
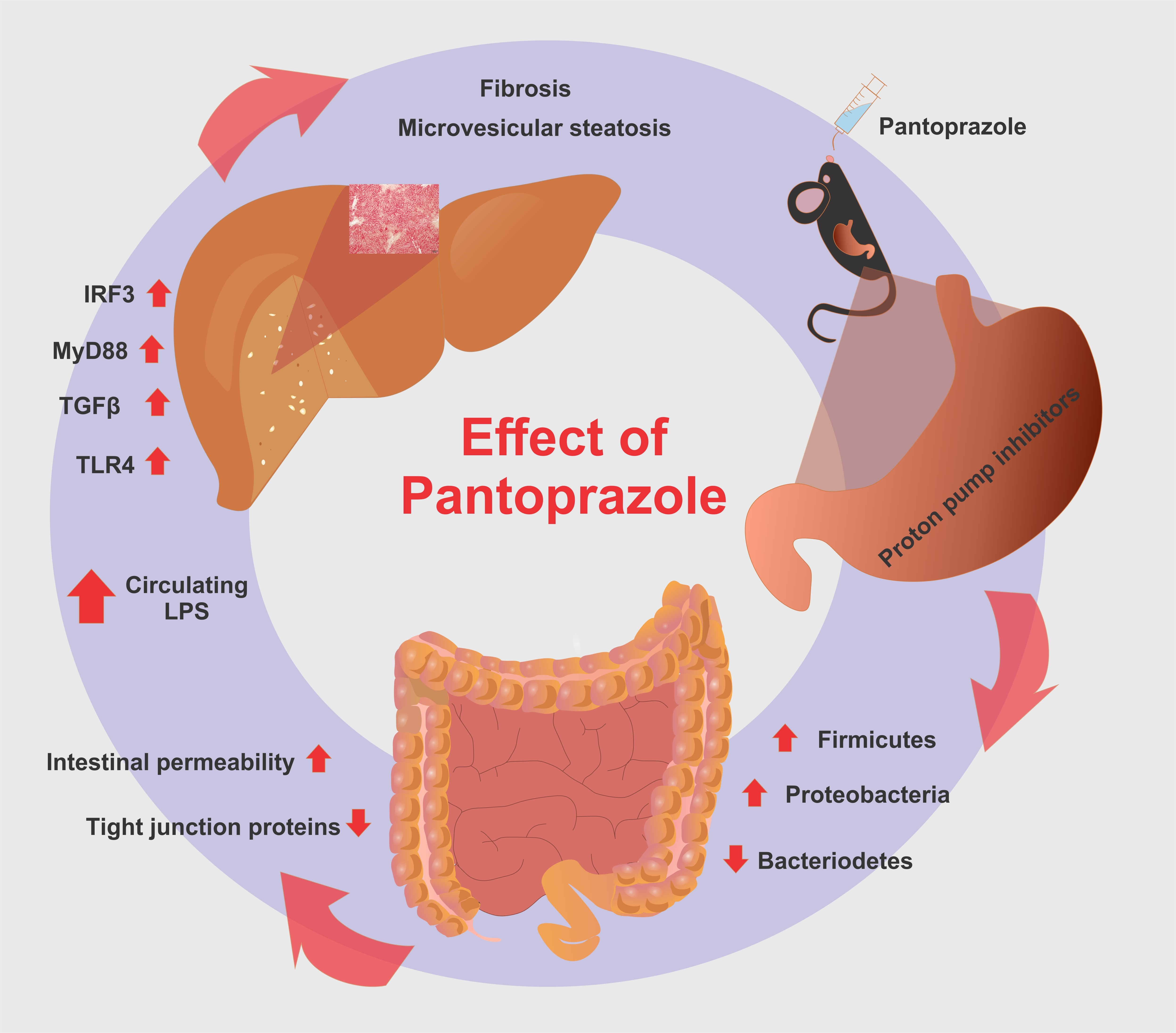
Proton Pump Inhibitor Pantoprazole Modulates Intestinal Microbiota and Induces TLR4 Signaling and Fibrosis in Mouse Liver

 , , , and
, , , and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
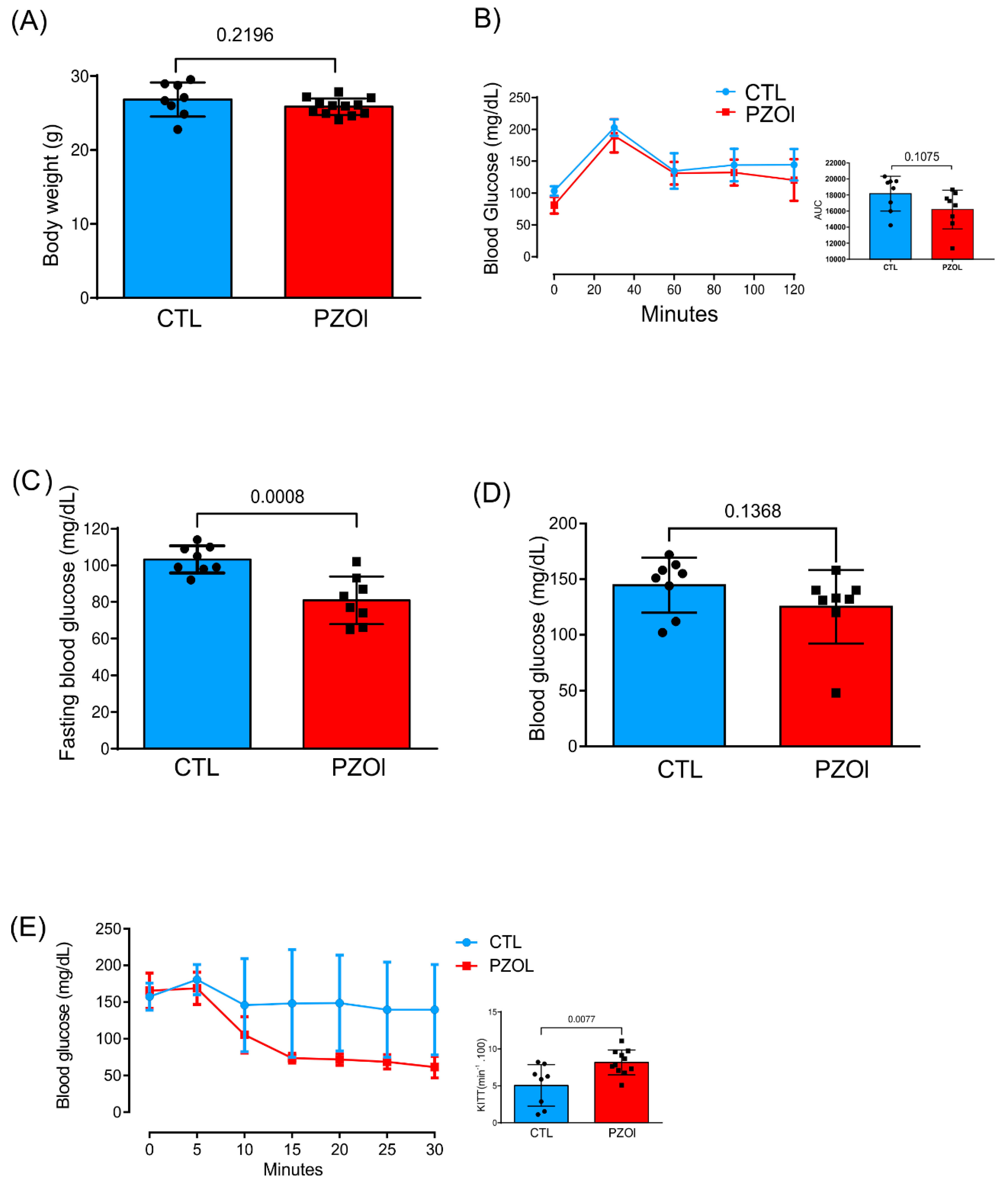
2.1. Animal Characterization
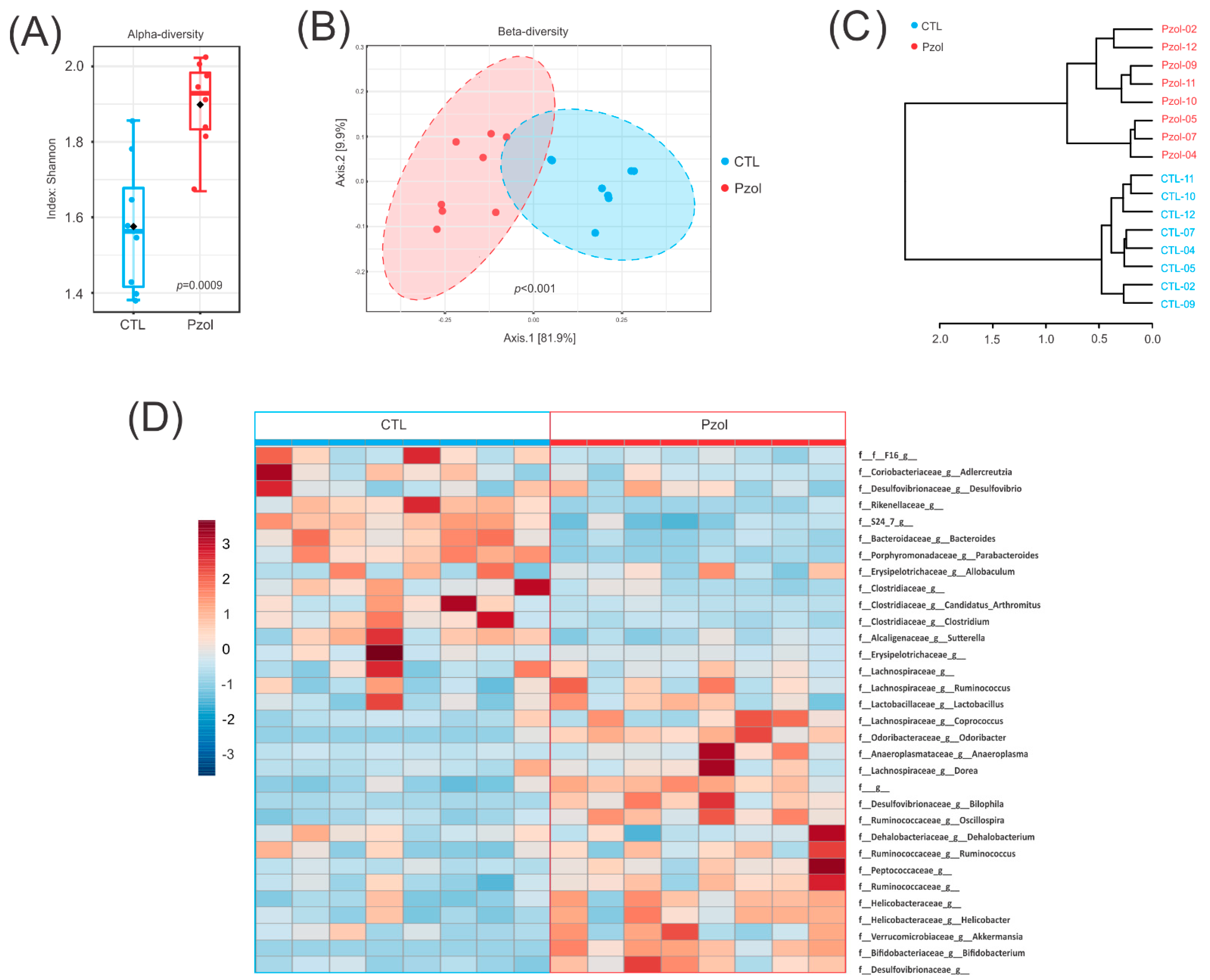
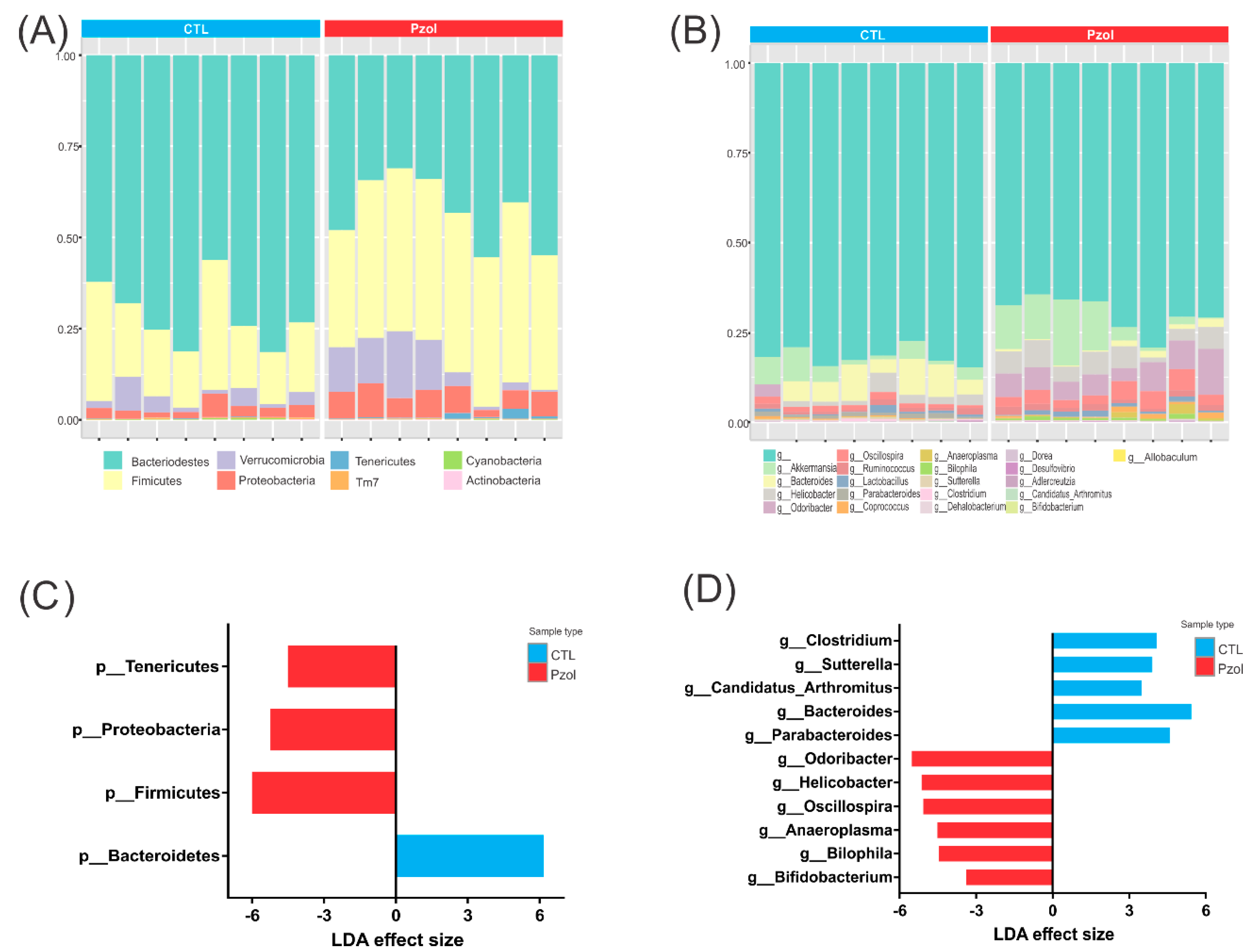
2.2. Microbiome Data Analysis after 60 Days of Pantoprazole
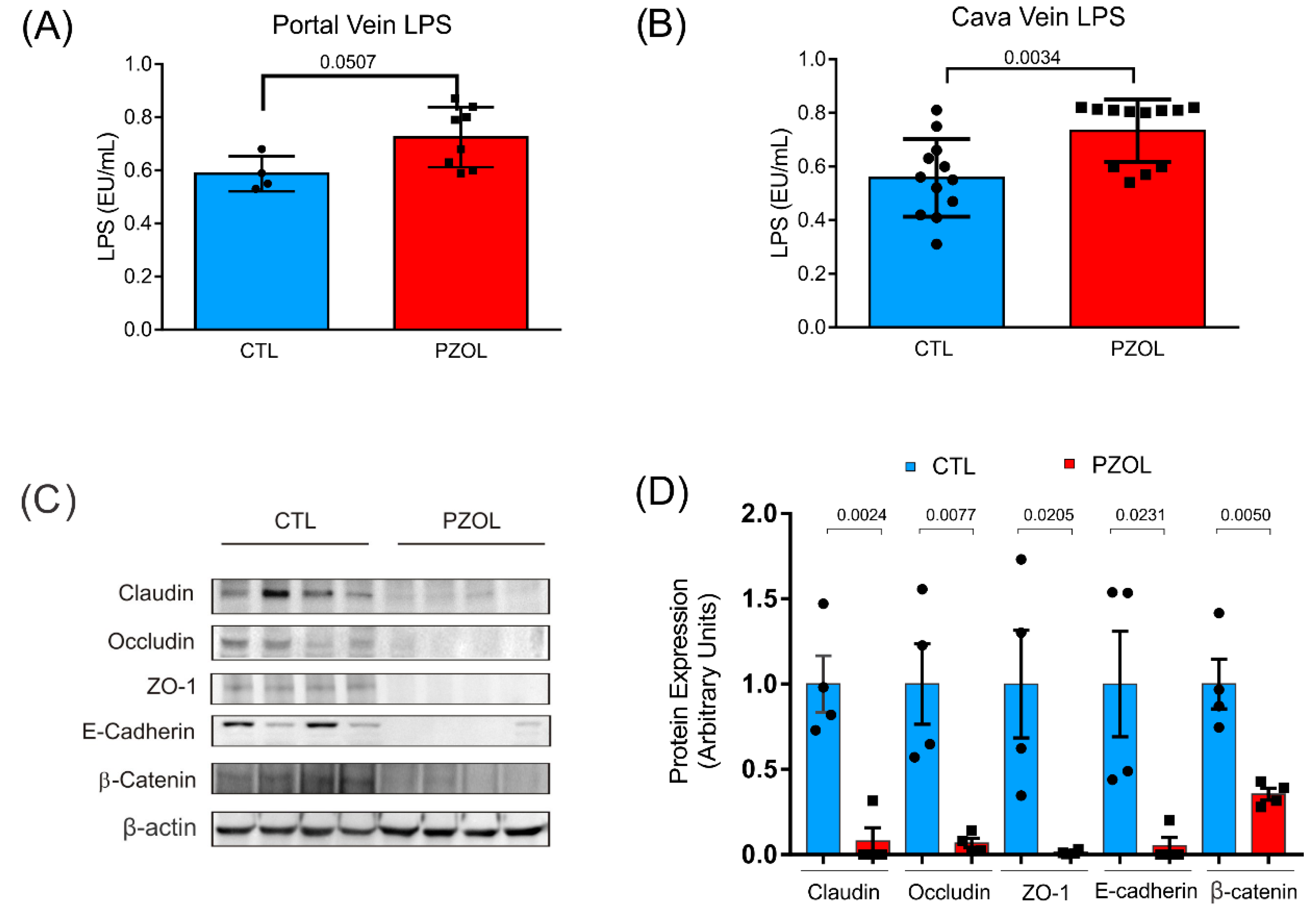
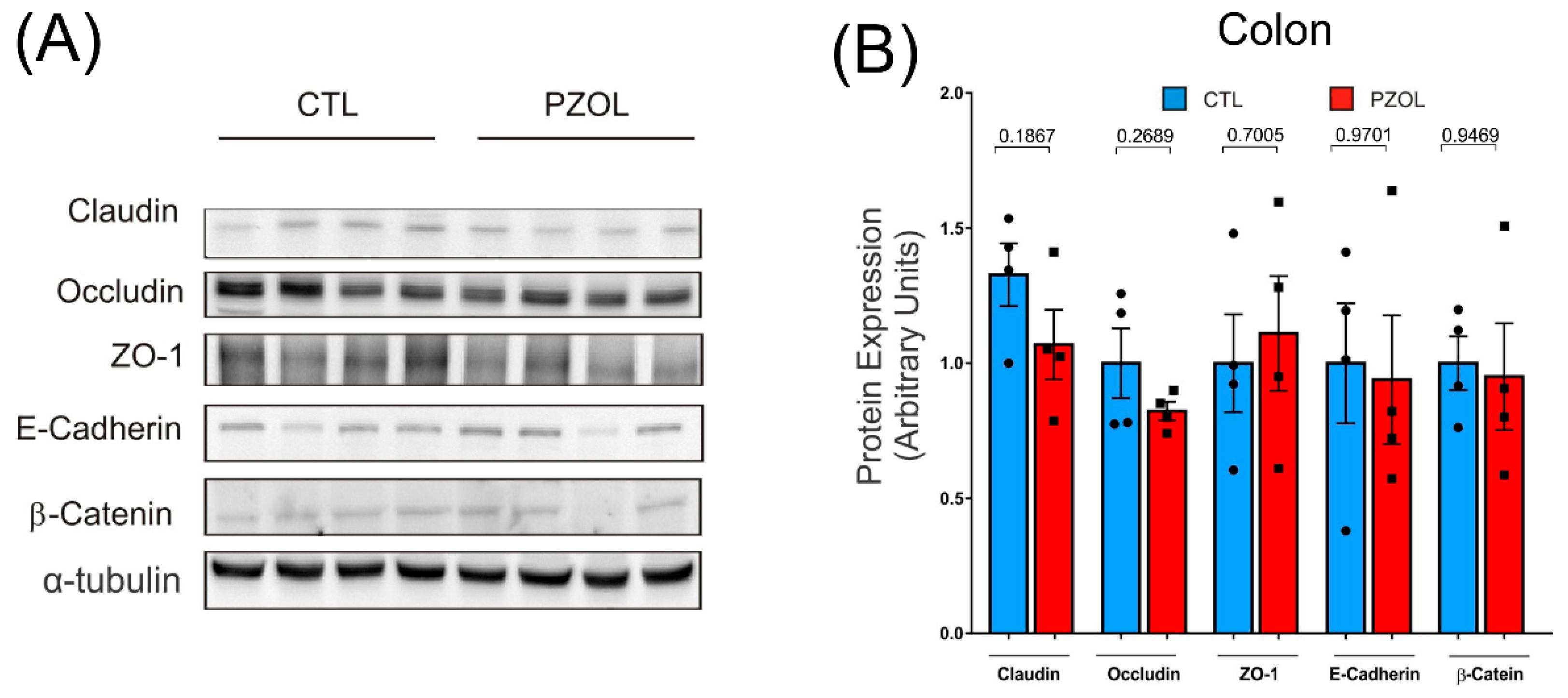
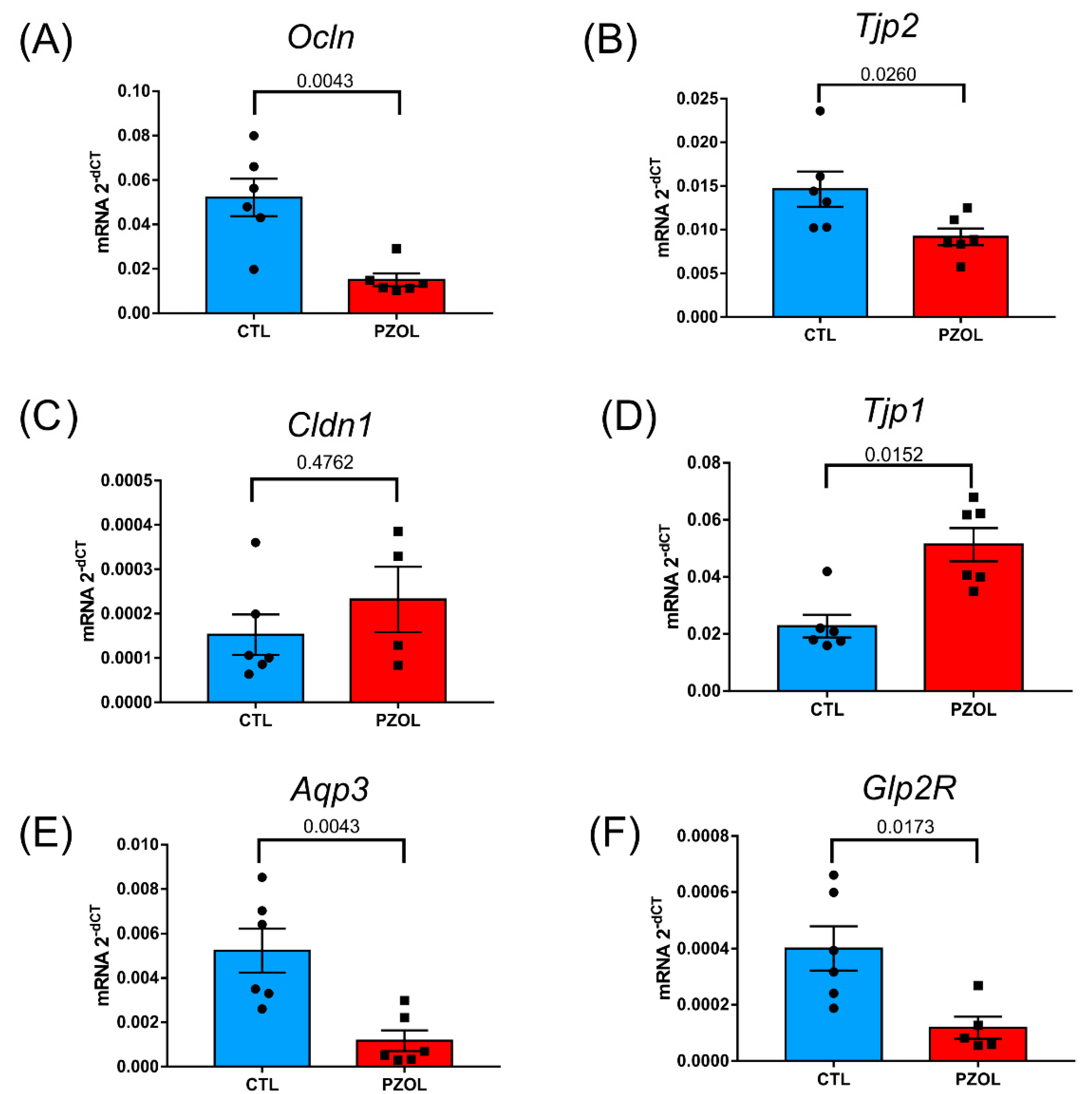
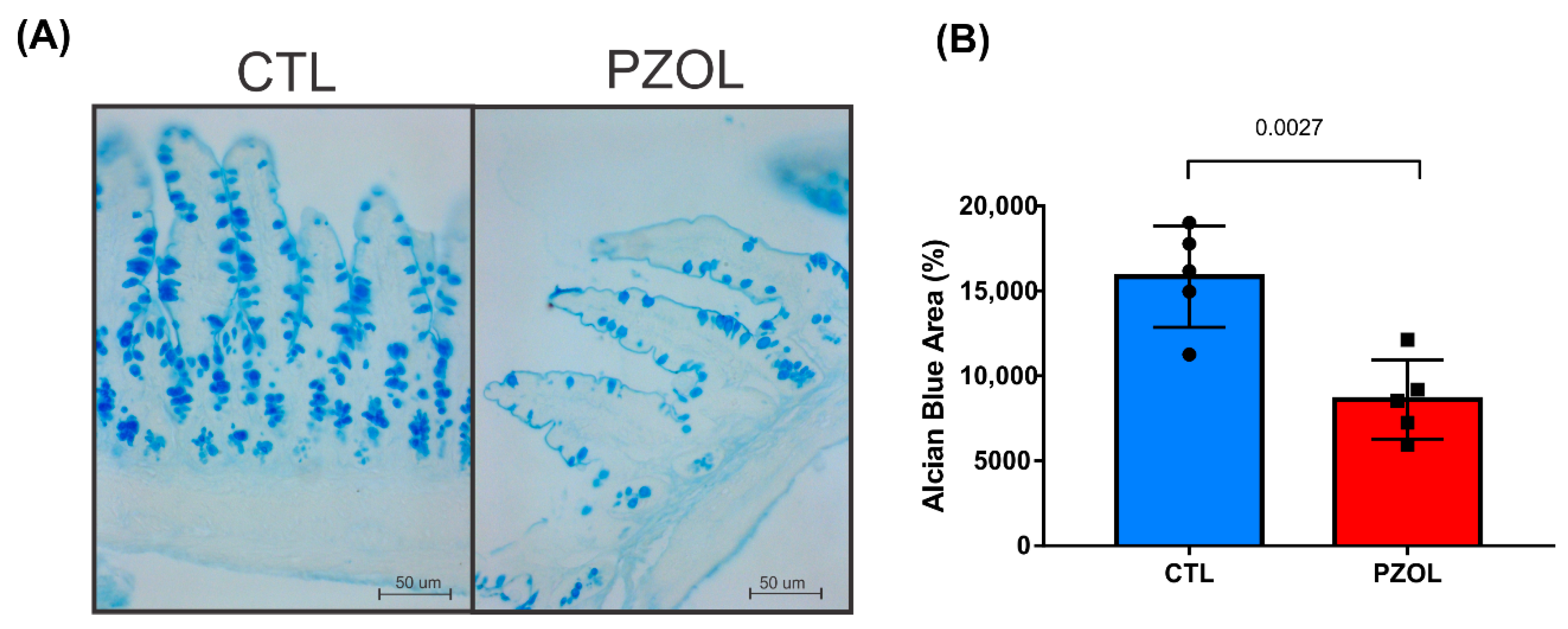
2.3. Pantoprazole-Induced Alterations in mRNA and Proteins of the Epithelial Barrier Integrity in Ileum but Not in Colon
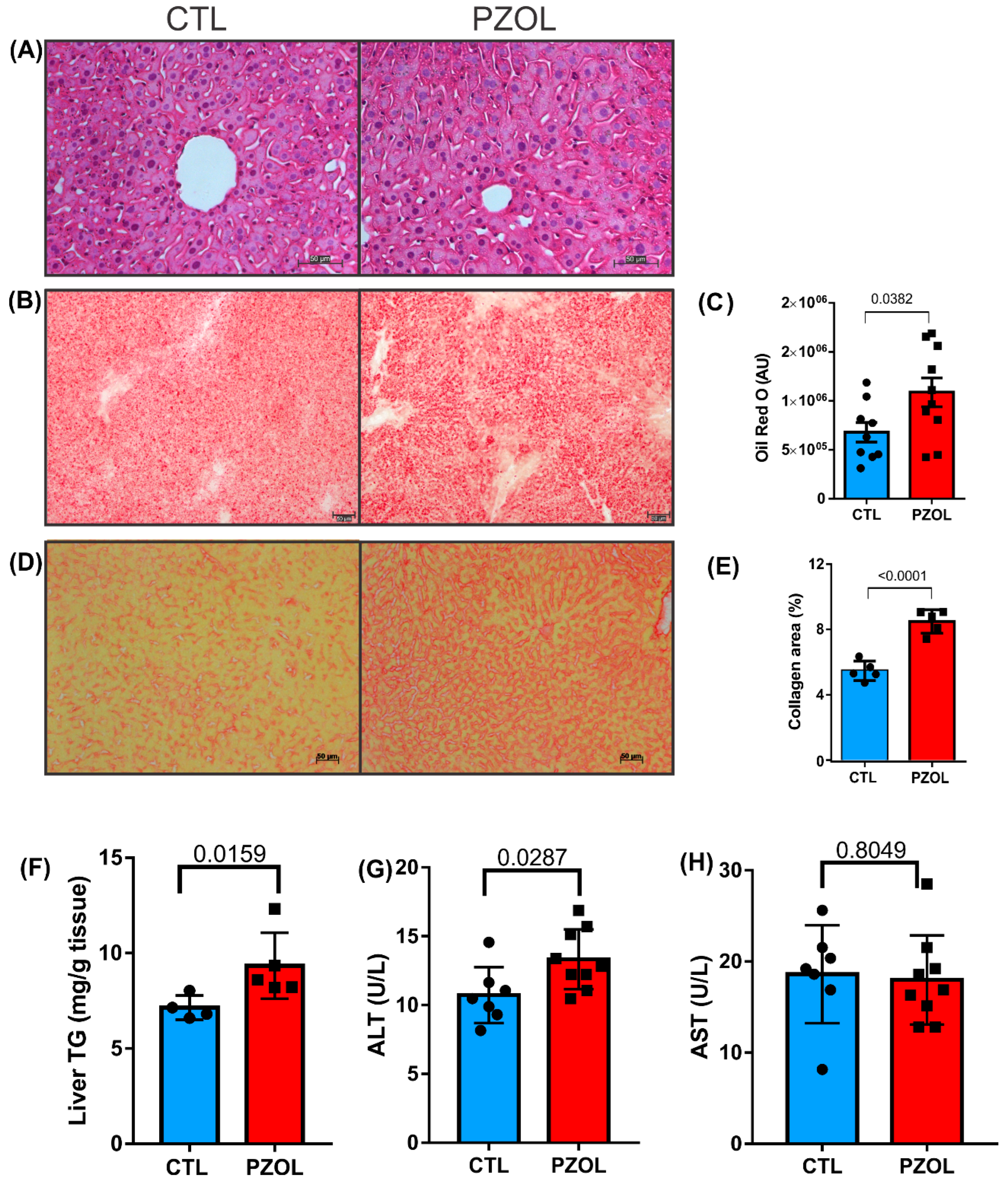
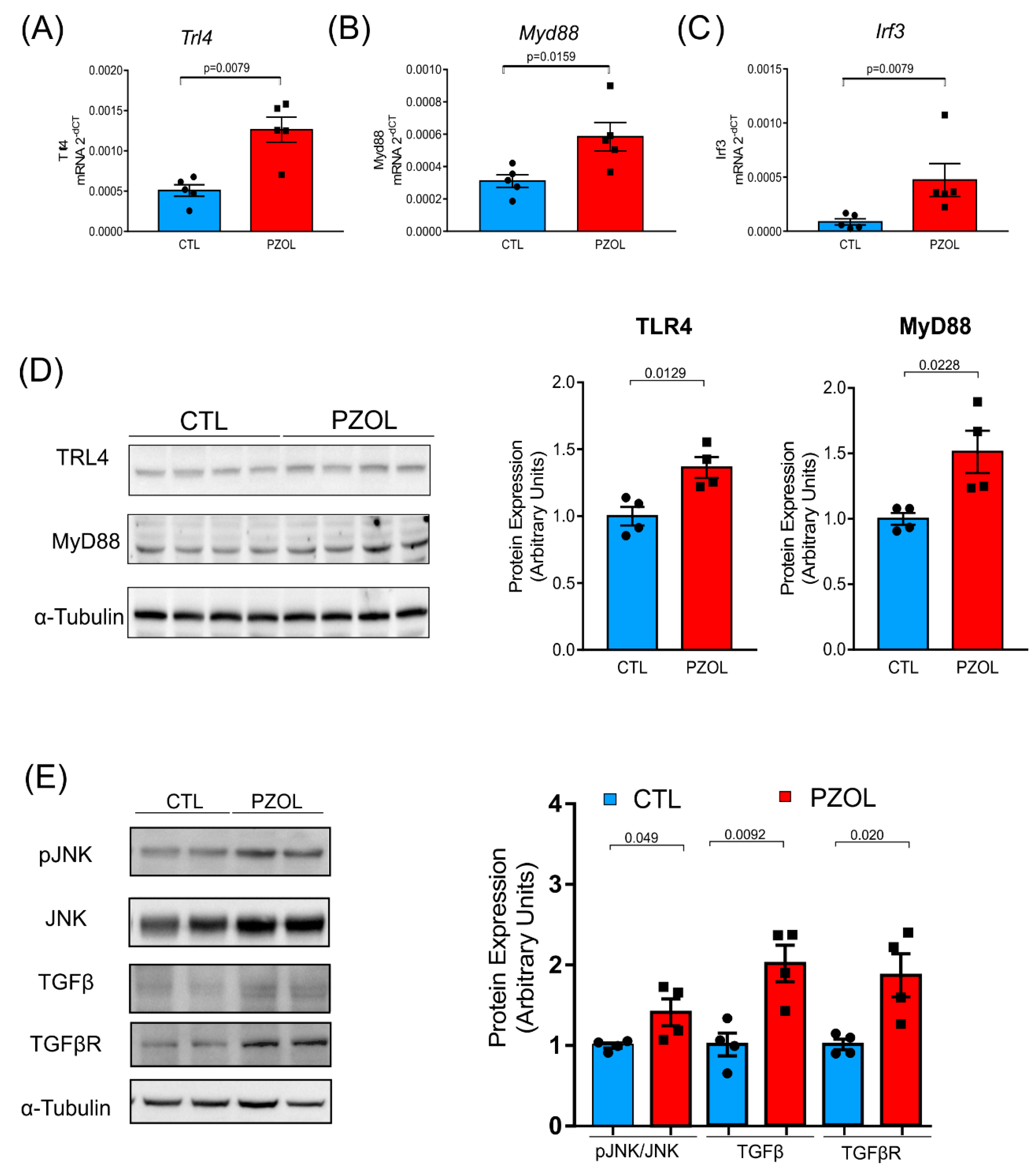
2.4. Pantoprazole-Induced Liver Fibrosis and Inflammation
3. Discussion
4. Materials and Methods
4.1. Animal Characterization
4.2. Glucose and Insulin Tolerance Test
4.3. Serum Dosage of LPS
4.4. TG, ALT, and AST Determination
4.5. Tissue Extraction for Immunoblotting
4.6. Histology and Morphometric Analysis
4.7. Microbiota Analysis
4.8. qPCR
4.9. Statistical Analyses
Suplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Scarpignato, C.; Pelosini, I.; Di Mario, F. Acid suppression therapy: Where do we go from here? Dig. Dis. 2006, 24, 11–46. [Google Scholar] [CrossRef]
- DeVault, K.R.; Castell, D.O.; Gastroenterology, A.C.o. Updated guidelines for the diagnosis and treatment of gastroesophageal reflux disease. Am. J. Gastroenterol. 2005, 100, 190–200. [Google Scholar] [CrossRef]
- Sharma, V.K.; Leontiadis, G.I.; Howden, C.W. Meta-analysis of randomized controlled trials comparing standard clinical doses of omeprazole and lansoprazole in erosive oesophagitis. Aliment. Pharmacol. Ther. 2001, 15, 227–231. [Google Scholar] [CrossRef]
- Suzuki, H.; Okada, S.; Hibi, T. Proton-pump inhibitors for the treatment of functional dyspepsia. Therap. Adv. Gastroenterol. 2011, 4, 219–226. [Google Scholar] [CrossRef]
- Bardou, M.; Toubouti, Y.; Benhaberou-Brun, D.; Rahme, E.; Barkun, A.N. Meta-analysis: Proton-pump inhibition in high-risk patients with acute peptic ulcer bleeding. Aliment. Pharmacol. Ther. 2005, 21, 677–686. [Google Scholar] [CrossRef]
- Islam, M.M.; Poly, T.N.; Walther, B.A.; Dubey, N.K.; Anggraini Ningrum, D.N.; Shabbir, S.A.; Jack Li, Y.C. Adverse outcomes of long-term use of proton pump inhibitors: A systematic review and meta-analysis. Eur. J. Gastroenterol. Hepatol. 2018, 30, 1395–1405. [Google Scholar] [CrossRef]
- de la Coba Ortiz, C.; Argüelles Arias, F.; Martín de Argila de Prados, C.; Júdez Gutiérrez, J.; Linares Rodríguez, A.; Ortega Alonso, A.; Rodríguez de Santiago, E.; Rodríguez-Téllez, M.; Vera Mendoza, M.I.; Aguilera Castro, L.; et al. Proton-pump inhibitors adverse effects: A review of the evidence and position statement by the Sociedad Española de Patología Digestiva. Rev. Esp. Enferm. Dig. 2016, 108, 207–224. [Google Scholar] [CrossRef]
- Eom, C.S.; Jeon, C.Y.; Lim, J.W.; Cho, E.G.; Park, S.M.; Lee, K.S. Use of acid-suppressive drugs and risk of pneumonia: A systematic review and meta-analysis. CMAJ 2011, 183, 310–319. [Google Scholar] [CrossRef]
- Malfertheiner, P.; Kandulski, A.; Venerito, M. Proton-pump inhibitors: Understanding the complications and risks. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 697–710. [Google Scholar] [CrossRef]
- Cheung, K.S.; Chan, E.W.; Wong, A.Y.S.; Chen, L.; Wong, I.C.K.; Leung, W.K. Long-term proton pump inhibitors and risk of gastric cancer development after treatment for. Gut 2018, 67, 28–35. [Google Scholar] [CrossRef]
- Song, H.; Zhu, J.; Lu, D. Long-term proton pump inhibitor (PPI) use and the development of gastric pre-malignant lesions. Cochrane Database Syst. Rev. 2014, 12, CD010623. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.; Kim, N.; Lee, J.; Hwang, Y.J.; Kim, H.W.; Chung, J.W.; Kim, J.W.; Lee, D.H. Associations among Gastric Juice pH, Atrophic Gastritis, Intestinal Metaplasia and. Gut Liver 2018, 12, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Correa, P. Helicobacter pylori and gastric carcinogenesis. Am. J. Surg. Pathol. 1995, 19 (Suppl. S1), S37–S43. [Google Scholar] [PubMed]
- Tsuda, A.; Suda, W.; Morita, H.; Takanashi, K.; Takagi, A.; Koga, Y.; Hattori, M. Influence of Proton-Pump Inhibitors on the Luminal Microbiota in the Gastrointestinal Tract. Clin. Transl. Gastroenterol. 2015, 6, e89. [Google Scholar] [CrossRef]
- Freedberg, D.E.; Toussaint, N.C.; Chen, S.P.; Ratner, A.J.; Whittier, S.; Wang, T.C.; Wang, H.H.; Abrams, J.A. Proton Pump Inhibitors Alter Specific Taxa in the Human Gastrointestinal Microbiome: A Crossover Trial. Gastroenterology 2015, 149, 883–885.e889. [Google Scholar] [CrossRef] [PubMed]
- Takagi, T.; Naito, Y.; Inoue, R.; Kashiwagi, S.; Uchiyama, K.; Mizushima, K.; Tsuchiya, S.; Okayama, T.; Dohi, O.; Yoshida, N.; et al. The influence of long-term use of proton pump inhibitors on the gut microbiota: An age-sex-matched case-control study. J. Clin. Biochem. Nutr. 2018, 62, 100–105. [Google Scholar] [CrossRef]
- Jandhyala, S.M.; Talukdar, R.; Subramanyam, C.; Vuyyuru, H.; Sasikala, M.; Nageshwar Reddy, D. Role of the normal gut microbiota. World. J. Gastroenterol. 2015, 21, 8787–8803. [Google Scholar] [CrossRef]
- Singh, A.; Cresci, G.A.; Kirby, D.F. Proton Pump Inhibitors: Risks and Rewards and Emerging Consequences to the Gut Microbiome. Nutr. Clin. Pract. 2018, 33, 614–624. [Google Scholar] [CrossRef]
- Minalyan, A.; Gabrielyan, L.; Scott, D.; Jacobs, J.; Pisegna, J.R. The Gastric and Intestinal Microbiome: Role of Proton Pump Inhibitors. Curr. Gastroenterol. Rep. 2017, 19, 42. [Google Scholar] [CrossRef]
- Imhann, F.; Vich Vila, A.; Bonder, M.J.; Lopez Manosalva, A.G.; Koonen, D.P.Y.; Fu, J.; Wijmenga, C.; Zhernakova, A.; Weersma, R.K. The influence of proton pump inhibitors and other commonly used medication on the gut microbiota. Gut Microbes 2017, 8, 351–358. [Google Scholar] [CrossRef]
- Bruno, G.; Zaccari, P.; Rocco, G.; Scalese, G.; Panetta, C.; Porowska, B.; Pontone, S.; Severi, C. Proton pump inhibitors and dysbiosis: Current knowledge and aspects to be clarified. World J. Gastroenterol. 2019, 25, 2706–2719. [Google Scholar] [CrossRef] [PubMed]
- Imhann, F.; Bonder, M.J.; Vich Vila, A.; Fu, J.; Mujagic, Z.; Vork, L.; Tigchelaar, E.F.; Jankipersadsing, S.A.; Cenit, M.C.; Harmsen, H.J.; et al. Proton pump inhibitors affect the gut microbiome. Gut 2016, 65, 740–748. [Google Scholar] [CrossRef] [PubMed]
- Saad, M.J.; Santos, A.; Prada, P.O. Linking Gut Microbiota and Inflammation to Obesity and Insulin Resistance. Physiology 2016, 31, 283–293. [Google Scholar] [CrossRef]
- Tsukumo, D.M.; Carvalho, B.M.; Carvalho Filho, M.A.; Saad, M.J. Translational research into gut microbiota: New horizons on obesity treatment: Updated 2014. Arch. Endocrinol. Metab. 2015, 59, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, B.M.; Saad, M.J. Influence of gut microbiota on subclinical inflammation and insulin resistance. Mediators Inflamm. 2013, 2013, 986734. [Google Scholar] [CrossRef]
- Jiao, N.; Baker, S.S.; Nugent, C.A.; Tsompana, M.; Cai, L.; Wang, Y.; Buck, M.J.; Genco, R.J.; Baker, R.D.; Zhu, R.; et al. Gut microbiome may contribute to insulin resistance and systemic inflammation in obese rodents: A meta-analysis. Physiol. Genomics 2018, 50, 244–254. [Google Scholar] [CrossRef]
- Zhi, C.; Huang, J.; Wang, J.; Cao, H.; Bai, Y.; Guo, J.; Su, Z. Connection between gut microbiome and the development of obesity. Eur. J. Clin. Microbiol. Infect. Dis. 2019, 38, 1987–1998. [Google Scholar] [CrossRef]
- Michelotti, G.A.; Machado, M.V.; Diehl, A.M. NAFLD, NASH and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 656–665. [Google Scholar] [CrossRef]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Kolodziejczyk, A.A.; Zheng, D.; Shibolet, O.; Elinav, E. The role of the microbiome in NAFLD and NASH. EMBO Mol. Med. 2019, 11, e9302. [Google Scholar] [CrossRef] [PubMed]
- Pyo, J.H.; Kim, T.J.; Lee, H.; Choi, S.C.; Cho, S.J.; Choi, Y.H.; Min, Y.W.; Min, B.H.; Lee, J.H.; Kang, M.; et al. Proton pump inhibitors use and the risk of fatty liver disease: A nationwide cohort study. J. Gastroenterol. Hepatol. 2021, 36, 1235–1243. [Google Scholar] [CrossRef] [PubMed]
- Llorente, C.; Jepsen, P.; Inamine, T.; Wang, L.; Bluemel, S.; Wang, H.J.; Loomba, R.; Bajaj, J.S.; Schubert, M.L.; Sikaroodi, M.; et al. Gastric acid suppression promotes alcoholic liver disease by inducing overgrowth of intestinal Enterococcus. Nat. Commun. 2017, 8, 837. [Google Scholar] [CrossRef]
- Lee, S.M.; Kim, N.; Nam, R.H.; Park, J.H.; Choi, S.I.; Park, Y.T.; Kim, Y.R.; Seok, Y.J.; Shin, C.M.; Lee, D.H. Gut microbiota and butyrate level changes associated with the long-term administration of proton pump inhibitors to old rats. Sci. Rep. 2019, 9, 6626. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Takashima, S.; Tanaka, F.; Kawaguchi, Y.; Usui, Y.; Fujimoto, K.; Nadatani, Y.; Otani, K.; Hosomi, S.; Nagami, Y.; Kamata, N.; et al. Proton pump inhibitors enhance intestinal permeability via dysbiosis of gut microbiota under stressed conditions in mice. Neurogastroenterol. Motil. 2020, 32, e13841. [Google Scholar] [CrossRef]
- Jackson, M.A.; Goodrich, J.K.; Maxan, M.E.; Freedberg, D.E.; Abrams, J.A.; Poole, A.C.; Sutter, J.L.; Welter, D.; Ley, R.E.; Bell, J.T.; et al. Proton pump inhibitors alter the composition of the gut microbiota. Gut 2016, 65, 749–756. [Google Scholar] [CrossRef]
- Amir, I.; Konikoff, F.M.; Oppenheim, M.; Gophna, U.; Half, E.E. Gastric microbiota is altered in oesophagitis and Barrett’s oesophagus and further modified by proton pump inhibitors. Environ. Microbiol. 2014, 16, 2905–2914. [Google Scholar] [CrossRef]
- Natividad, J.M.; Lamas, B.; Pham, H.P.; Michel, M.L.; Rainteau, D.; Bridonneau, C.; da Costa, G.; van Hylckama Vlieg, J.; Sovran, B.; Chamignon, C.; et al. Bilophila wadsworthia aggravates high fat diet induced metabolic dysfunctions in mice. Nat. Commun. 2018, 9, 2802. [Google Scholar] [CrossRef]
- Chen, C.; Zhang, C.; Wang, X.; Zhang, F.; Zhang, Z.; Ma, P.; Feng, S. Helicobacter pylori infection may increase the severity of nonalcoholic fatty liver disease via promoting liver function damage, glycometabolism, lipid metabolism, inflammatory reaction and metabolic syndrome. Eur. J. Gastroenterol. Hepatol. 2020, 32, 857–866. [Google Scholar] [CrossRef]
- Ierardi, E.; Losurdo, G.; Fortezza, R.F.; Principi, M.; Barone, M.; Leo, A.D. Optimizing proton pump inhibitors in. World J. Gastroenterol. 2019, 25, 5097–5104. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.C.; Tu, Y.K.; Lee, G.Y.; Chang, R.H.; Huang, Y.; Bukhari, K.; Tsai, Y.C.; Fu, Y.; Huang, H.K.; Munir, K.M. Effects of Proton Pump Inhibitors on Glycemic Control and Incident Diabetes: A Systematic Review and Meta-Analysis. J. Clin. Endocrinol. Metab. 2021, 106, 3354–3366. [Google Scholar] [CrossRef]
- Singh, P.; Indaram, A.; Greenberg, R.; Visvalingam, V.; Bank, S. Long term omeprazole therapy for reflux esophagitis:follow-up in serum gastrin levels, EC cell hyperplasia and neoplasia. World J. Gastroenterol. 2000, 6, 789–792. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Pinzon, W.L.; Lakey, J.R.; Rabinovitch, A. Combination therapy with glucagon-like peptide-1 and gastrin induces beta-cell neogenesis from pancreatic duct cells in human islets transplanted in immunodeficient diabetic mice. Cell Transplant. 2008, 17, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Ajdić, D.; McShan, W.M.; McLaughlin, R.E.; Savić, G.; Chang, J.; Carson, M.B.; Primeaux, C.; Tian, R.; Kenton, S.; Jia, H.; et al. Genome sequence of Streptococcus mutans UA159, a cariogenic dental pathogen. Proc. Natl. Acad. Sci. USA 2002, 99, 14434–14439. [Google Scholar] [CrossRef] [PubMed]
- Zihni, C.; Mills, C.; Matter, K.; Balda, M.S. Tight junctions: From simple barriers to multifunctional molecular gates. Nat. Rev. Mol. Cell Biol. 2016, 17, 564–580. [Google Scholar] [CrossRef]
- Cazzo, E.; Gestic, M.A.; Utrini, M.P.; Chaim, F.D.; Geloneze, B.; Pareja, J.C.; Chaim, E.A.; Magro, D.O. GLP-2: A poorly understood mediator enrolled in various bariatric/metabolic surgery-related pathophysiologic mechanisms. Arq. Bras. Cir. Dig. 2016, 29, 272–275. [Google Scholar] [CrossRef]
- Covasa, M.; Stephens, R.W.; Toderean, R.; Cobuz, C. Intestinal Sensing by Gut Microbiota: Targeting Gut Peptides. Front. Endocrinol. 2019, 10, 82. [Google Scholar] [CrossRef]
- Tandra, S.; Yeh, M.M.; Brunt, E.M.; Vuppalanchi, R.; Cummings, O.W.; Ünalp-Arida, A.; Wilson, L.A.; Chalasani, N. Presence and significance of microvesicular steatosis in nonalcoholic fatty liver disease. J. Hepatol. 2011, 55, 654–659. [Google Scholar] [CrossRef]
- Henderson, N.C.; Rieder, F.; Wynn, T.A. Fibrosis: From mechanisms to medicines. Nature 2020, 587, 555–566. [Google Scholar] [CrossRef]
- Seki, E.; De Minicis, S.; Osterreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef] [PubMed]
- Paik, Y.H.; Schwabe, R.F.; Bataller, R.; Russo, M.P.; Jobin, C.; Brenner, D.A. Toll-like receptor 4 mediates inflammatory signaling by bacterial lipopolysaccharide in human hepatic stellate cells. Hepatology 2003, 37, 1043–1055. [Google Scholar] [CrossRef]
- Sharifnia, T.; Antoun, J.; Verriere, T.G.; Suarez, G.; Wattacheril, J.; Wilson, K.T.; Peek, R.M.; Abumrad, N.N.; Flynn, C.R. Hepatic TLR4 signaling in obese NAFLD. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G270–G278. [Google Scholar] [CrossRef] [PubMed]
- Bezerra, R.M.; Ueno, M.; Silva, M.S.; Tavares, D.Q.; Carvalho, C.R.; Saad, M.J.; Gontijo, J.A. A high-fructose diet induces insulin resistance but not blood pressure changes in normotensive rats. Braz. J. Med. Biol. Res. 2001, 34, 1155–1160. [Google Scholar] [CrossRef] [PubMed]
- Guadagnini, D.; Rocha, G.Z.; Santos, A.; Assalin, H.B.; Hirabara, S.M.; Curi, R.; Oliveira, A.G.; Prada, P.O.; Saad, M.J.A. Microbiota determines insulin sensitivity in TLR2-KO mice. Life Sci. 2019, 234, 116793. [Google Scholar] [CrossRef]
- de Souza, D.N.; Teixeira, C.J.; Veronesi, V.B.; Murata, G.M.; Santos-Silva, J.C.; Hecht, F.B.; Vicente, J.M.; Bordin, S.; Anhê, G.F. Dexamethasone programs lower fatty acid absorption and reduced PPAR-γ and fat/CD36 expression in the jejunum of the adult rat offspring. Life Sci. 2021, 265, 118765. [Google Scholar] [CrossRef]
- Hecht, F.B.; Teixeira, C.J.; de Souza, D.N.; Mesquita, F.P.N.; Roso, R.E.D.V.; Sodré, F.S.; Veronesi, V.B.; da Rocha, D.F.; Menezes, Y.G.D.; Pioli, M.R.; et al. Antenatal corticosteroid therapy modulates hepatic AMPK phosphorylation and maternal lipid metabolism in early lactating rats. Biomed. Pharmacother. 2021, 144, 112355. [Google Scholar] [CrossRef]
- Tobar, N.; Oliveira, A.G.; Guadagnini, D.; Bagarolli, R.A.; Rocha, G.Z.; Araújo, T.G.; Santos-Silva, J.C.; Zollner, R.L.; Boechat, L.H.; Carvalheira, J.B.; et al. Diacerhein improves glucose tolerance and insulin sensitivity in mice on a high-fat diet. Endocrinology 2011, 152, 4080–4093. [Google Scholar] [CrossRef]
- Veronesi, V.B.; Pioli, M.R.; de Souza, D.N.; Teixeira, C.J.; Murata, G.M.; Santos-Silva, J.C.; Hecht, F.B.; Vicente, J.M.; Bordin, S.; Anhê, G.F. Agomelatine reduces circulating triacylglycerides and hepatic steatosis in fructose-treated rats. Biomed. Pharmacother. 2021, 141, 111807. [Google Scholar] [CrossRef]
- Vicente, J.M.; Teixeira, C.J.; Santos-Silva, J.C.; de Souza, D.N.; Tobar, N.; Furtuoso, F.S.; Adabo, I.G.; Sodré, F.S.; Murata, G.; Bordin, S.; et al. The absence of lactation after pregnancy induces long-term lipid accumulation in maternal liver of mice. Life Sci. 2019, 217, 261–270. [Google Scholar] [CrossRef]
- Bialkowska, A.B.; Ghaleb, A.M.; Nandan, M.O.; Yang, V.W. Improved Swiss-rolling Technique for Intestinal Tissue Preparation for Immunohistochemical and Immunofluorescent Analyses. J. Vis. Exp. 2016, 113, e54161. [Google Scholar] [CrossRef] [PubMed]
- Illumina 16S metagenomic sequencing library preparation (Illumina Technical Note 15044223). Illumina. 2014. Available online: https://support.illumina.com/downloads/16s_metagenomic_sequencing_library_preparation.html (accessed on 19 June 2018).
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [PubMed]
- Alishum, A. DADA2 formatted 16S rRNA gene sequences for both bacteria & archaea. Res. Data 2019. [Google Scholar] [CrossRef]
- Dhariwal, A.; Chong, J.; Habib, S.; King, I.L.; Agellon, L.B.; Xia, J. MicrobiomeAnalyst: A web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 2017, 45, W180–W188. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.; Liu, P.; Zhou, G.; Xia, J. Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protoc. 2020, 15, 799–821. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Assalin, H.B.; De Almeida, K.C.G.; Guadagnini, D.; Santos, A.; Teixeira, C.J.; Bordin, S.; Rocha, G.Z.; Saad, M.J.A. Proton Pump Inhibitor Pantoprazole Modulates Intestinal Microbiota and Induces TLR4 Signaling and Fibrosis in Mouse Liver. Int. J. Mol. Sci. 2022, 23, 13766. https://doi.org/10.3390/ijms232213766
Assalin HB, De Almeida KCG, Guadagnini D, Santos A, Teixeira CJ, Bordin S, Rocha GZ, Saad MJA. Proton Pump Inhibitor Pantoprazole Modulates Intestinal Microbiota and Induces TLR4 Signaling and Fibrosis in Mouse Liver. International Journal of Molecular Sciences. 2022; 23(22):13766. https://doi.org/10.3390/ijms232213766
Chicago/Turabian StyleAssalin, Heloisa B., Kelly Cristiane Gabriel De Almeida, Dioze Guadagnini, Andrey Santos, Caio J. Teixeira, Silvana Bordin, Guilherme Z. Rocha, and Mario J. A. Saad. 2022. "Proton Pump Inhibitor Pantoprazole Modulates Intestinal Microbiota and Induces TLR4 Signaling and Fibrosis in Mouse Liver" International Journal of Molecular Sciences 23, no. 22: 13766. https://doi.org/10.3390/ijms232213766
APA StyleAssalin, H. B., De Almeida, K. C. G., Guadagnini, D., Santos, A., Teixeira, C. J., Bordin, S., Rocha, G. Z., & Saad, M. J. A. (2022). Proton Pump Inhibitor Pantoprazole Modulates Intestinal Microbiota and Induces TLR4 Signaling and Fibrosis in Mouse Liver. International Journal of Molecular Sciences, 23(22), 13766. https://doi.org/10.3390/ijms232213766