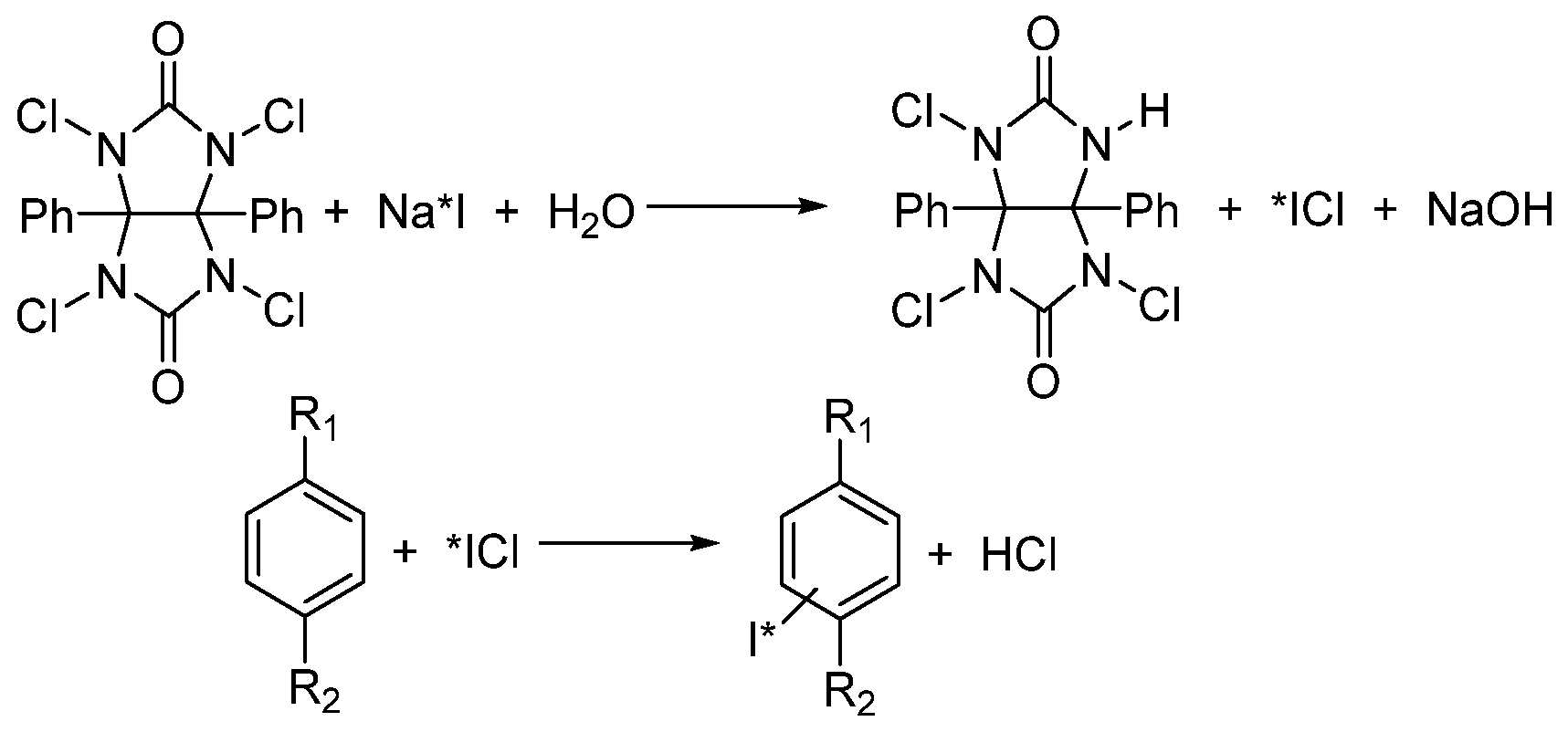
- Mertens, J.; Gysemans, M.; Bossuyt-Piron, C.; Thomas, M. High yield preparation of pure 2-radioiodo-ketanserin of high specific activity, a serotonin S2 receptor tracer for spect. J. Label. Compd. Radiopharm. 1990, 28, 731–738. [Google Scholar] [CrossRef]
- Mushtaq, S.; Jeon, J.; Shaheen, A.; Jang, B.S.; Park, S.H. Critical analysis of radioiodination techniques for micro and macro organic molecules. J. Radioanal. Nucl. Chem. 2016, 309, 859–889. [Google Scholar] [CrossRef]
- Cavina, L.; van der Born, D.; Klaren, P.H.M.; Feiters, M.C.; Boerman, O.C.; Rutjes, F.P.J.T. Design of Radioiodinated Pharmaceuticals: Structural Features Affecting Metabolic Stability towards in Vivo Deiodination. Eur. J. Org. Chem. 2017, 2017, 3387–3414. [Google Scholar] [CrossRef]
- Adam, M.J.; Wilbur, D.S. Radiohalogens for imaging and therapy. Chem. Soc. Rev. 2005, 34, 153. [Google Scholar] [CrossRef]
- Seevers, R.H.; Counsell, R.E. Radioiodination techniques for small organic molecules. Chem. Rev. 1982, 82, 575–590. [Google Scholar] [CrossRef]
- Audi, G.; Bersillon, O.; Blachot, J.; Wapstra, A.H. The Nubase evaluation of nuclear and decay properties. Nucl. Phys. A 2003, 729, 3–128. [Google Scholar] [CrossRef]
- Goldsmith, S.J. Targeted Radionuclide Therapy: A Historical and Personal Review. Semin. Nucl. Med. 2020, 50, 87–97. [Google Scholar] [CrossRef] [PubMed]
- El-Azony, K.M.; El-Mohty, A.A.; Killa, H.M.; Seddik, U.; Khater, S.I. An investigation of the 125I-radioiodination of colchicine for medical purposes. J. Label. Compd. Radiopharm. 2009, 52, 1–5. [Google Scholar] [CrossRef]
- Akanji, A.G.; Muramoto, E.; Caldeira Filho, J.d.S.; Couto, R.M.; de Araújo, E.B. Radiolabeling and biodistribution of monoclonal antibody (MAb) anti-CD20 with iodine-131. Braz. Arch. Biol. Technol. 2005, 48, 69–72. [Google Scholar] [CrossRef][Green Version]
- Daruwati, I.; Hanafiah, A.; Retnoningrum, D.S.; Rachmawati, H. Labeling of the Recombinant Streptokinase Using Iodine-131 as a New Thrombolytic Agent. At. Indones. 2013, 38, 106. [Google Scholar] [CrossRef]
- Kleinova, V.; Chaloupkova, H.; Svecova, H.; Fiser, M. Radioiodination and biodistribution of the monoclonal antibody TU-20 and its scFv fragment. J. Radioanal. Nucl. Chem. 2010, 286, 847–851. [Google Scholar] [CrossRef]
- Robinson, M.K.; Doss, M.; Shaller, C.; Narayanan, D.; Marks, J.D.; Adler, L.P.; González Trotter, D.E.; Adams, G.P. Quantitative Immuno-Positron Emission Tomography Imaging of HER2-Positive Tumor Xenografts with an Iodine-124 Labeled Anti-HER2 Diabody. Cancer Res. 2005, 65, 1471–1478. [Google Scholar] [CrossRef]
- Cherry, S.R.; Dahlbom, M. PET: Physics, Instrumentation, and Scanners. In PET; Springer: New York, NY, USA, 2006; pp. 1–117. ISBN 978-0-387-34946-6. [Google Scholar]
- Madsen, M.T. Recent Advances in SPECT Imaging. J. Nucl. Med. 2007, 48, 661–673. [Google Scholar] [CrossRef]
- Vassaux, G.; Groot-Wassink, T. In Vivo Noninvasive Imaging for Gene Therapy. J. Biomed. Biotechnol. 2003, 2003, 92–101. [Google Scholar] [CrossRef]
- Arbizu, J.; Luquin, M.R.; Abella, J.; de la Fuente-Fernández, R.; Fernandez-Torrón, R.; García-Solís, D.; Garrastachu, P.; Jiménez-Hoyuela, J.M.; Llaneza, M.; Lomeña, F.; et al. Functional neuroimaging in the diagnosis of patients with parkinsonism: Update and recommendations for clinical use. Rev. Española Med. Nucl. e Imagen Mol. 2014, 33, 215–226. [Google Scholar] [CrossRef]
- Silberstein, E.B.; Alavi, A.; Balon, H.R.; Clarke, S.E.M.; Divgi, C.; Gelfand, M.J.; Goldsmith, S.J.; Jadvar, H.; Marcus, C.S.; Martin, W.H.; et al. The SNMMI Practice Guideline for Therapy of Thyroid Disease with 131I 3.0. J. Nucl. Med. 2012, 53, 1633–1651. [Google Scholar] [CrossRef]
- Pacini, F.; Schlumberger, M.; Dralle, H.; Elisei, R.; Smit, J.W.A.; Wiersinga, W. European consensus for the management of patients with differentiated thyroid carcinoma of the follicular epithelium. Eur. J. Endocrinol. 2006, 154, 787–803. [Google Scholar] [CrossRef] [PubMed]
- Stöcklin, G. Bromine-77 and iodine-123 radiopharmaceuticals. Int. J. Appl. Radiat. Isot. 1977, 28, 131–148. [Google Scholar] [CrossRef]
- Kumar, K.; Ghosh, A. Radiochemistry, Production Processes, Labeling Methods, and ImmunoPET Imaging Pharmaceuticals of Iodine-124. Molecules 2021, 26, 414. [Google Scholar] [CrossRef]
- Adelstein, S.J.; Kassis, A.I.; Bodei, L.; Mariani, G. Radiotoxicity of Iodine-125 and Other Auger-Electron-Emitting Radionuclides: Background to Therapy. Cancer Biother. Radiopharm. 2003, 18, 301–316. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, M.C.; Correia, J.D.G. Biomedical applications of radioiodinated peptides. Eur. J. Med. Chem. 2019, 179, 56–77. [Google Scholar] [CrossRef] [PubMed]
- Darcourt, J.; Booij, J.; Tatsch, K.; Varrone, A.; Vander Borght, T.; Kapucu, O.L.; Någren, K.; Nobili, F.; Walker, Z.; Van Laere, K. EANM procedure guidelines for brain neurotransmission SPECT using (123)I-labelled dopamine transporter ligands, version 2. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 443–450. [Google Scholar] [CrossRef]
- Djang, D.S.W.; Janssen, M.J.R.; Bohnen, N.; Booij, J.; Henderson, T.A.; Herholz, K.; Minoshima, S.; Rowe, C.C.; Sabri, O.; Seibyl, J.; et al. SNM Practice Guideline for Dopamine Transporter Imaging with 123I-Ioflupane SPECT 1.0. J. Nucl. Med. 2012, 53, 154–163. [Google Scholar] [CrossRef]
- Kostelnik, T.I.; Orvig, C. Radioactive Main Group and Rare Earth Metals for Imaging and Therapy. Chem. Rev. 2019, 119, 902–956. [Google Scholar] [CrossRef]
- Saha, G.B. Fundamentals of Nuclear Pharmacy; Springer International Publishing: Cham, Switzerland, 2018; ISBN 978-3-319-57579-7. [Google Scholar]
- Synowiecki, M.A.; Perk, L.R.; Nijsen, J.F.W. Production of novel diagnostic radionuclides in small medical cyclotrons. EJNMMI Radiopharm. Chem. 2018, 3, 3. [Google Scholar] [CrossRef]
- Starovoitova, V.N.; Tchelidze, L.; Wells, D.P. Production of medical radioisotopes with linear accelerators. Appl. Radiat. Isot. 2014, 85, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Coenen, H.H.; Gee, A.D.; Adam, M.; Antoni, G.; Cutler, C.S.; Fujibayashi, Y.; Jeong, J.M.; Mach, R.H.; Mindt, T.L.; Pike, V.W.; et al. Consensus nomenclature rules for radiopharmaceutical chemistry—Setting the record straight. Nucl. Med. Biol. 2017, 55, v–xi. [Google Scholar] [CrossRef] [PubMed]
- Pimlott, S.L.; Sutherland, A. Molecular tracers for the PET and SPECT imaging of disease. Chem. Soc. Rev. 2011, 40, 149–162. [Google Scholar] [CrossRef] [PubMed]
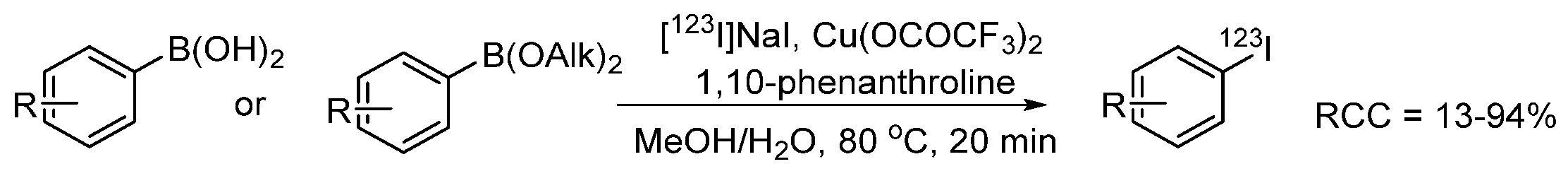
- Webster, S.; O’Rourke, K.M.; Fletcher, C.; Pimlott, S.L.; Sutherland, A.; Lee, A.L. Rapid Iododeboronation with and without Gold Catalysis: Application to Radiolabelling of Arenes. Chem. A Eur. J. 2018, 24, 937–943. [Google Scholar] [CrossRef]
- Kim, G.; Weiss, S.J.; Levine, R.L. Methionine oxidation and reduction in proteins. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 901–905. [Google Scholar] [CrossRef] [PubMed]
- Cant, A.A.; Champion, S.; Bhalla, R.; Pimlott, S.L.; Sutherland, A. Nickel-Mediated Radioiodination of Aryl and Heteroaryl Bromides: Rapid Synthesis of Tracers for SPECT Imaging. Angew. Chemie Int. Ed. 2013, 52, 7829–7832. [Google Scholar] [CrossRef]
- Zhang, P.; Zhuang, R.; Guo, Z.; Su, X.; Chen, X.; Zhang, X. A Highly Efficient Copper-Mediated Radioiodination Approach Using Aryl Boronic Acids. Chem. A Eur. J. 2016, 22, 16783–16786. [Google Scholar] [CrossRef]
- Wilson, T.C.; McSweeney, G.; Preshlock, S.; Verhoog, S.; Tredwell, M.; Cailly, T.; Gouverneur, V. Radiosynthesis of SPECT tracers via a copper mediated 123I iodination of (hetero)aryl boron reagents. Chem. Commun. 2016, 52, 13277–13280. [Google Scholar] [CrossRef]
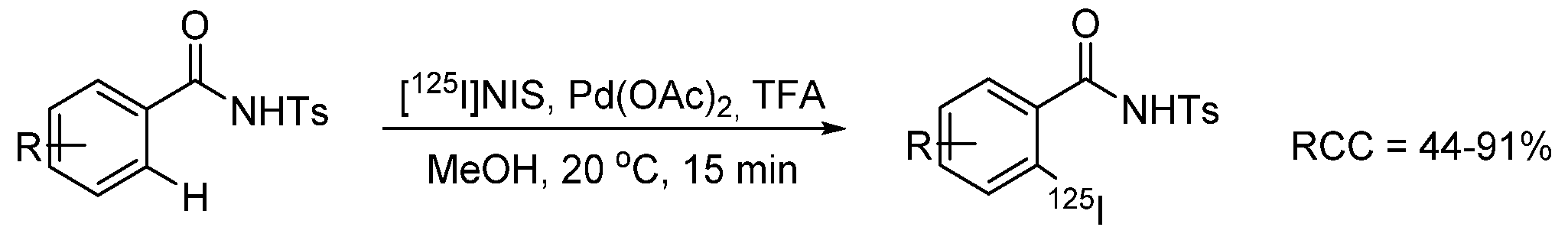
- Dubost, E.; Babin, V.; Benoist, F.; Hébert, A.; Barbey, P.; Chollet, C.; Bouillon, J.-P.; Manrique, A.; Pieters, G.; Fabis, F.; et al. Palladium-Mediated Site-Selective C–H Radio-iodination. Org. Lett. 2018, 20, 6302–6305. [Google Scholar] [CrossRef] [PubMed]
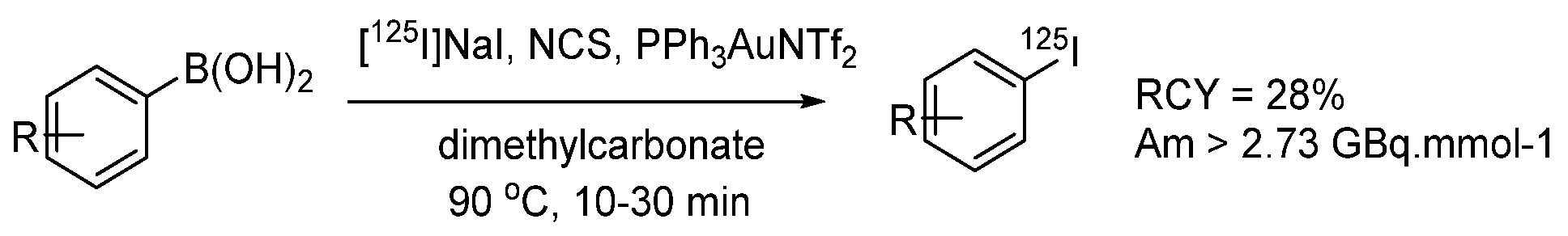
- Molloy, J.J.; O’Rourke, K.M.; Frias, C.P.; Sloan, N.L.; West, M.J.; Pimlott, S.L.; Sutherland, A.; Watson, A.J.B. Mechanism of Cu-Catalyzed Aryl Boronic Acid Halodeboronation Using Electrophilic Halogen: Development of a Base-Catalyzed Iododeboronation for Radiolabeling Applications. Org. Lett. 2019, 21, 2488–2492. [Google Scholar] [CrossRef] [PubMed]
- Cant, A.A.; Bhalla, R.; Pimlott, S.L.; Sutherland, A. Nickel-catalysed aromatic Finkelstein reaction of aryl and heteroaryl bromides. Chem. Commun. 2012, 48, 3993. [Google Scholar] [CrossRef]
- Adam, M.J.; Ponce, Y.Z.; Berry, J.M. Synthesis of L-6-[123I]iodo-m-tyrosine a potential spect brain imaging agent. J. Label. Compd. Radiopharm. 1990, 28, 1065–1072. [Google Scholar] [CrossRef]
- Kabalka, G.W.; Yao, M.-L. No-carrier-added radiohalogenations utilizing organoboranes: The synthesis of iodine-123 labeled curcumin. J. Organomet. Chem. 2009, 694, 1638–1641. [Google Scholar] [CrossRef]
- Cailly, T.; Dumas, N.; Millet, P.; Lemaître, S.; Fabis, F.; Charnay, Y.; Rault, S. Synthesis and characterization of a iodine-125-labeled pyrrolo[1,2-a]thieno[3,2-e]pyrazine and evaluation as a potential 5-HT4R SPECT tracer. Eur. J. Med. Chem. 2010, 45, 5465–5467. [Google Scholar] [CrossRef]
- Neto, C.; Oliveira, M.; Gano, L.; Marques, F.; Santos, I.; Morais, G.; Yasuda, T.; Thiemann, T.; Botelho, F.; Oliveira, C. Evaluation of Novel Radioiodinated C7-substituted Δ6,7—estradiol Derivatives for Molecular Recognition of ER-Positive Breast Tumours. Curr. Radiopharm. 2009, 2, 83–91. [Google Scholar] [CrossRef]
- Lalut, J.; Tournier, B.B.; Cailly, T.; Lecoutey, C.; Corvaisier, S.; Davis, A.; Ballandonne, C.; Since, M.; Millet, P.; Fabis, F.; et al. Synthesis and evaluation of novel serotonin 4 receptor radiotracers for single photon emission computed tomography. Eur. J. Med. Chem. 2016, 116, 90–101. [Google Scholar] [CrossRef]
- Samnick, S.; Bader, J.B.; Müller, M.; Chapot, C.; Richter, S.; Schaefer, A.; Sax, B.; Kirsch, C.-M. Improved labelling of no-carrier-added 123I-MIBG and preliminary clinical evaluation in patients with ventricular arrhythmias. Nucl. Med. Commun. 1999, 20, 537–546. [Google Scholar] [CrossRef]
- Chitneni, S.K.; Reitman, Z.J.; Spicehandler, R.; Gooden, D.M.; Yan, H.; Zalutsky, M.R. Synthesis and evaluation of radiolabeled AGI-5198 analogues as candidate radiotracers for imaging mutant IDH1 expression in tumors. Bioorg. Med. Chem. Lett. 2018, 28, 694–699. [Google Scholar] [CrossRef]
- Neumeyer, J.L.; Wang, S.; Milius, R.A.; Baldwin, R.M.; Zea-Ponce, Y.; Hoffer, P.B.; Sybirska, E.; Al-Tikriti, M.; Charney, D.S. [123I]-2.beta.-carbomethoxy-3.beta.-(4-iodophenyl)tropane: High-affinity SPECT (single photon emission computed tomography) radiotracer of monoamine reuptake sites in brain. J. Med. Chem. 1991, 34, 3144–3146. [Google Scholar] [CrossRef]
- Donovan, A.; Forbes, J.; Dorff, P.; Schaffer, P.; Babich, J.; Valliant, J.F. A New Strategy for Preparing Molecular Imaging and Therapy Agents Using Fluorine-Rich (Fluorous) Soluble Supports. J. Am. Chem. Soc. 2006, 128, 3536–3537. [Google Scholar] [CrossRef]
- Rajerison, H.; Faye, D.; Roumesy, A.; Louaisil, N.; Boeda, F.; Faivre-Chauvet, A.; Gestin, J.-F.; Legoupy, S. Ionic liquid supported organotin reagents to prepare molecular imaging and therapy agents. Org. Biomol. Chem. 2016, 14, 2121–2126. [Google Scholar] [CrossRef]
- Vaidyanathan, G.; Zalutsky, M.R. No-carrier-added synthesis of meta-[131I]iodobenzylguanidine. Appl. Radiat. Isot. 1993, 44, 621–628. [Google Scholar] [CrossRef]
- Nakagawa, C.; Toyama, M.; Takeuchi, R.; Takahashi, T.; Tanaka, H. Synthesis of [123I]-iodometomidate from a polymer-supported precursor with a large excluded volume. RSC Adv. 2016, 6, 12215–12218. [Google Scholar] [CrossRef]
- Srivastava, P.C.; Callahan, A.P.; Cunningham, E.B.; Knapp, F.F. Potential cerebral perfusion agents: Synthesis and evaluation of a radioiodinated vinylalkylbarbituric acid analog. J. Med. Chem. 1983, 26, 742–746. [Google Scholar] [CrossRef]
- Kabalka, G.W.; Akula, M.R.; Zhang, J. Synthesis of radioiodinated aryl iodides via boronate precursors. Nucl. Med. Biol. 2002, 29, 841–843. [Google Scholar] [CrossRef]
- Akula, M.R.; Yao, M.-L.; Kabalka, G.W. Triolborates: Water-soluble complexes of arylboronic acids as precursors to iodoarenes. Tetrahedron Lett. 2010, 51, 1170–1171. [Google Scholar] [CrossRef]
- Yong, L.; Yao, M.-L.; Green, J.F.; Kelly, H.; Kabalka, G.W. Syntheses and characterization of polymer-supported organotrifluoroborates: Applications in radioiodination reactions. Chem. Commun. 2010, 46, 2623. [Google Scholar] [CrossRef]
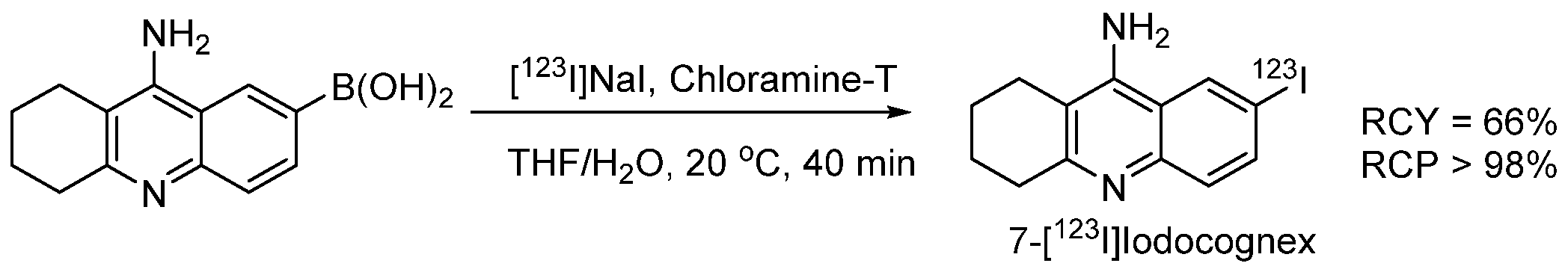
- Akula, M.R.; Zhang, J.H.; Kabalka, G.W. [123I]Iodocognex, a potent spect agent to map acetylcholinesterase via a boronic acid precursor. J. Label. Compd. Radiopharm. 2001, 44, S260–S261. [Google Scholar] [CrossRef]
- Moustapha, M.E.; Motaleb, M.A.; Ibrahim, I.T.; Moustafa, M.E. Oxidative radioiodination of aripiprazole by chloramine-T as a route to a potential brain imaging agent: A mechanistic approach. Radiochemistry 2013, 55, 116–122. [Google Scholar] [CrossRef]
- Abdel-Bary, H.M.; Moustafa, K.A.; Abdel-Ghaney, I.Y.; Sallam, K.M.; Shamsel-Din, H.A. Synthesis and radioiodination of new dipeptide coupled with biologically active pyridine moiety. J. Radioanal. Nucl. Chem. 2013, 298, 9–18. [Google Scholar] [CrossRef]
- EL-Ghany, E.A.; Amine, A.M.; EL-Sayed, A.S.; EL-Kolaly, M.T.; Abdel-Gelil, F. Radiochemical and biological characteristics of radioactive iodine labeled indomethacin for imaging of inflammation. J. Radioanal. Nucl. Chem. 2005, 266, 117–124. [Google Scholar] [CrossRef]
- El-Azony, K.M.; El-Mohty, A.A.; Seddik, U.; Khater, S.I. Radioiodination and bioevaluation of nitrofurantoin for urinary tract imaging. J. Label. Compd. Radiopharm. 2012, 55, 315–319. [Google Scholar] [CrossRef]
- Bapat, K.; Chintalwar, G.J.; Pandey, U.; Thakur, V.S.; Sarma, H.D.; Samuel, G.; Pillai, M.R.A.; Chattopadhyay, S.; Venkatesh, M. Preparation and in vitro evaluation of radioiodinated bakuchiol as an anti tumor agent. Appl. Radiat. Isot. 2005, 62, 389–393. [Google Scholar] [CrossRef]
- Sadri, K.; Gandomkar, M.; Babaei, M.H.; Najafi, R.; Zakavi, S.R.; Sadat Ebrahimi, S.E. Synthesis and biodistribution studies of iodine-131 D-amino acid YYK peptide as a potential therapeutic agent for labeling an anti-CD20 antibody. J. Label. Compd. Radiopharm. 2009, 52, 289–294. [Google Scholar] [CrossRef]
- Amin, A.M.; Soliman, S.E.; El-Aziz, H.A. Preparation and biodistribution of [125I]Melphalan: A potential radioligand for diagnostic and therapeutic applications. J. Label. Compd. Radiopharm. 2009, 53, 1–5. [Google Scholar] [CrossRef]
- Amin, A.M.; Soliman, S.E.; El-Aziz, H.A.; El-Enein, S.A.A. Radioiodination of Zaleplon and Its in-vivo Biologic Behavior in Mice: An Imaging Probe for Brain. Int. J. Chem. 2013, 6, 17. [Google Scholar] [CrossRef]
- Mounetou, E.; Miot-Noirault, E.; Gaudreault, R.C.; Madelmont, J.C. N-4-iodophenyl-N′-2-chloroethylurea, a novel potential anticancer agent with colon-specific accumulation: Radioiodination and comparative in vivo biodistribution profiles. Investig. New Drugs 2010, 28, 124–131. [Google Scholar] [CrossRef]
- El-Kawy, O.A.; Hashem, A.M.; Amin, M.A.; El-Wetery, A.S. Preparation and evaluation of [125I]troxacitabine: L-nucleoside model of a potential agent for tumor diagnosis and radiotherapy. J. Label. Compd. Radiopharm. 2011, 54, 98–104. [Google Scholar] [CrossRef]
- Jeon, J.; Ma, S.-Y.; Choi, D.S.; Jang, B.-S.; Kang, J.A.; Nam, Y.R.; Yoon, S.; Park, S.H. Radiosynthesis of 123I-labeled hesperetin for biodistribution study of orally administered hesperetin. J. Radioanal. Nucl. Chem. 2015, 306, 437–443. [Google Scholar] [CrossRef]
- Al-Momani, E.; Zlatopolskiy, B.D.; Solbach, C.; Reske, S.N.; Machulla, H.-J. Synthesis of 15-(4-[131I]iodophenyl)pentadecanoic acid (p-IPPA) via tin-precursor using Chloramine-T as an oxidant. J. Radioanal. Nucl. Chem. 2010, 286, 231–234. [Google Scholar] [CrossRef]
- Efimova, Y.M.; Wierczinski, B.; Haemers, S.; van Well, A.A. Changes in the secondary structure of proteins labeled with 125I: CD spectroscopy and enzymatic activity studies. J. Radioanal. Nucl. Chem. 2005, 264, 91–96. [Google Scholar] [CrossRef]
- Kung, H.F.; Kasliwal, R.; Pan, S.; Kung, M.P.; Mach, R.H.; Guo, Y.Z. Dopamine D-2 receptor imaging radiopharmaceuticals: Synthesis, radiolabeling and in vitro binding of (R)-(+)- and (S)-(-)-3-iodo-2-hydroxy-6-methoxy-N-[(1-ethyl-2-pyrrolidinyl)methyl]benzamide. J. Med. Chem. 1988, 31, 1039–1043. [Google Scholar] [CrossRef]
- Koziorowski, J.; Henssen, C.; Weinreich, R. A new convenient route to radioiodinated N-succinimidyl 3- and 4-iodobenzoate, two reagents for radioiodination of proteins. Appl. Radiat. Isot. 1998, 49, 955–959. [Google Scholar] [CrossRef]
- Lee, D.S.C.; Griffiths, B.W. Comparative studies of iodo-bead and chloramine-T methods for the radioiodination of human alpha-fetoprotein. J. Immunol. Methods 1984, 74, 181–189. [Google Scholar] [CrossRef]
- Hussain, A.A.; Dittert, L.W. Non-Destructive Method for Radiolabelling Biomolecules by Halogenation. U.S. Patent 5,424,402, 13 June 1995. [Google Scholar]
- Amin, A.M.; El-bary, A.A.; El-Mohty, A.A.; Saad, S.M.; El-Sharawy, D.M. Radioiodination and biological evaluation of valsartan as a tracer for cardiovascular disorder detection. Nat. Sci. 2013, 5, 526–531. [Google Scholar] [CrossRef]
- Yeşilağaç, R.; Ünak, P.; Medine, E.İ.; İçhedef, Ç.A.; Ertay, T.; Müftüler, F.Z.B. Enzymatic synthesis of 125/131I labeled 8-hydroxyquinoline glucuronide and in vitro/in vivo evaluation of biological influence. Appl. Radiat. Isot. 2011, 69, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Hussien, H.; Goud, A.A.; Amin, A.M.; EL-Sheikh, R.; Seddik, U. Comparative study between chloramine-T and iodogen to prepare radioiodinated etodolac for inflammation imaging. J. Radioanal. Nucl. Chem. 2011, 288, 9–15. [Google Scholar] [CrossRef]
- Krummeich, C.; Holschbach, M.; Stöcklin, G. Direct n.c.a. electrophilic radioiodination of tyrosine analogues; their in vivo stability and brain-uptake in mice. Appl. Radiat. Isot. 1994, 45, 929–935. [Google Scholar] [CrossRef]
- Siaens, R.; Eijsink, V.G.H.; Dierckx, R.; Slegers, G. (123)I-Labeled chitinase as specific radioligand for in vivo detection of fungal infections in mice. J. Nucl. Med. 2004, 45, 1209–1216. [Google Scholar] [PubMed]
- Avcıbaşı, U.; Avcıbaşı, N.; Akalın, H.A.; Ediz, M.; Demiroğlu, H.; Gümüşer, F.G.; Özçalışkan, E.; Türkcan, C.; Uygun, D.A.; Akgöl, S. Synthesis and biodistribution of novel magnetic-poly(HEMA–APH) nanopolymer radiolabeled with iodine-131 and investigation its fate in vivo for cancer therapy. J. Nanoparticle Res. 2013, 15, 2021. [Google Scholar] [CrossRef]
- Wang, K.; Adelstein, S.J.; Kassis, A.I. DMSO increases radioiodination yield of radiopharmaceuticals. Appl. Radiat. Isot. 2008, 66, 50–59. [Google Scholar] [CrossRef] [PubMed]
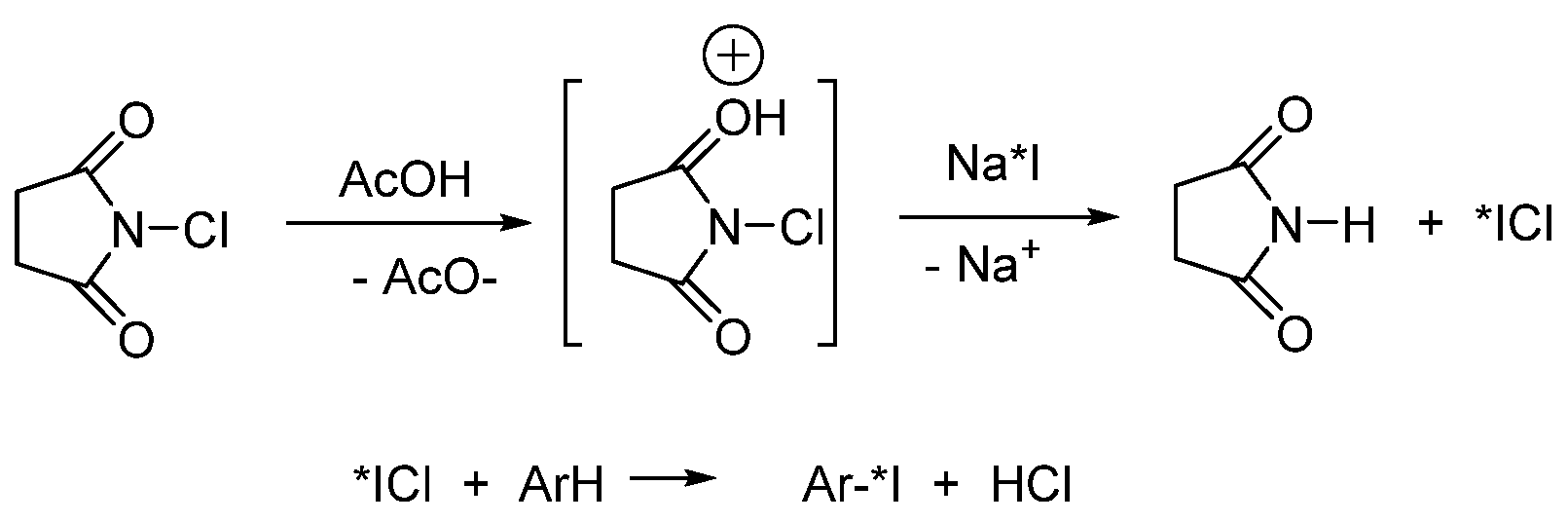
- Yamamoto, T.; Toyota, K.; Morita, N. An efficient and regioselective iodination of electron-rich aromatic compounds using N-chlorosuccinimide and sodium iodide. Tetrahedron Lett. 2010, 51, 1364–1366. [Google Scholar] [CrossRef]
- Li, G.; Kakarla, R.; Gerritz, S.W. A fast and efficient bromination of isoxazoles and pyrazoles by microwave irradiation. Tetrahedron Lett. 2007, 48, 4595–4599. [Google Scholar] [CrossRef]
- Prakash, G.K.S.; Mathew, T.; Hoole, D.; Esteves, P.M.; Wang, Q.; Rasul, G.; Olah, G.A. N -Halosuccinimide/BF3H2O, Efficient Electrophilic Halogenating Systems for Aromatics. J. Am. Chem. Soc. 2004, 126, 15770–15776. [Google Scholar] [CrossRef]
- Mattner, F.; Mardon, K.; Katsifis, A. Pharmacological evaluation of [123I]-CLINDE: A radioiodinated imidazopyridine-3-acetamide for the study of peripheral benzodiazepine binding sites (PBBS). Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 779–789. [Google Scholar] [CrossRef]
- Xu, Y.-P.; Yang, M.; Pan, D.-H.; Wang, L.-Z. Preparation of 131I–betulinic acid and its biodistribution in murine model of hepatocellular tumor. J. Radioanal. Nucl. Chem. 2011, 288, 157–161. [Google Scholar] [CrossRef]
- Wilbur, D.S. Radiohalogenation of proteins: An overview of radionuclides, labeling methods and reagents for conjugate labeling. Bioconjug. Chem. 1992, 3, 433–470. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; El-Emir, E.; Rajkumar, V.; Robson, M.; Jathoul, A.P.; Pedley, R.B.; Årstad, E. One-Pot Synthesis of an 125I-Labeled Trifunctional Reagent for Multiscale Imaging with Optical and Nuclear Techniques. Angew. Chem. Int. Ed. 2011, 50, 6793–6795. [Google Scholar] [CrossRef]
- Yan, R.; Sander, K.; Galante, E.; Rajkumar, V.; Badar, A.; Robson, M.; El-Emir, E.; Lythgoe, M.F.; Pedley, R.B.; Årstad, E. A One-Pot Three-Component Radiochemical Reaction for Rapid Assembly of 125I-Labeled Molecular Probes. J. Am. Chem. Soc. 2013, 135, 703–709. [Google Scholar] [CrossRef]
- El-Wetery, A.S.; El-Mohty, A.A.; Ayyoub, S.; Raieh, M. Catalytic effect of copper(II) chloride on the radioiodination of L-p-iodophenylalanine. J. Label. Compd. Radiopharm. 1997, 39, 631–644. [Google Scholar] [CrossRef]
- Wafelman, A.R.; Konings, M.C.P.; Hoefnagel, C.A.; Maes, R.A.A.; Beijnen, J.H. Synthesis, radiolabelling and stability of radioiodinated m-iodobenzylguanidine, a review. Appl. Radiat. Isot. 1994, 45, 997–1007. [Google Scholar] [CrossRef]
- Chattopadhyay, S.; Das, M.; Sarkar, B..; Prabhakar, G.; Mehra, K.S.; Ramamoorthy, N. Stabilisation of [131I]meta-iodobenzylguanidine at room temperature as organic extract in ethyl acetate/chloroform. Appl. Radiat. Isot. 2001, 54, 241–244. [Google Scholar] [CrossRef]
- Kuge, Y.; Katada, Y.; Shimonaka, S.; Temma, T.; Kimura, H.; Kiyono, Y.; Yokota, C.; Minematsu, K.; Seki, K.; Tamaki, N.; et al. Synthesis and evaluation of radioiodinated cyclooxygenase-2 inhibitors as potential SPECT tracers for cyclooxygenase-2 expression. Nucl. Med. Biol. 2006, 33, 21–27. [Google Scholar] [CrossRef]
- Kiyono, Y.; Sugita, T.; Ueda, M.; Kawashima, H.; Kanegawa, N.; Kuge, Y.; Fujibayashi, Y.; Saji, H. Evaluation of radioiodinated (2S,αS)-2-(α-(2-iodophenoxy)benzyl)morpholine as a radioligand for imaging of norepinephrine transporter in the heart. Nucl. Med. Biol. 2008, 35, 213–218. [Google Scholar] [CrossRef]
- Farouk, N. Radioiodination of (N-diethylaminoethyl)-4-iodobenzamide (IBZA) as a new potent melanoma imaging and therapeutic agent via isotopic exchange reaction. J. Radioanal. Nucl. Chem. 2011, 289, 7–11. [Google Scholar] [CrossRef]
- Eersels, J.L.H.; Mertens, J.; Herscheid, J.D.M. Optimization of the labeling yield by determination of the Cu+–acetonitrile complex constant in Cu+-catalyzed nucleophilic exchange reactions in mixed solvent conditions. J. Radioanal. Nucl. Chem. 2011, 288, 291–296. [Google Scholar] [CrossRef][Green Version]
- Eersels, J.L.H.; Mertens, J.; Herscheid, J.D.M. The Cu+-assisted radioiodination Kit: Mechanistic study of unexplored parameters concerning the acidity and redox properties of the reaction medium. Appl. Radiat. Isot. 2010, 68, 309–313. [Google Scholar] [CrossRef] [PubMed]
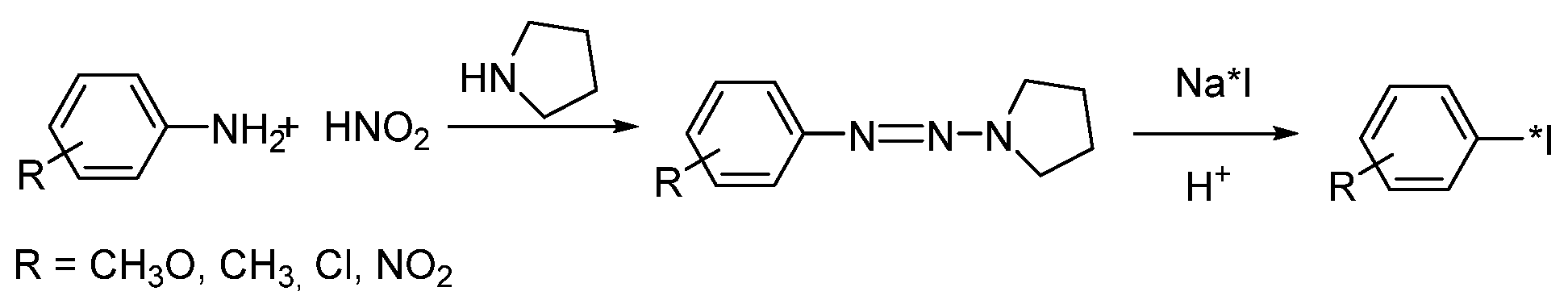
- Foster, N.I.; Dannals, R.; Burns, H.D.; Heindel, N.D. A condition variation study for radioiodination via triazene intermediates. J. Radioanal. Chem. 1981, 65, 95–105. [Google Scholar] [CrossRef]
- Karimi Zarchi, M.A.; Ebrahimi, N. Iodination of stable aromatic diazonium salt using crosslinked poly (4-vinylpyridine)-supported iodide. J. Appl. Polym. Sci. 2012, 124, 2807–2813. [Google Scholar] [CrossRef]
- Sloan, N.L.; Luthra, S.K.; McRobbie, G.; Pimlott, S.L.; Sutherland, A. A one-pot radioiodination of aryl amines via stable diazonium salts: Preparation of 125I-imaging agents. Chem. Commun. 2017, 53, 11008–11011. [Google Scholar] [CrossRef]
- Khalaj, A.; Beiki, D.; Rafiee, H.; Najafi, R. A new and simple synthesis of N-succinimidyl-4-[127/125I]iodobenzoate involving a microwave—accelerated iodination step. J. Label. Compd. Radiopharm. 2001, 44, 235–240. [Google Scholar] [CrossRef]
- Vivier, M.; Rapp, M.; Papon, J.; Labarre, P.; Galmier, M.-J.; Sauzière, J.; Madelmont, J.-C. Synthesis, Radiosynthesis, and Biological Evaluation of New Proteasome Inhibitors in a Tumor Targeting Approach. J. Med. Chem. 2008, 51, 1043–1047. [Google Scholar] [CrossRef] [PubMed]
- Navarro, L.; Berdal, M.; Chérel, M.; Pecorari, F.; Gestin, J.-F.; Guérard, F. Prosthetic groups for radioiodination and astatination of peptides and proteins: A comparative study of five potential bioorthogonal labeling strategies. Bioorg. Med. Chem. 2019, 27, 167–174. [Google Scholar] [CrossRef]
- Guérard, F.; Lee, Y.-S.; Baidoo, K.; Gestin, J.-F.; Brechbiel, M.W. Unexpected Behavior of the Heaviest Halogen Astatine in the Nucleophilic Substitution of Aryliodonium Salts. Chem. A Eur. J. 2016, 22, 12332–12339. [Google Scholar] [CrossRef]
- Guérard, F.; Navarro, L.; Lee, Y.-S.; Roumesy, A.; Alliot, C.; Chérel, M.; Brechbiel, M.W.; Gestin, J.-F. Bifunctional aryliodonium salts for highly efficient radioiodination and astatination of antibodies. Bioorg. Med. Chem. 2017, 25, 5975–5980. [Google Scholar] [CrossRef]
- Reilly, S.W.; Makvandi, M.; Xu, K.; Mach, R.H. Rapid Cu-Catalyzed [211At]Astatination and [125I]Iodination of Boronic Esters at Room Temperature. Org. Lett. 2018, 20, 1752–1755. [Google Scholar] [CrossRef]
- Dubost, E.; Babin, V.; Benoist, F.; Hébert, A.; Pigrée, G.; Bouillon, J.-P.; Fabis, F.; Cailly, T. Improvements of C–H Radio-Iodination of N-Acylsulfonamides toward Implementation in Clinics. Synthesis 2019, 51, 4393–4400. [Google Scholar] [CrossRef]
- Lu, Z.; Pham, T.T.; Rajkumar, V.; Yu, Z.; Pedley, R.B.; Årstad, E.; Maher, J.; Yan, R. A Dual Reporter Iodinated Labeling Reagent for Cancer Positron Emission Tomography Imaging and Fluorescence-Guided Surgery. J. Med. Chem. 2018, 61, 1636–1645. [Google Scholar] [CrossRef] [PubMed]
- Bolton, A.E.; Hunter, W.M. The labelling of proteins to high specific radioactivities by conjugation to a 125I-containing acylating agent. Application to the radioimmunoassay. Biochem. J. 1973, 133, 529–538. [Google Scholar] [CrossRef]
- Russell, J.; O’Donoghue, J.A.; Finn, R.; Koziorowski, J.; Ruan, S.; Humm, J.L.; Ling, C.C. Iodination of annexin V for imaging apoptosis. J. Nucl. Med. 2002, 43, 671–677. [Google Scholar]
- Vaidyanathan, G.; Zalutsky, M.R. Preparation of N-succinimidyl 3-[*I]iodobenzoate: An agent for the indirect radioiodination of proteins. Nat. Protoc. 2006, 1, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Garg, S.; Garg, P.K.; Zalutsky, M.R. N-Succinimidyl 5-(trialkylstannyl)-3-pyridinecarboxylates: A new class of reagents for protein radioiodination. Bioconjug. Chem. 1991, 2, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Chen, J.; Quinn, T.P.; Jurisson, S.S. Radioiodination of Rhenium Cyclized α-Melanocyte-Stimulating Hormone Resulting in Enhanced Radioactivity Localization and Retention in Melanoma. Cancer Res. 2004, 64, 1411–1418. [Google Scholar] [CrossRef] [PubMed]
- Vaidyanathan, G.; Zalutsky, M.R. Synthesis of N-succinimidyl 4-guanidinomethyl-3-[*I]iodobenzoate: A radio-iodination agent for labeling internalizing proteins and peptides. Nat. Protoc. 2007, 2, 282–286. [Google Scholar] [CrossRef]
- Khawli, L.A.; Van Den Abbeele, A.D.; Kassis, A.I. N-(m-[125I]iodophenyl)maleimide: An agent for high yield radiolabeling of antibodies. Int. J. Radiat. Appl. Instrum. Part B Nucl. Med. Biol. 1992, 19, 289–295. [Google Scholar] [CrossRef]
- Bhojani, M.S.; Ranga, R.; Luker, G.D.; Rehemtulla, A.; Ross, B.D.; Van Dort, M.E. Synthesis and Investigation of a Radioiodinated F3 Peptide Analog as a SPECT Tumor Imaging Radioligand. PLoS ONE 2011, 6, e22418. [Google Scholar] [CrossRef]
- Billaud, E.M.F.; Vidal, A.; Vincenot, A.; Besse, S.; Bouchon, B.; Debiton, E.; Miot-Noirault, E.; Miladi, I.; Rbah-Vidal, L.; Auzeloux, P.; et al. Development and Preliminary Evaluation of TFIB, a New Bimodal Prosthetic Group for Bioactive Molecule Labeling. ACS Med. Chem. Lett. 2015, 6, 168–172. [Google Scholar] [CrossRef]
- Orlova, A.; Bruskin, A.; Sivaev, I.; Sjöberg, S.; Lundqvist, H.; Tolmachev, V. Radio-iodination of monoclonal antibody using potassium [125I]-(4-isothiocyanatobenzylammonio)-iodo-decahydro-closo-dodecaborate (iodo-DABI). Anticancer Res. 2006, 26, 1217–1223. [Google Scholar]
- Lin, R.; Liu, N.; Yang, Y.; Li, B.; Liao, J.; Jin, J. Radioiodination of protein using 2,3,5,6-tetrafluorophenyl 3-(nido-carboranyl) propionate (TCP) as a potential bi-functional linker: Synthesis and biodistribution in mice. Appl. Radiat. Isot. 2009, 67, 83–87. [Google Scholar] [CrossRef]
- Wilbur, D.S.; Chyan, M.-K.; Hamlin, D.K.; Nguyen, H.; Vessella, R.L. Reagents for Astatination of Biomolecules. 5. Evaluation of Hydrazone Linkers in 211At- and 125I-Labeled closo -Decaborate(2-) Conjugates of Fab′ as a Means of Decreasing Kidney Retention. Bioconjug. Chem. 2011, 22, 1089–1102. [Google Scholar] [CrossRef]
- Lewis, J.S.; Windhorst, A.D.; Zeglis, B.M. (Eds.) Radiopharmaceutical Chemistry; Springer International Publishing: Cham, Switzerland, 2019; ISBN 978-3-319-98946-4. [Google Scholar]
- Janabi, M.; Pollock, C.M.; Chacko, A.-M.; Hunter, D.H. Resin-supported arylstannanes as precursors for radiolabeling with iodine: Benzaldehydes, benzoic acids, benzamides, and NHS esters. Can. J. Chem. 2015, 93, 207–217. [Google Scholar] [CrossRef]
- Kondo, N.; Temma, T.; Shimizu, Y.; Ono, M.; Saji, H. Radioiodinated Peptidic Imaging Probes for in vivo Detection of Membrane Type-1 Matrix Metalloproteinase in Cancers. Biol. Pharm. Bull. 2015, 38, 1375–1382. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.H.; Shim, H.E.; Yun, S.-J.; Kim, H.R.; Mushtaq, S.; Lee, C.H.; Park, S.H.; Choi, D.S.; Lee, D.-E.; Byun, E.-B.; et al. Highly efficient method for 125I-radiolabeling of biomolecules using inverse-electron-demand Diels–Alder reaction. Bioorg. Med. Chem. 2016, 24, 2589–2594. [Google Scholar] [CrossRef] [PubMed]
- Neumann, S.; Biewend, M.; Rana, S.; Binder, W.H. The CuAAC: Principles, Homogeneous and Heterogeneous Catalysts, and Novel Developments and Applications. Macromol. Rapid Commun. 2020, 41, 1900359. [Google Scholar] [CrossRef]
- Kenry; Liu, B. Bio-orthogonal Click Chemistry for In Vivo Bioimaging. Trends Chem. 2019, 1, 763–778. [Google Scholar] [CrossRef]
- Choi, M.H.; Shim, H.E.; Nam, Y.R.; Kim, H.R.; Kang, J.A.; Lee, D.E.; Park, S.H.; Choi, D.S.; Jang, B.S.; Jeon, J. Synthesis and evaluation of an125I-labeled azide prosthetic group for efficient and bioorthogonal radiolabeling of cyclooctyne-group containing molecules using copper-free click reaction. Bioorganic Med. Chem. Lett. 2016, 26, 875–878. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, B.L.; Guo, Z.; Bernardes, G.J.L. Inverse electron demand Diels–Alder reactions in chemical biology. Chem. Soc. Rev. 2017, 46, 4895–4950. [Google Scholar] [CrossRef]
- Mangner, T.J.; Wu, J.L.; Wieland, D.M. Solid-phase exchange radioiodination of aryl iodides. Facilitation by ammonium sulfate. J. Org. Chem. 1982, 47, 1484–1488. [Google Scholar] [CrossRef]
- Wieland, D.M.; Wu, J.; Brown, L.E.; Mangner, T.J.; Swanson, D.P.; Beierwaltes, W.H. Radiolabeled adrenergi neuron-blocking agents: Adrenomedullary imaging with [131I]iodobenzylguanidine. J. Nucl. Med. 1980, 21, 349–353. [Google Scholar]
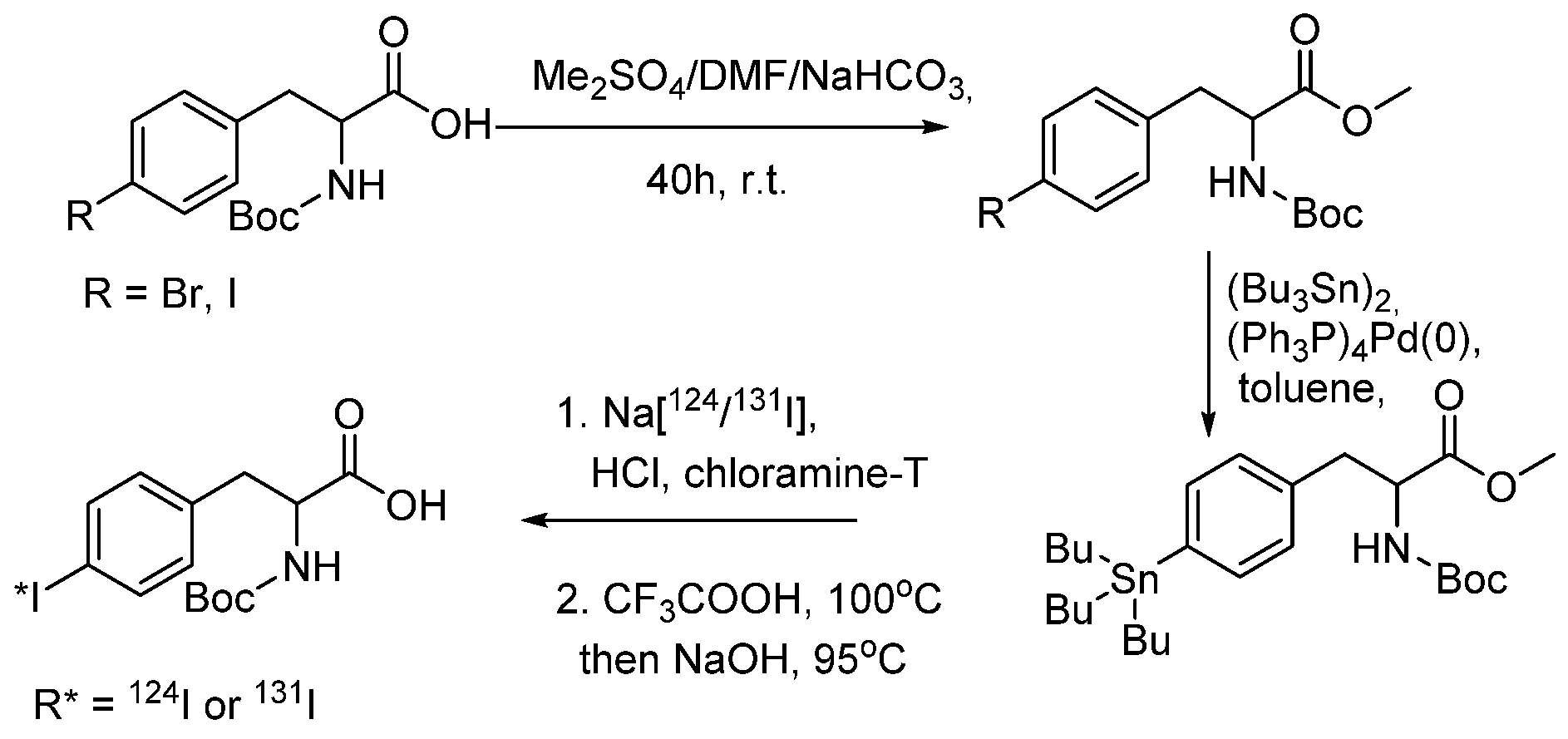
- Donovan, A.C.; Valliant, J.F. A convenient solution-phase method for the preparation of meta-iodobenzylguanidine in high effective specific activity. Nucl. Med. Biol. 2008, 35, 741–746. [Google Scholar] [CrossRef]
- Jager, P.L.; Vaalburg, W.; Pruim, J.; de Vries, E.G.; Langen, K.J.; Piers, D.A. Radiolabeled amino acids: Basic aspects and clinical applications in oncology. J. Nucl. Med. 2001, 42, 432–445. [Google Scholar] [PubMed]
- Samnick, S.; Bader, J.B.; Hellwig, D.; Moringlane, J.R.; Alexander, C.; Romeike, B.F.M.; Feiden, W.; Kirsch, C.-M. Clinical Value of Iodine-123-Alpha-Methyl-l-Tyrosine Single-Photon Emission Tomography in the Differential Diagnosis of Recurrent Brain Tumor in Patients Pretreated for Glioma at Follow-Up. J. Clin. Oncol. 2002, 20, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Samnick, S.; Hellwig, D.; Bader, J.B.; Romeike, B.F.; Moringlane, J.R.; Feiden, W.; Kirsch, C.-M. Initial evaluation of the feasibility of single photon emission tomography with p-[123I]iodo-L-phenylalanine for routine brain tumour imaging. Nucl. Med. Commun. 2002, 23, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Farmakis, G.; Brandau, W.; Hellwig, D.; Wollenweber, F.; Schaefer, A.; Kirsch, C.-M.; Samnick, S. PET Imaging With p-[I-124]iodo-l-phenylalanine as a New Tool for Diagnosis and Postoperative Control in Patients With Glioma. Clin. Nucl. Med. 2008, 33, 441–442. [Google Scholar] [CrossRef] [PubMed]
- Samnick, S.; Richter, S.; Romeike, B.F.; Heimann, A.; Feiden, W.; Kempski, O.; Kirsch, C.-M. Investigation of iodine-123-labelled amino acid derivatives for imaging cerebral gliomas: Uptake in human glioma cells and evaluation in stereotactically implanted C6 glioma rats. Eur. J. Nucl. Med. 2000, 27, 1543–1551. [Google Scholar] [CrossRef]
- Samnick, S.; Romeike, B.F.; Lehmann, T.; Israel, I.; Rübe, C.; Mautes, A.; Reiners, C.; Kirsch, C.-M. Efficacy of Systemic Radionuclide Therapy with p-131I-Iodo-l-Phenylalanine Combined with External Beam Photon Irradiation in Treating Malignant Gliomas. J. Nucl. Med. 2009, 50, 2025–2032. [Google Scholar] [CrossRef][Green Version]
- Israel, I.; Brandau, W.; Farmakis, G.; Samnick, S. Improved synthesis of no-carrier-added p-[124I]iodo-l-phenylalanine and p-[131I]iodo-l-phenylalanine for nuclear medicine applications in malignant gliomas. Appl. Radiat. Isot. 2008, 66, 513–522. [Google Scholar] [CrossRef]
- Langen, K.-J.; Pauleit, D.; Coenen, H.H. 3-[123I]Iodo-α-methyl-L-tyrosine: Uptake mechanisms and clinical applications. Nucl. Med. Biol. 2002, 29, 625–631. [Google Scholar] [CrossRef]
- Kloss, G.; Leven, M. Accumulation of radioiodinated tyrosine derivatives in the adrenal medulla and in melanomas. Eur. J. Nucl. Med. 1979, 4, 179–186. [Google Scholar] [CrossRef]
- Trivedi, M.A.; Carnochan, P. An improved synthetic procedure for L-3-iodo-α-methyl tyrosine suitable for preparation in kit form. J. Label. Compd. Radiopharm. 1993, 33, 61–64. [Google Scholar] [CrossRef]
- Kloster, G.; Bockslaff, H. L-3-123I-α-methyltyrosine for melanoma detection: A comparative evaluation. Int. J. Nucl. Med. Biol. 1982, 9, 259–269. [Google Scholar] [CrossRef]
- Krummeich, C.; Holschbach, M. Kit preparation of n.c.a. 3-[123I]Iodo-l-α-methyltyrosine [123I]IMT, [123I]OMIT and [123I]OMIMT using Sep-PakTM C-18 cartridges. Appl. Radiat. Isot. 1995, 46, 917–921. [Google Scholar] [CrossRef]
- Luurtsema, G.; Jager, P.L.; Piers, A.; de Hooge, M.N. An automated synthesis module for preparation of l-3-[123I]iodo-alpha-methyl tyrosine. Appl. Radiat. Isot. 2001, 55, 783–788. [Google Scholar] [CrossRef]
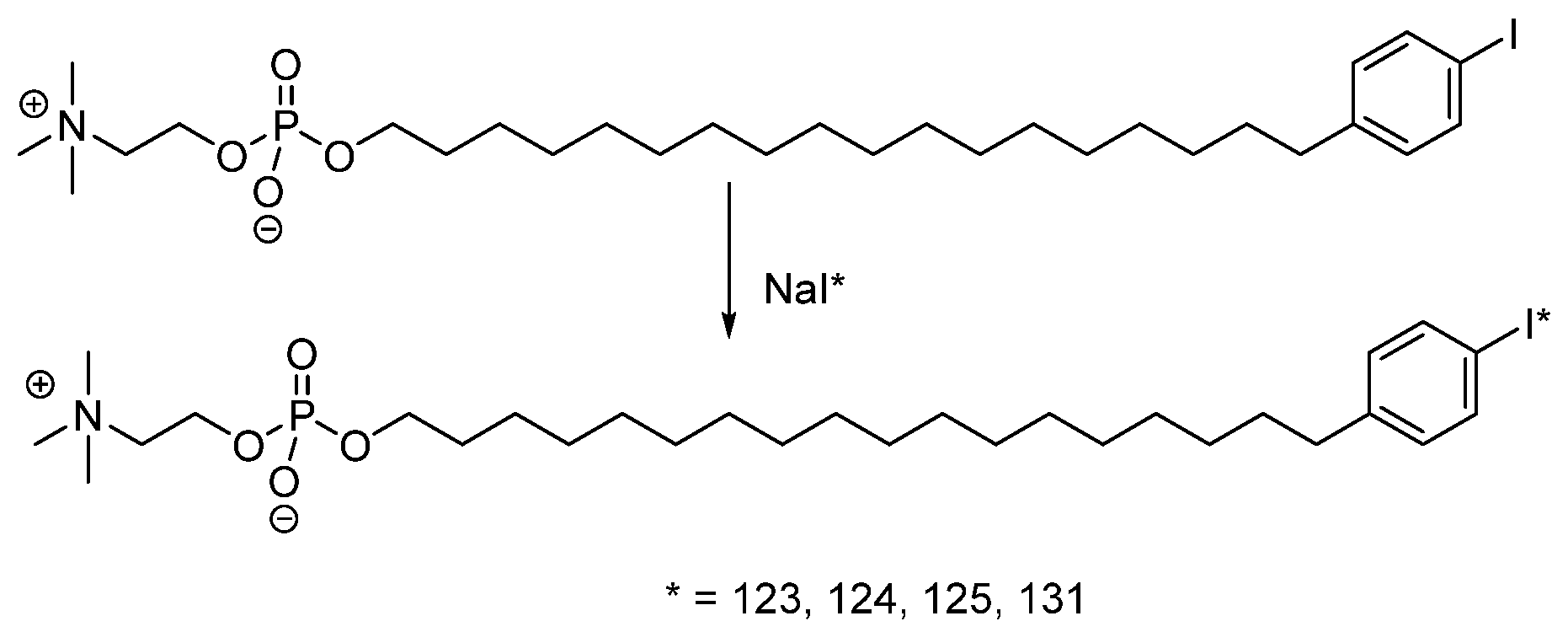
- Ailawadhi, S.; Stiff, P.; Ibrahim, E.; Green, D.J.; Lipe, B.; Cull, E.H.; Callander, N.S.; Friend, J.; Longcor, J.; Oliver, K. CLR 131 (Iopofosine I-131) Treatment in Triple Class Refractory and Beyond Multiple Myeloma Patients: Preliminary Efficacy and Safety Results from the Phase 2 Clover-1 Trial. Blood 2021, 138, 1652. [Google Scholar] [CrossRef]
- Rampy, M.A.; Chou, T.S.; Pinchuk, A.N.; Skinner, R.W.S.; Gross, M.D.; Fisher, S.; Wahl, R.; Counsell, R.E. Synthesis and biological evaluation of radioiodinated phospholipid ether analogs. Nucl. Med. Biol. 1995, 22, 505–512. [Google Scholar] [CrossRef]
- Pinchuk, A.N.; Rampy, M.A.; Longino, M.A.; Skinner, R.W.S.; Gross, M.D.; Weichert, J.P.; Counsell, R.E. Synthesis and Structure−Activity Relationship Effects on the Tumor Avidity of Radioiodinated Phospholipid Ether Analogues. J. Med. Chem. 2006, 49, 2155–2165. [Google Scholar] [CrossRef]
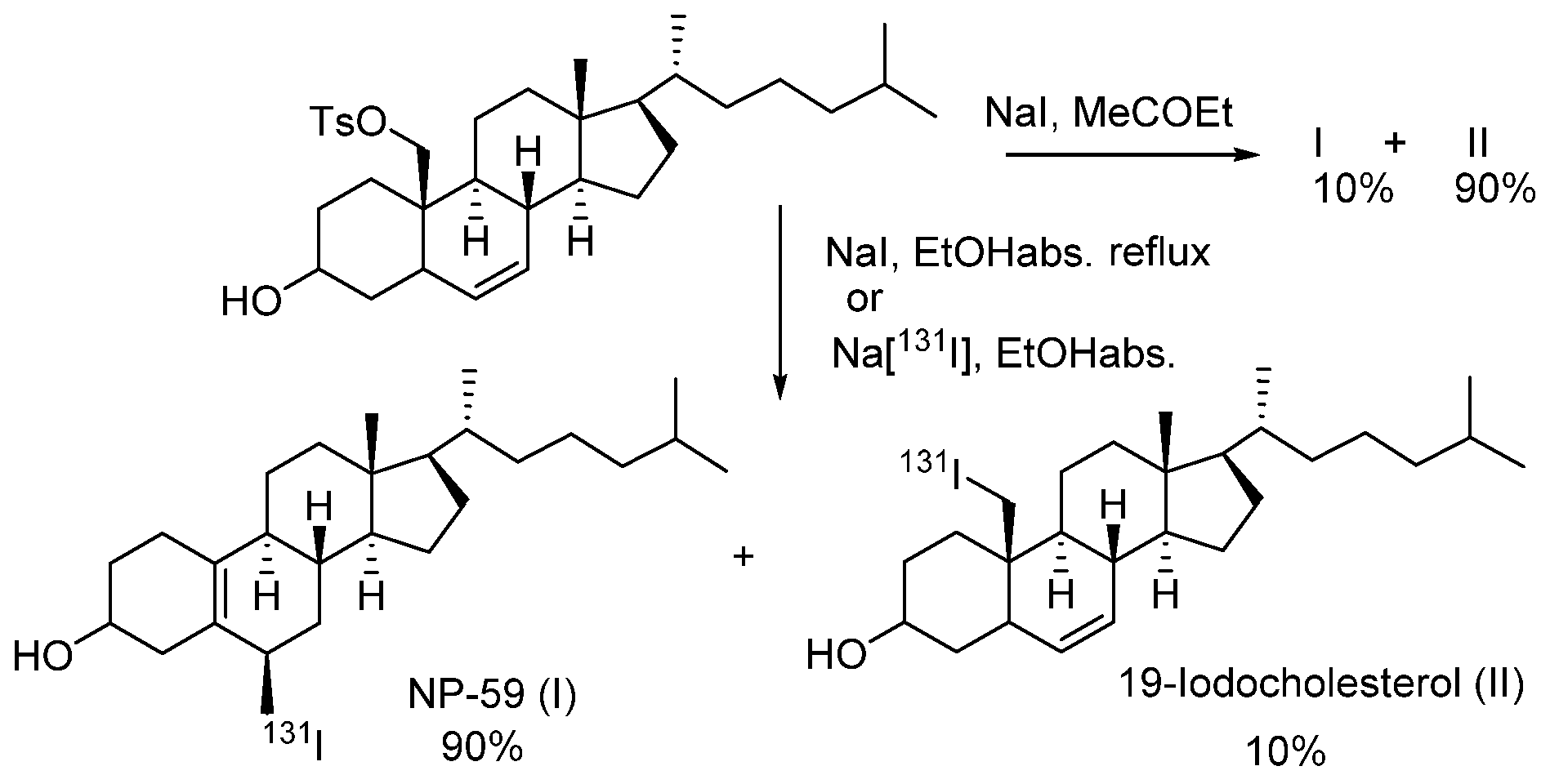
- Lee, J.; Ha, J.; Lee, S.-K.; Park, H.L.; Kim, S.-H.; Lim, D.-J.; Lee, J.M.; Chang, S.-A.; Kang, M.I.; Kim, M.-H. Feasibility of Iodine-131 6β-Methyl-Iodo-19 Norcholesterol (NP-59) Scintigraphy to Complement Adrenal Venous Sampling in Management of Primary Aldosteronism: A Case Series. Int. J. Gen. Med. 2021, 14, 673–680. [Google Scholar] [CrossRef]
- Basmadjian, G.P.; Hetzel, K.R.; Ice, R.D.; Beierwaltes, W.H. Synthesis of a new adrenal cortex imaging agent 6β-131I-iodomethyl-19-nor cholest -5(10)-en-3β-ol (NP-59). J. Labelled Compd. 1975, 11, 427–434. [Google Scholar] [CrossRef]
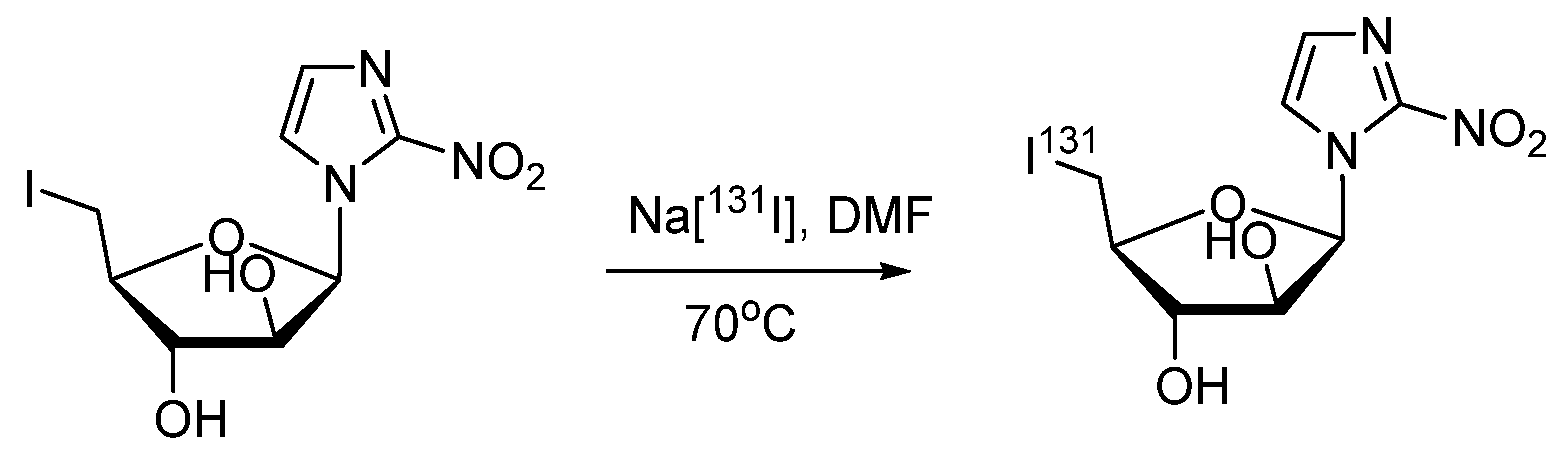
- Mercer, J.; McEwan, A.; Wiebe, L. [131I]IAZA as a Molecular Radiotherapeutic (MRT) Drug: Wash-Out with Cold IAZA Accelerates Clearance in a Murine Tumor Model. Curr. Radiopharm. 2013, 6, 87–91. [Google Scholar] [CrossRef]
- Mannan, R.H.; Somayaji, V.V.; Lee, J.; Mercer, J.R.; Chapman, J.D.; Wiebe, L.I. Radioiodinated 1-(5-iodo-5-deoxy-beta-D-arabinofuranosyl)-2-nitroimidazole (iodoazomycin arabinoside: IAZA): A novel marker of tissue hypoxia. J. Nucl. Med. 1991, 32, 1764–1770. [Google Scholar]
- Stypinski, D.; Wiebe, L.; Mercer, J. A rapid and simple assay to determine the blood and urine concentrations of 1-(5-[123/125I]iodo-5-deoxyarabinofuranosyl)-2-nitroimidazole, a hypoxic cell marker. J. Pharm. Biomed. Anal. 1998, 16, 1067–1073. [Google Scholar] [CrossRef]
- Goodman, M.M.; Chen, P.; Plisson, C.; Martarello, L.; Galt, J.; Votaw, J.R.; Kilts, C.D.; Malveaux, G.; Camp, V.M.; Shi, B.; et al. Synthesis and Characterization of Iodine-123 Labeled 2β-Carbomethoxy-3β-(4‘-((Z)-2-iodoethenyl)phenyl)nortropane. A Ligand for in Vivo Imaging of Serotonin Transporters by Single-Photon-Emission Tomography. J. Med. Chem. 2003, 46, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Tamagnan, G.D.; Alagille, D.; Da Costa, H. Compounds and Amyloid Probes Thereof for Therapeutic and Imaging Uses. U.S. Patent 7,700,616, 20 April 2010. [Google Scholar]
- DeGrado, T.; Holden, J.; Ng, C.; Raffel, D.; John Gatley, S. Comparison of 16-iodohexadecanoic acid (IHDA) and 15-p-iodophenylpentadecanoic acid (IPPA) metabolism and kinetics in the isolated rat heart. Eur. J. Nucl. Med. 1988, 14, 600–606. [Google Scholar] [CrossRef]
- Culbert, P.A.; Lu, J.; Adam, M.J. Regiospecific synthesis of 15-(4-[123I]iodophenyl)pentadecanoic acid-IPPA via methyl 15-(4-trimethylstannylphenyl)pentadecanoate. Appl. Radiat. Isot. 1997, 48, 745–747. [Google Scholar] [CrossRef]
- Goodman, M.M.; Kirsch, G.; Knapp, F.F. Synthesis and evaluation of radioiodinated terminal para-iodophenyl-substituted.alpha.- and.beta.-methyl-branched fatty acids. J. Med. Chem. 1984, 27, 390–397. [Google Scholar] [CrossRef]
- Pirovano, G.; Jannetti, S.A.; Carter, L.M.; Sadique, A.; Kossatz, S.; Guru, N.; Demétrio De Souza França, P.; Maeda, M.; Zeglis, B.M.; Lewis, J.S.; et al. Targeted Brain Tumor Radiotherapy Using an Auger Emitter. Clin. Cancer Res. 2020, 26, 2871–2881. [Google Scholar] [CrossRef]
- Wilson, T.C.; Jannetti, S.A.; Guru, N.; Pillarsetty, N.; Reiner, T.; Pirovano, G. Improved radiosynthesis of 123I-MAPi, an auger theranostic agent. Int. J. Radiat. Biol. 2020, 1–7. [Google Scholar] [CrossRef]
- Kung, M.-P.; Hou, C.; Zhuang, Z.-P.; Zhang, B.; Skovronsky, D.; Trojanowski, J.Q.; Lee, V.M.-Y.; Kung, H.F. IMPY: An improved thioflavin-T derivative for in vivo labeling of β-amyloid plaques. Brain Res. 2002, 956, 202–210. [Google Scholar] [CrossRef]
- Zhuang, Z.-P.; Kung, M.-P.; Hou, C.; Skovronsky, D.M.; Gur, T.L.; Plössl, K.; Trojanowski, J.Q.; Lee, V.M.-Y.; Kung, H.F. Radioiodinated Styrylbenzenes and Thioflavins as Probes for Amyloid Aggregates. J. Med. Chem. 2001, 44, 1905–1914. [Google Scholar] [CrossRef]
- Mazère, J.; Lamare, F.; Allard, M.; Fernandez, P.; Mayo, W. 123I-Iodobenzovesamicol SPECT Imaging of Cholinergic Systems in Dementia with Lewy Bodies. J. Nucl. Med. 2017, 58, 123–128. [Google Scholar] [CrossRef]
- Van Dort, M.E.; Jung, Y.-W.; Gildersleeve, D.L.; Hagen, C.A.; Kuhl, D.E.; Wieland, D.M. Synthesis of the 123I- and 125I-labeled cholinergic nerve marker (-)-5-iodobenzovesamicol. Nucl. Med. Biol. 1993, 20, 929–937. [Google Scholar] [CrossRef]
- Fahn, S.; Oakes, D.; Shoulson, I.; Kieburtz, K.; Rudolph, A.; Lang, A.; Olanow, C.W.; Tanner, C.; Marek, K. Parkinson Study Group Levodopa and the progression of Parkinson’s disease. N. Engl. J. Med. 2004, 351, 2498–2508. [Google Scholar] [CrossRef] [PubMed]
- De Bruyne, S.; Boos, T.L.; Wyffels, L.; Goeman, J.L.; Rice, K.C.; De Vos, F. Synthesis, radiosynthesis and in vivo evaluation of [ 123I]-4-(2-(bis(4-fluorophenyl)methoxy)ethyl)-1-(4-iodobenzyl)piperidine as a selective tracer for imaging the dopamine transporter. J. Label. Compd. Radiopharm. 2009, 52, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Tuma Santos, C.A.; Wallace, W.D.; Kim, S.; Vijayakumar, V. Pitfalls and Artifacts of 123I-Ioflupane SPECT in Parkinsonian Syndromes: A Quality Improvement Teaching Tool. J. Nucl. Med. Technol. 2021, 49, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Plhak, E.; Gößnitzer, E.; Aigner, R.M.; Kvaternik, H. Radioiodination and Purification of [131I]β-CIT and [131I]FP-CIT with an Automated Radiosynthesizer. Pharmaceuticals 2022, 15, 96. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Bensimon, C.; Lockwood, J.; Yan, X.; Fernando, P.; Wells, R.G.; Duan, Y.; Chen, Y.-X.; Redshaw, J.R.; Covitz, P.A.; et al. Synthesis and characterization of 123I-CMICE-013: A potential SPECT myocardial perfusion imaging agent. Bioorg. Med. Chem. 2013, 21, 2903–2911. [Google Scholar] [CrossRef]
- Thivat, E.; Rouanet, J.; Auzeloux, P.; Sas, N.; Jouberton, E.; Levesque, S.; Billoux, T.; Mansard, S.; Molnar, I.; Chanchou, M.; et al. Phase I study of [131I] ICF01012, a targeted radionuclide therapy, in metastatic melanoma: MELRIV-1 protocol. BMC Cancer 2022, 22, 417. [Google Scholar] [CrossRef]
- Viallard, C.; Perrot, Y.; Boudhraa, Z.; Jouberton, E.; Miot-Noirault, E.; Bonnet, M.; Besse, S.; Mishellany, F.; Cayre, A.; Maigne, L.; et al. [123I]ICF01012 melanoma imaging and [131I]ICF01012 dosimetry allow adapted internal targeted radiotherapy in preclinical melanoma models. Eur. J. Dermatol. 2015, 25, 29–35. [Google Scholar] [CrossRef]
- Chezal, J.-M.; Papon, J.; Labarre, P.; Lartigue, C.; Galmier, M.-J.; Decombat, C.; Chavignon, O.; Maublant, J.; Teulade, J.-C.; Madelmont, J.-C.; et al. Evaluation of Radiolabeled (Hetero)Aromatic Analogues of N -(2-diethylaminoethyl)-4-iodobenzamide for Imaging and Targeted Radionuclide Therapy of Melanoma. J. Med. Chem. 2008, 51, 3133–3144. [Google Scholar] [CrossRef]
- Bonnet-Duquennoy, M.; Papon, J.; Mishellany, F.; Labarre, P.; Guerquin-Kern, J.-L.; Wu, T.-D.; Gardette, M.; Maublant, J.; Penault-Llorca, F.; Miot-Noirault, E.; et al. Targeted radionuclide therapy of melanoma: Anti-tumoural efficacy studies of a new 131I labelled potential agent. Int. J. Cancer 2009, 125, 708–716. [Google Scholar] [CrossRef]
- Maresca, K.P.; Hillier, S.M.; Femia, F.J.; Keith, D.; Barone, C.; Joyal, J.L.; Zimmerman, C.N.; Kozikowski, A.P.; Barrett, J.A.; Eckelman, W.C.; et al. A Series of Halogenated Heterodimeric Inhibitors of Prostate Specific Membrane Antigen (PSMA) as Radiolabeled Probes for Targeting Prostate Cancer. J. Med. Chem. 2009, 52, 347–357. [Google Scholar] [CrossRef]
- Yerrabelli, R.S.; He, P.; Fung, E.K.; Kramer, K.; Zanzonico, P.B.; Humm, J.L.; Guo, H.; Pandit-Taskar, N.; Larson, S.M.; Cheung, N.-K.V. IntraOmmaya compartmental radioimmunotherapy using 131I-omburtamab—Pharmacokinetic modeling to optimize therapeutic index. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 1166–1177. [Google Scholar] [CrossRef] [PubMed]
- Pandit-Taskar, N.; Zanzonico, P.B.; Kramer, K.; Grkovski, M.; Fung, E.K.; Shi, W.; Zhang, Z.; Lyashchenko, S.K.; Fung, A.M.; Pentlow, K.S.; et al. Biodistribution and Dosimetry of Intraventricularly Administered 124I-Omburtamab in Patients with Metastatic Leptomeningeal Tumors. J. Nucl. Med. 2019, 60, 1794–1801. [Google Scholar] [CrossRef] [PubMed]
- Matthews, D.C.; Appelbaum, F.R.; Eary, J.F.; Fisher, D.R.; Durack, L.D.; Hui, T.E.; Martin, P.J.; Mitchell, D.; Press, O.W.; Storb, R.; et al. Phase I Study of 131I-Anti-CD45 Antibody Plus Cyclophosphamide and Total Body Irradiation for Advanced Acute Leukemia and Myelodysplastic Syndrome. Blood 1999, 94, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- Pagel, J.M.; Gooley, T.A.; Rajendran, J.; Fisher, D.R.; Wilson, W.A.; Sandmaier, B.M.; Matthews, D.C.; Deeg, H.J.; Gopal, A.K.; Martin, P.J.; et al. Allogeneic hematopoietic cell transplantation after conditioning with 131I–anti-CD45 antibody plus fludarabine and low-dose total body irradiation for elderly patients with advanced acute myeloid leukemia or high-risk myelodysplastic syndrome. Blood 2009, 114, 5444–5453. [Google Scholar] [CrossRef]
- Geyer, M.B.; Cadzin, B.; Halton, E.; Kane, P.; Senechal, B.; Geoghegan, E.M.; Reddy, V.; Berger, M.S.; Ludwig, D.; Pandit-Taskar, N.; et al. Iomab with Adoptive Cellular Therapy (Iomab-ACT): A Pilot Study of 131-I Apamistamab Followed By CD19-Targeted CAR T-Cell Therapy for Patients with Relapsed or Refractory B-Cell Acute Lymphoblastic Leukemia or Diffuse Large B-Cell Lymphoma. Blood 2021, 138, 4810. [Google Scholar] [CrossRef]





















































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
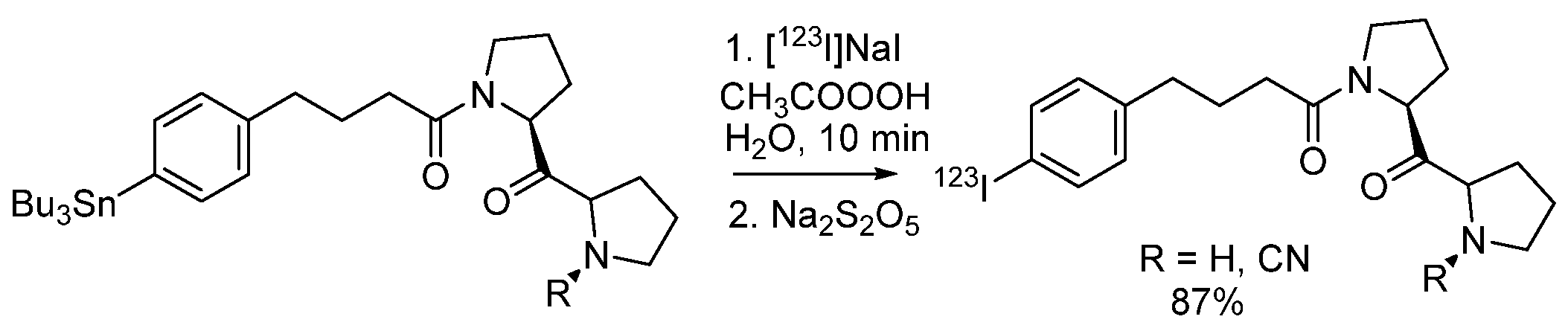{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
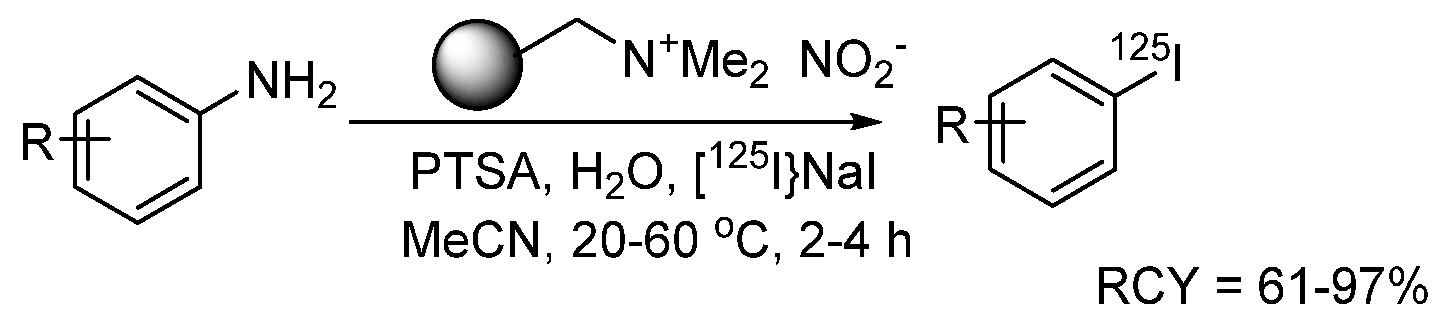{kind=link}
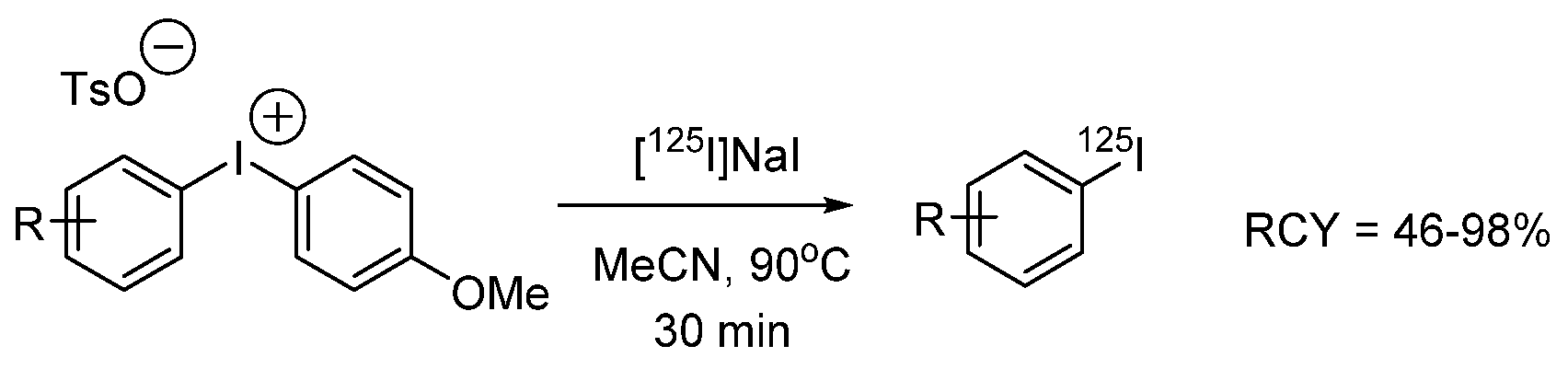{kind=link}
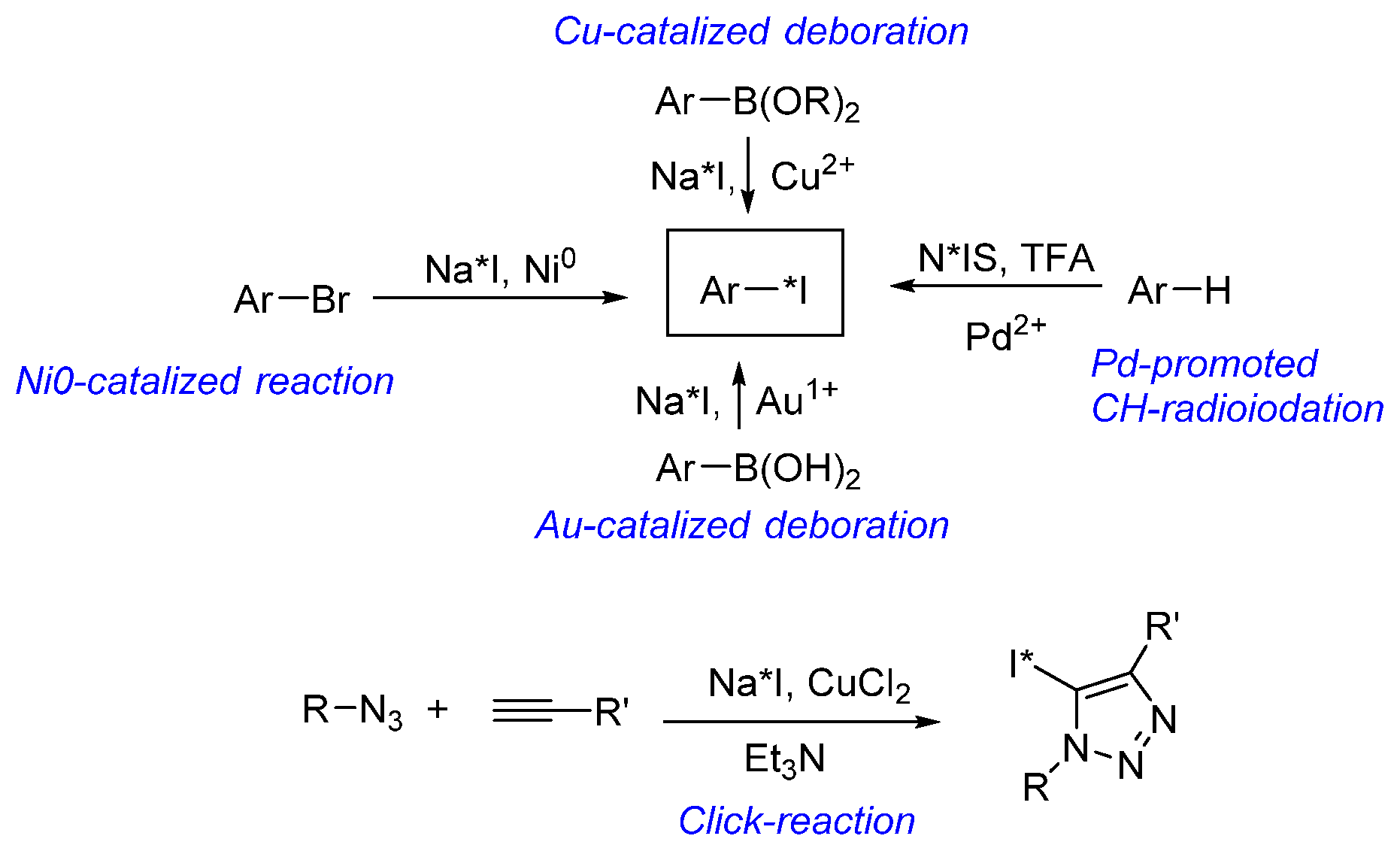{kind=link}
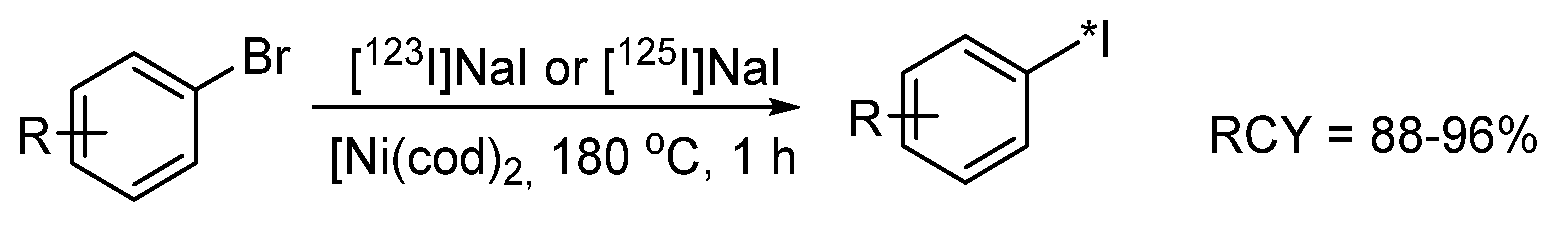{kind=link}
{kind=link}
{kind=link}
{kind=link}
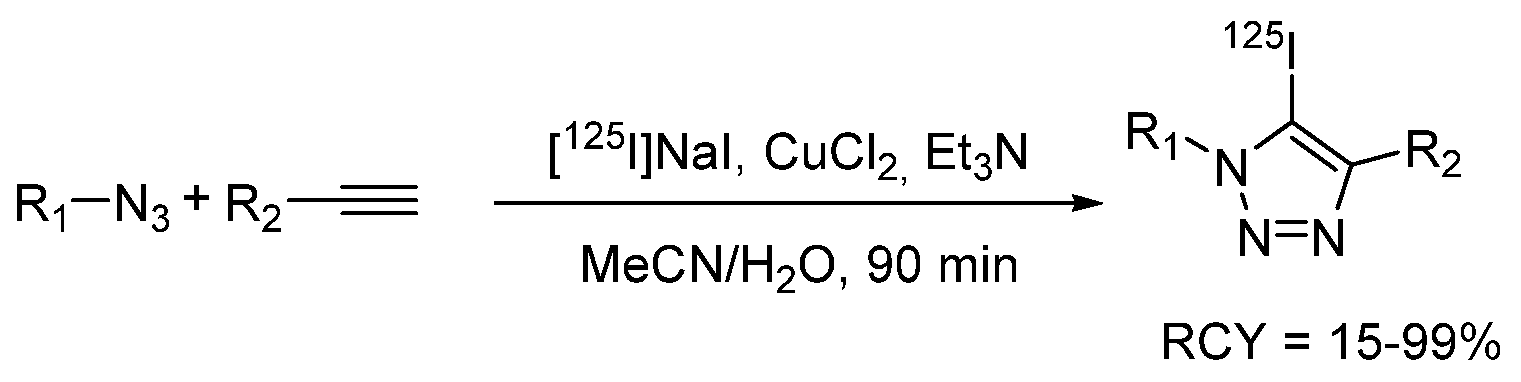{kind=link}
{kind=link}
{kind=link}
{kind=link}
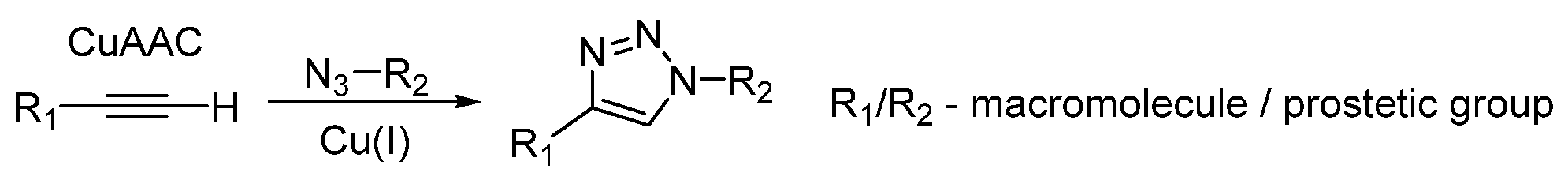{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
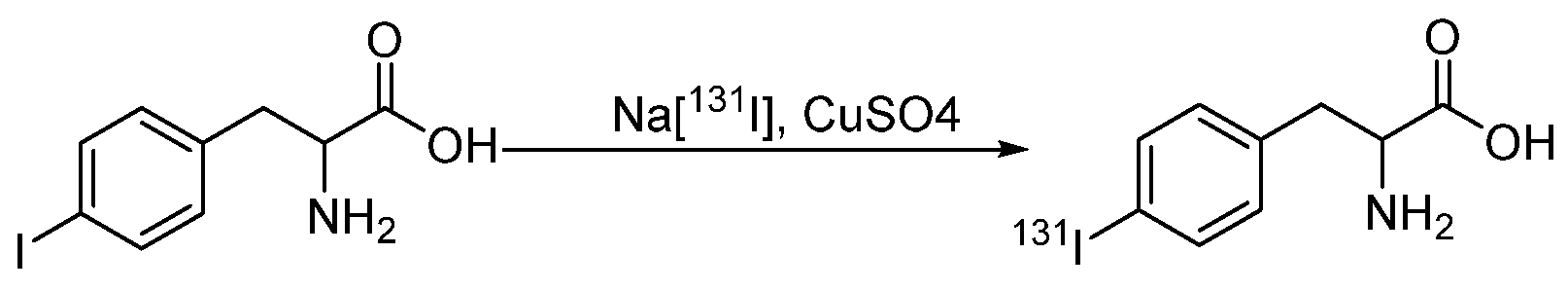{kind=link}
{kind=link}
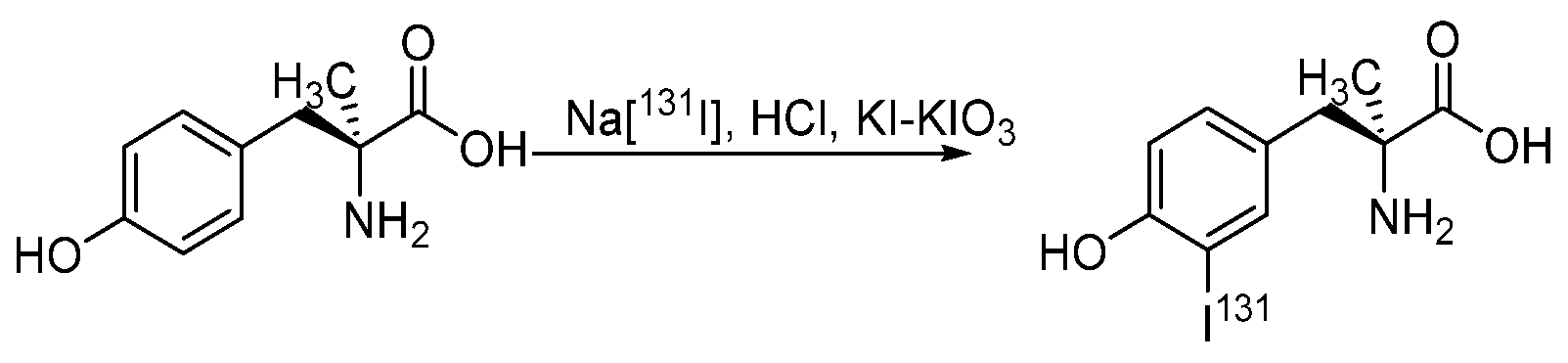{kind=link}
{kind=link}
{kind=link}
{kind=link}
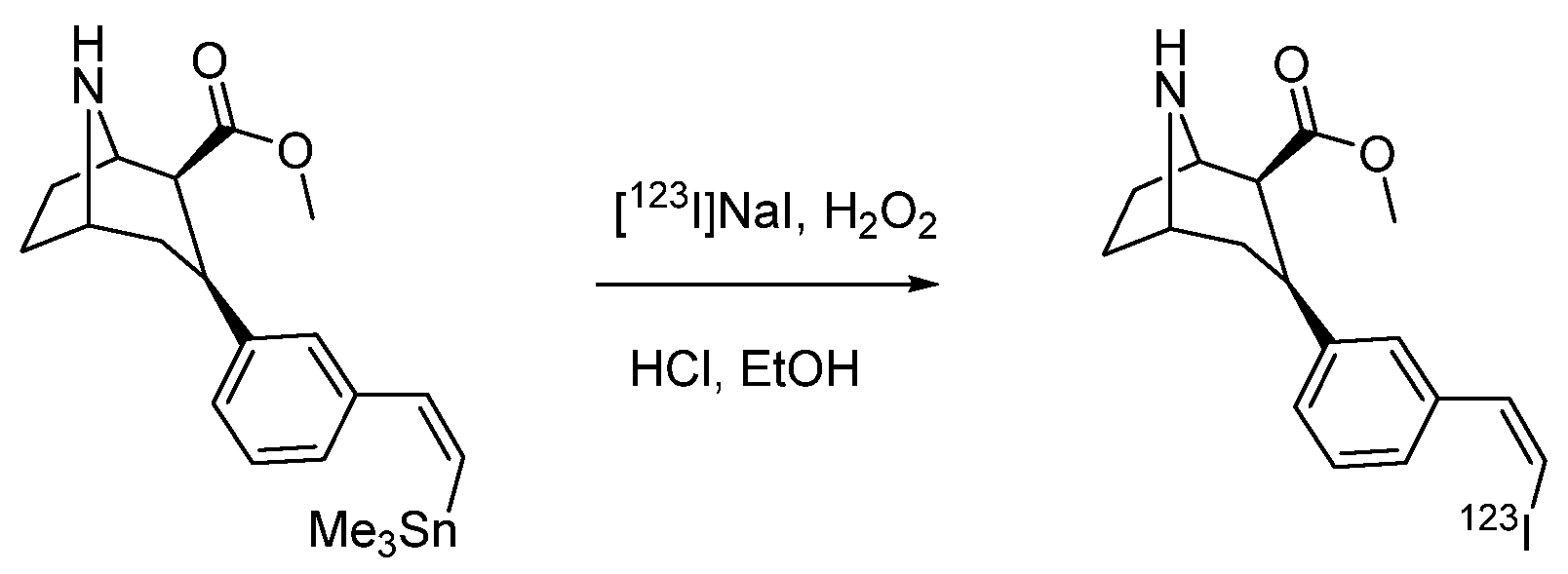{kind=link}
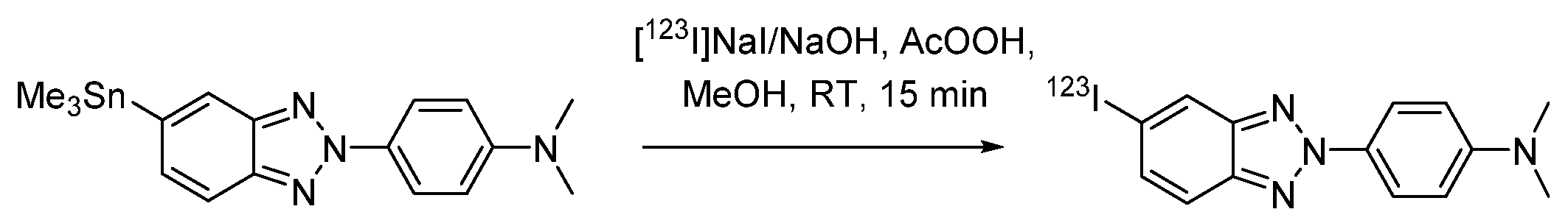{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
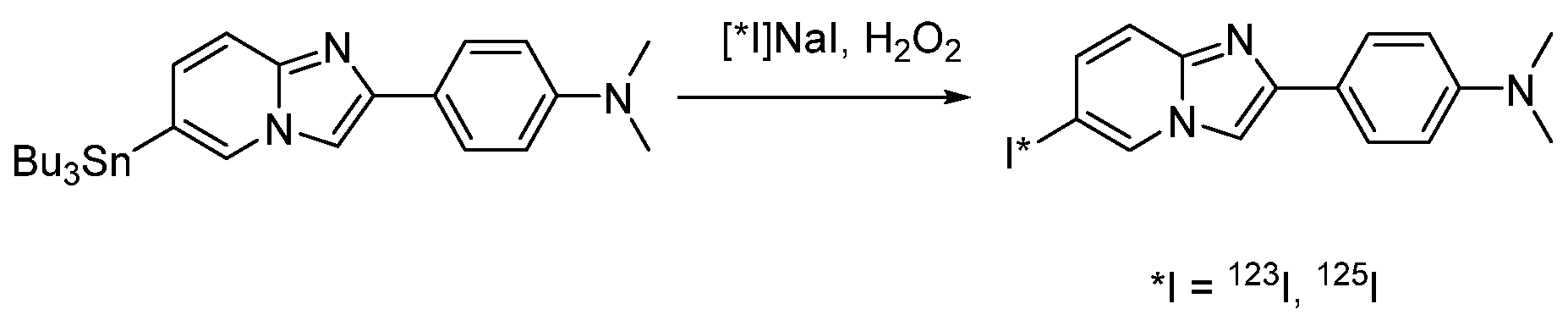{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | SPECT | PET |
|---|---|---|
| Equipment | γ-Camera | Tomograph |
| Radioisotopes | γ-Emitters | β+-emitters |
| Average procedure duration | 30–40 min | 10–20 min |
| Image reconstruction | Automatic | automatic |
| Spatial resolution | 12–16 mm | 4–6 mm |
| Sensitivity | Lower than that of PET | High |
| Multimodality | It is possible to combine SPECT-CT and SPECT-MRI | It is possible to combine PET-CT and PET-MRI |
| Availability | Wide | Less available than SPECT |
| Resulting image | 2D or 3D reconstruction | 3D |
| Quantification | Only semiquantitative assessment | Possible |
| Dose load on tissues | Lower than that of PET | Higher, but balanced by higher sensitivity |
| Radioisotope | 123I | 124I | 125I | 131I |
|---|---|---|---|---|
| Half-lifetime T1/2 | 13.22 h | 4.18 days | 59.39 days | 8.02 days |
| Decay type | EC (100%) | EC (77%) β+ (23%) | EC (100%) | β− (90%) EC (10%) |
| Energy of emitted particles * | γ (159 keV), 28.4 e−/decay (Auger electrons) | γ (603 keV) γ (511 keV upon annihilation of β+ with an electron) β+ (213.8 keV) | γ (35.5 keV), 19.5 e−/decay (Auger electrons) | γ (364 keV), β− (606 keV) |
| Production ** | Cyclotron: 124Xe(p, pn) 123Xe → 123I 124Xe(p, 2n) 123Cs→ 123Xe → 123I | Cyclotron: 124Te(p, n) → 124I | Nuclear reactor: 124Xe(n, γ) → 125mXe → 125I; 124Xe (n, γ) → 125gXe → 125I | Nuclear reactor: 131Te(n, γ) → 131I or 235U fission |
| Application | SPECT, radiotherapy | PET | Radiotherapy in vitro and in vivo experiments on small animals | Radiotherapy, SPECT |
| Ref. | [25] | [26] | [27] | [28] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrov, S.A.; Yusubov, M.S.; Beloglazkina, E.K.; Nenajdenko, V.G. Synthesis of Radioiodinated Compounds. Classical Approaches and Achievements of Recent Years. Int. J. Mol. Sci. 2022, 23, 13789. https://doi.org/10.3390/ijms232213789
Petrov SA, Yusubov MS, Beloglazkina EK, Nenajdenko VG. Synthesis of Radioiodinated Compounds. Classical Approaches and Achievements of Recent Years. International Journal of Molecular Sciences. 2022; 23(22):13789. https://doi.org/10.3390/ijms232213789
Chicago/Turabian StylePetrov, Stanislav A., Mekhman S. Yusubov, Elena K. Beloglazkina, and Valentine G. Nenajdenko. 2022. "Synthesis of Radioiodinated Compounds. Classical Approaches and Achievements of Recent Years" International Journal of Molecular Sciences 23, no. 22: 13789. https://doi.org/10.3390/ijms232213789
APA StylePetrov, S. A., Yusubov, M. S., Beloglazkina, E. K., & Nenajdenko, V. G. (2022). Synthesis of Radioiodinated Compounds. Classical Approaches and Achievements of Recent Years. International Journal of Molecular Sciences, 23(22), 13789. https://doi.org/10.3390/ijms232213789