
Influence of the Microbiome Metagenomics and Epigenomics on Gastric Cancer

 , , , ,
, , , ,
Abstract
1. Introduction
2. Gastric Cancer Subtypes
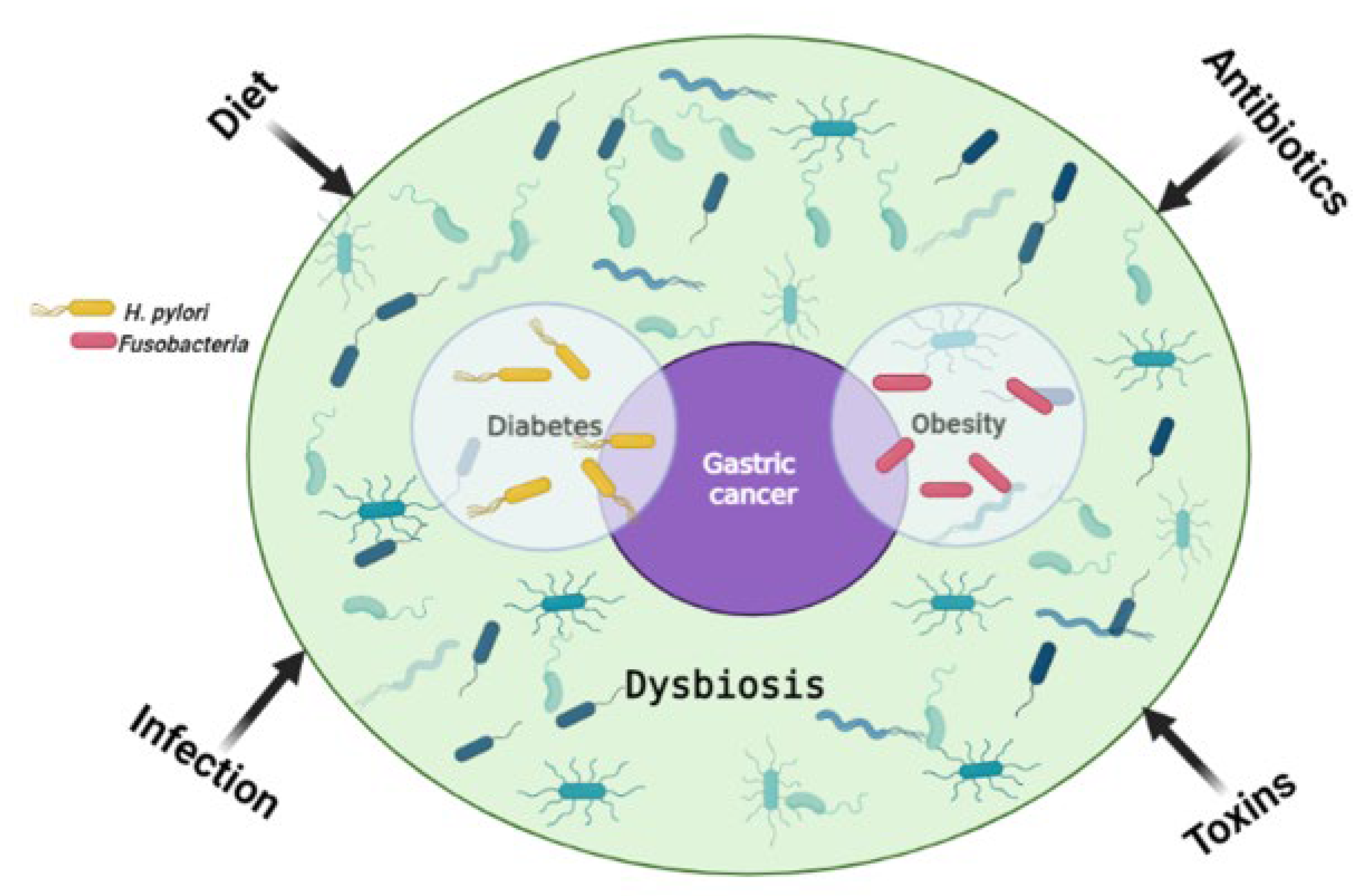
3. The Link between Gut Microbiome and Gastric Cancer Risk Factors
3.1. Obesity
3.2. Diabetes
3.3. Acid Reflux-Related Disorders
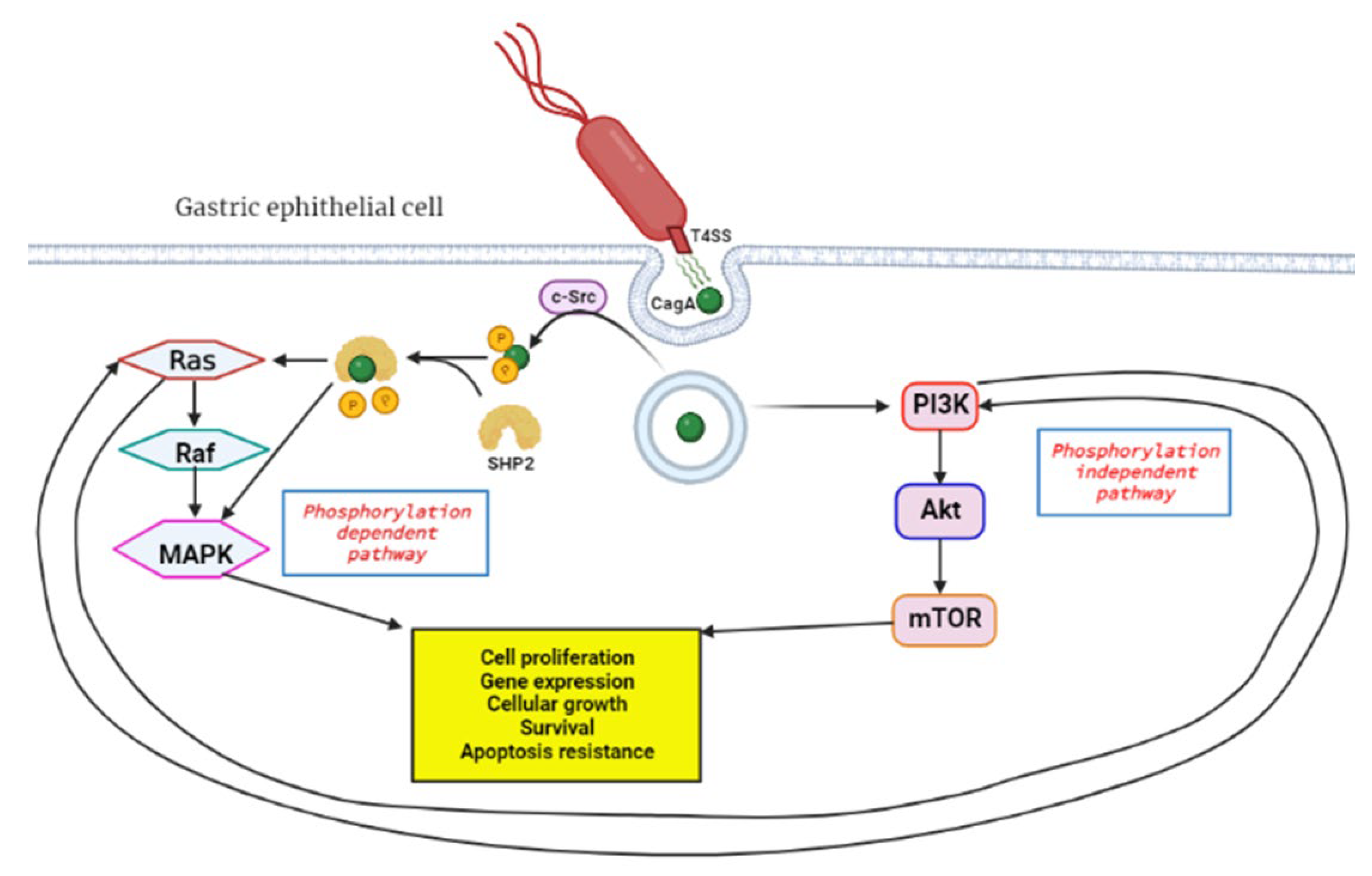
3.4. Chronic Infection and Inflammation
4. Other Microbes Implicated in GC Pathogenesis
4.1. The Boas-Oppler Bacillus
4.2. Mycoplasma
5. Compounds Linked with GC Induction
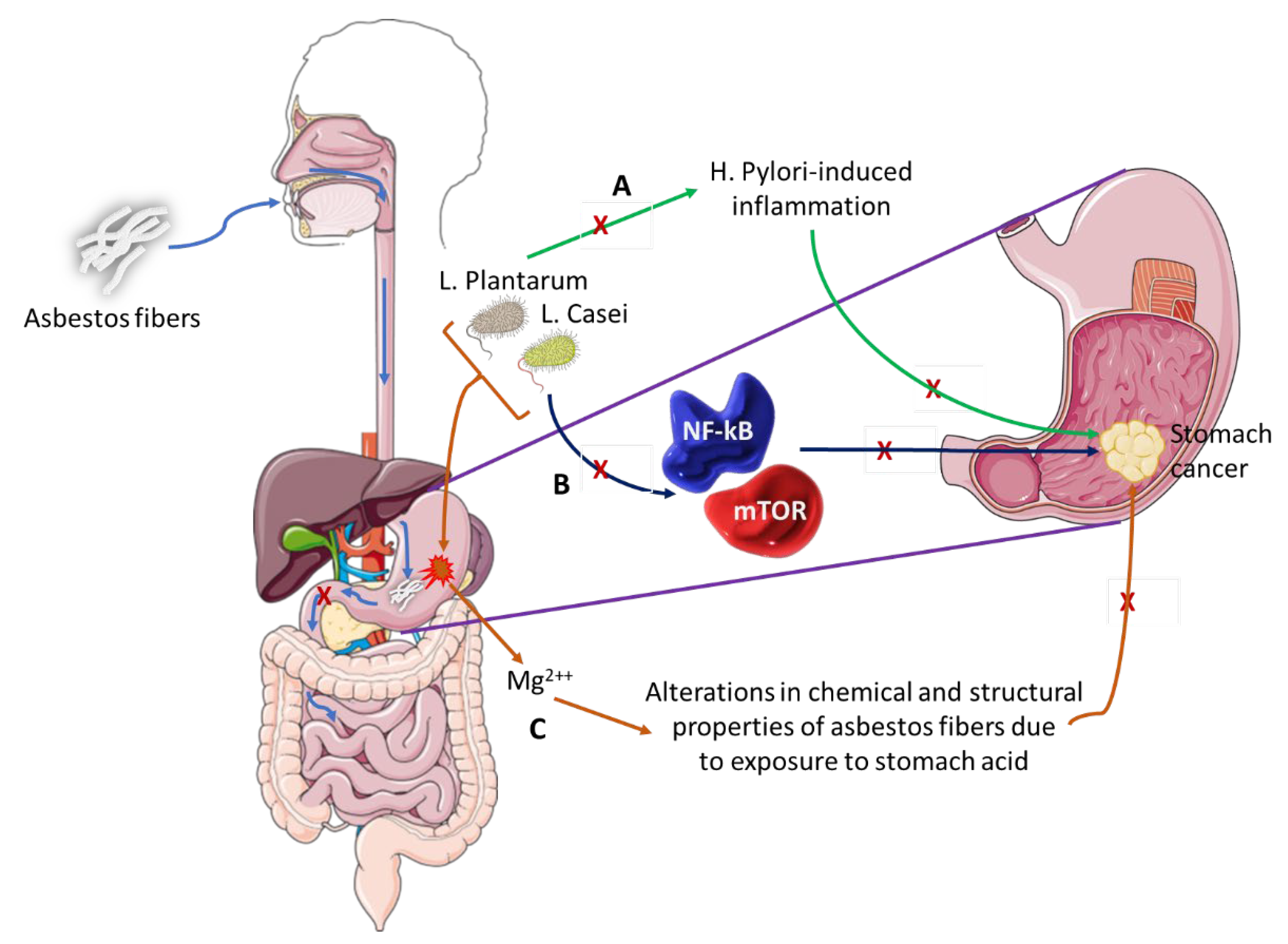
5.1. Contribution of Microbes in Asbestos-Induced GC
- (A)
- L. Plantarum has the ability to block H. Pylori-induced inflammation that is associated with GC development.
- (B)
- L. Casei bacterium downregulates pro-oncogenic signaling pathways (NF-kB and mTOR) thus inhibiting cancer development and progression.
- (C)
- This pair of bacteria can alter the chemical and structural properties of white asbestos by the removal of magnesium ions, a process that could be explored as preventative therapy in individuals exposed to asbestos fibers or as therapeutic intervention in asbestos-induced GC.
5.2. Enterobacteriaceae and Nitrosamines Production
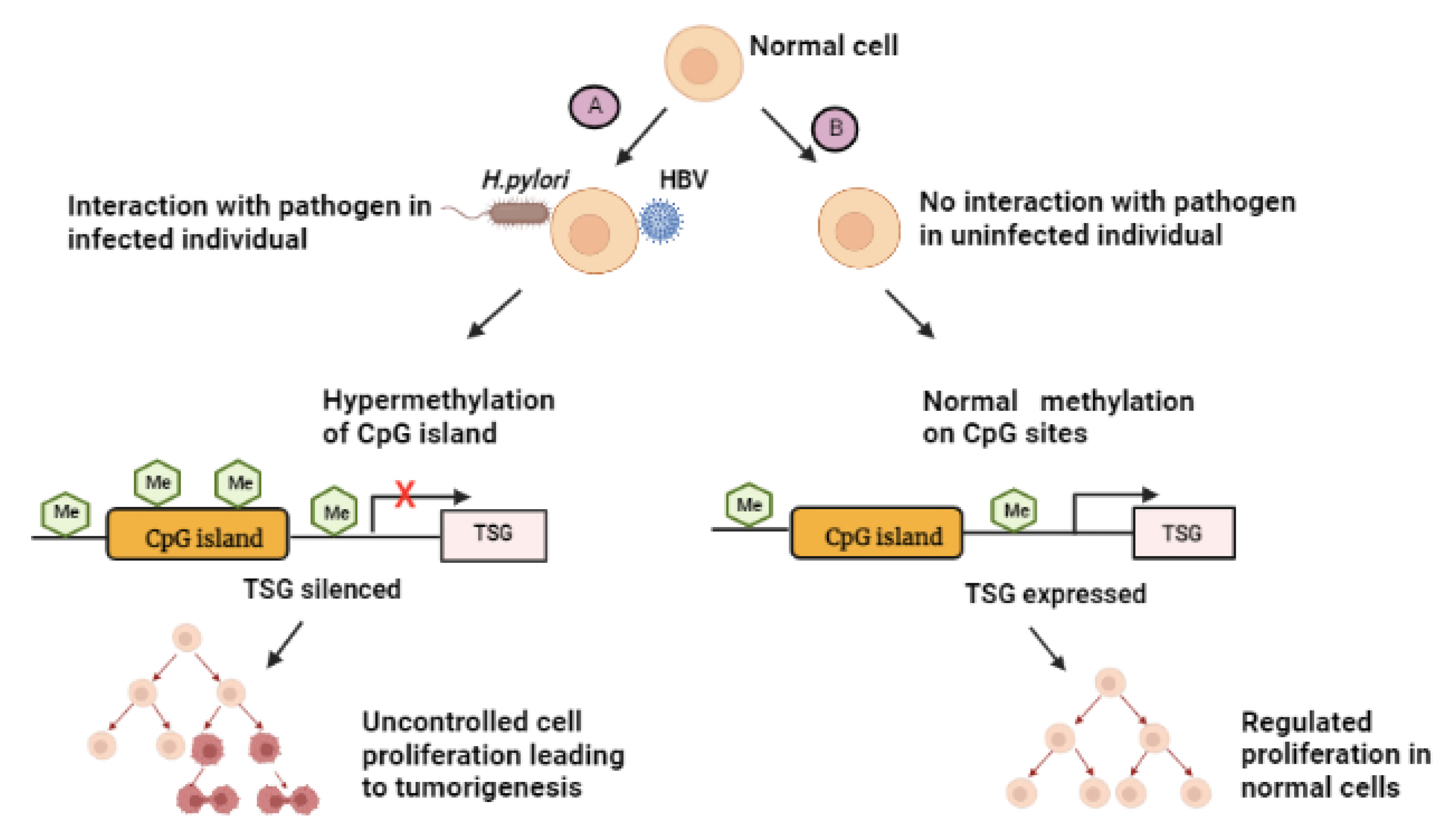
6. The Role of the Gut Microbiome on the Epigenomics of GC
7. The Effect of Proton Pump Inhibitors on the Gut Microbiome
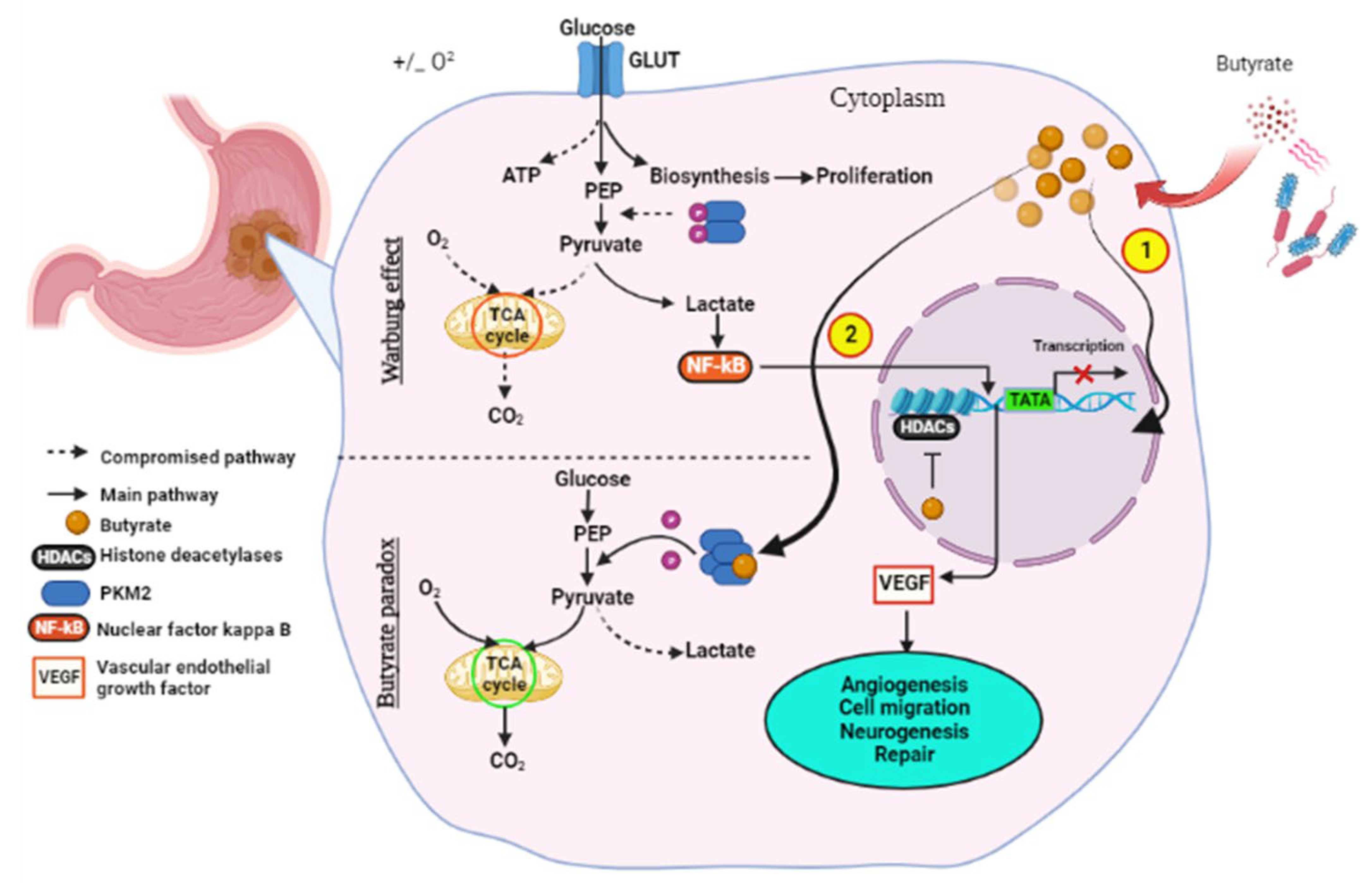
8. Gut Microbiome in Metabolic Rewiring
9. Gut Microbiome and its Products in Gastric Cancer Therapy
9.1. Bacteriotherapy
9.2. Butyrate-Based Therapy
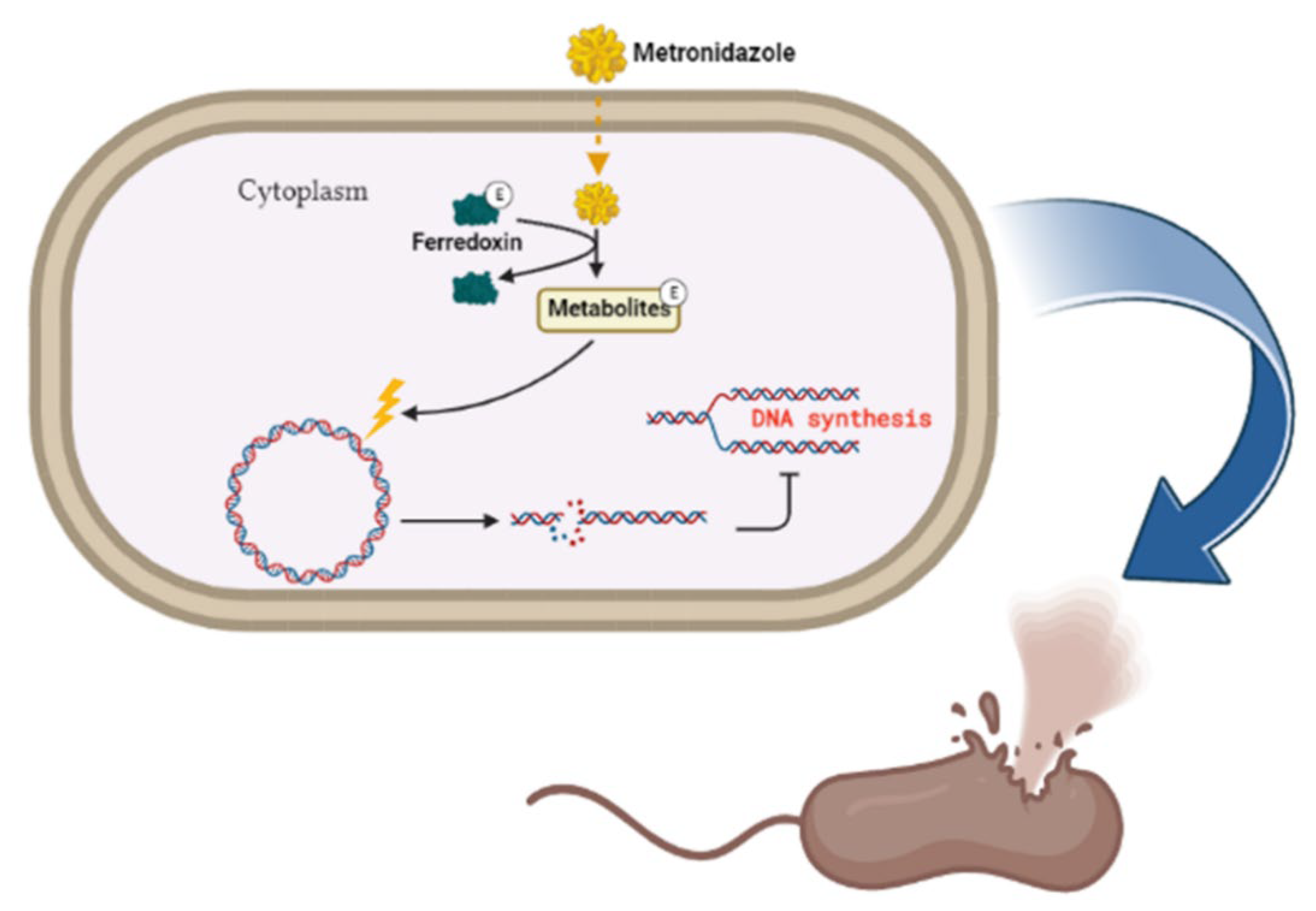
9.3. F. Nucleatum as Potential Therapeutic Target
9.4. Anti-Mycoplasma Therapy
9.5. CRISPR/Cas9
9.6. PI3k/Akt/mTOR Signaling Pathway Targets
9.7. Microbial Ablation
10. Conclusions and Future Considerations
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Lou, L.; Wang, L.; Zhang, Y.; Chen, G.; Lin, L.; Jin, X.; Huang, Y.; Chen, J. Sex difference in incidence of gastric cancer: An international comparative study based on the Global Burden of Disease Study 2017. BMJ Open 2020, 10, e033323. [Google Scholar] [CrossRef] [PubMed]
- Smyth, E.C.; Nilsson, M.; Grabsch, H.I.; van Grieken, N.C.; Lordick, F. Gastric cancer. Lancet 2020, 396, 635–648. [Google Scholar] [CrossRef]
- Hu, H.-M.; Tsai, H.-J.; Ku, H.-Y.; Lo, S.-S.; Shan, Y.-S.; Chang, H.-C.; Chao, Y.; Chen, J.-S.; Chen, S.-C.; Chiang, C.-J. Survival outcomes of management in metastatic gastric adenocarcinoma patients. Sci. Rep. 2021, 11, 23142. [Google Scholar] [CrossRef]
- Andreeva, N.V.; Gabbasova, R.R.; Grivennikov, S.I. Microbiome in cancer progression and therapy. Curr. Opin. Microbiol. 2020, 56, 118–126. [Google Scholar] [CrossRef]
- Gong, M.; Meng, L.; Jiang, B.; Zhang, J.; Yang, H.; Wu, J.; Shou, C. p37 from Mycoplasma hyorhinis promotes cancer cell invasiveness and metastasis through activation of MMP-2 and followed by phosphorylation of EGFR. Molecular cancer therapeutics 2008, 7, 530–537. [Google Scholar] [CrossRef]
- Higashi, H.; Nakaya, A.; Tsutsumi, R.; Yokoyama, K.; Fujii, Y.; Ishikawa, S.; Higuchi, M.; Takahashi, A.; Kurashima, Y.; Teishikata, Y. Helicobacter pylori CagA induces Ras-independent morphogenetic response through SHP-2 recruitment and activation. J. Biol. Chem. 2004, 279, 17205–17216. [Google Scholar] [CrossRef] [PubMed]
- Serna, G.; Ruiz-Pace, F.; Hernando, J.; Alonso, L.; Fasani, R.; Landolfi, S.; Comas, R.; Jimenez, J.; Elez, E.; Bullman, S. Fusobacterium nucleatum persistence and risk of recurrence after preoperative treatment in locally advanced rectal cancer. Ann. Oncol. 2020, 31, 1366–1375. [Google Scholar] [CrossRef]
- Xu, Y.; Li, H.; Chen, W.; Yao, X.; Xing, Y.; Wang, X.; Zhong, J.; Meng, G. Mycoplasma hyorhinis activates the NLRP3 inflammasome and promotes migration and invasion of gastric cancer cells. PLoS ONE 2013, 8, e77955. [Google Scholar] [CrossRef]
- Martin, A.M.; Sun, E.W.; Rogers, G.B.; Keating, D.J. The influence of the gut microbiome on host metabolism through the regulation of gut hormone release. Front. Physiol. 2019, 10, 428. [Google Scholar] [CrossRef]
- Martinez, K.B.; Pierre, J.F.; Chang, E.B. The gut microbiota: The gateway to improved metabolism. Gastroenterol. Clin. 2016, 45, 601–614. [Google Scholar] [CrossRef] [PubMed]
- Rongrui, L.; Na, H.; Zongfang, L.; Fanpu, J.; Shiwen, J. Epigenetic mechanism involved in the HBV/HCV-related hepatocellular carcinoma tumorigenesis. Curr. Pharm. Des. 2014, 20, 1715–1725. [Google Scholar] [CrossRef]
- Yuza, K.; Nagahashi, M.; Watanabe, S.; Takabe, K.; Wakai, T. Hypermutation and microsatellite instability in gastrointestinal cancers. Oncotarget 2017, 8, 112103. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.; Barnes, S.; Demark-Wahnefried, W.; Morrow, C.; Salvador, C.; Skibola, C.; Tollefsbol, T.O. Influences of diet and the gut microbiome on epigenetic modulation in cancer and other diseases. Clin. Epigenet. 2015, 7, 112. [Google Scholar] [CrossRef]
- Engstrand, L.; Graham, D.Y. Microbiome and gastric cancer. Dig. Dis. Sci. 2020, 65, 865–873. [Google Scholar] [CrossRef]
- Lauren, P. The two histological main types of gastric carcinoma: Diffuse and so-called intestinal-type carcinoma: An attempt at a histo-clinical classification. Acta Pathol. Microbiol. Scand. 1965, 64, 31–49. [Google Scholar] [CrossRef] [PubMed]
- Bosman, F.T.; Carneiro, F.; Hruban, R.H.; Theise, N.D. WHO Classification of Tumours of the Digestive System; World Health Organization: Geneva, Switzerland, 2010. [Google Scholar]
- Chiang, C.Y.; Huang, K.H.; Fang, W.L.; Wu, C.W.; Chen, J.H.; Lo, S.S.; Hsieh, M.C.; Shen, K.H.; Li, A.F.Y.; Niu, D.M.; et al. Factors associated with recurrence within 2 years after curative surgery for gastric adenocarcinoma. World J. Surg. 2011, 35, 2472–2478. [Google Scholar] [CrossRef]
- Tan, I.B.; Ivanova, T.; Lim, K.H.; Ong, C.W.; Deng, N.; Lee, J.; Tan, S.H.; Wu, J.; Lee, M.H.; Ooi, C.H. Intrinsic subtypes of gastric cancer, based on gene expression pattern, predict survival and respond differently to chemotherapy. Gastroenterology 2011, 141, 476–485.e11. [Google Scholar] [CrossRef]
- Sohn, B.H.; Hwang, J.-E.; Jang, H.-J.; Lee, H.-S.; Oh, S.C.; Shim, J.-J.; Lee, K.-W.; Kim, E.H.; Yim, S.Y.; Lee, S.H. Clinical significance of four molecular subtypes of gastric cancer identified by the cancer genome atlas project. Clin. Cancer Res. 2017, 23, 4441–4449. [Google Scholar] [CrossRef]
- Lei, Z.; Tan, I.B.; Das, K.; Deng, N.; Zouridis, H.; Pattison, S.; Chua, C.; Feng, Z.; Guan, Y.K.; Ooi, C.H. Identification of molecular subtypes of gastric cancer with different responses to PI3-kinase inhibitors and 5-fluorouracil. Gastroenterology 2013, 145, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Network, C.G.A.R. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202. [Google Scholar]
- Cristescu, R.; Lee, J.; Nebozhyn, M.; Kim, K.-M.; Ting, J.C.; Wong, S.S.; Liu, J.; Yue, Y.G.; Wang, J.; Yu, K. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat. Med. 2015, 21, 449–456. [Google Scholar] [CrossRef]
- Ahn, S.; Lee, S.-J.; Kim, Y.; Kim, A.; Shin, N.; Choi, K.U.; Lee, C.-H.; Huh, G.Y.; Kim, K.-M.; Setia, N. High-throughput protein and mRNA expression–based classification of gastric cancers can identify clinically distinct subtypes, concordant with recent molecular classifications. Am. J. Surg. Pathol. 2017, 41, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Setia, N.; Agoston, A.T.; Han, H.S.; Mullen, J.T.; Duda, D.G.; Clark, J.W.; Deshpande, V.; Mino-Kenudson, M.; Srivastava, A.; Lennerz, J.K. A protein and mRNA expression-based classification of gastric cancer. Mod. Pathol. 2016, 29, 772–784. [Google Scholar] [CrossRef]
- Ding, S.Z.; Smith, M.F., Jr.; Goldberg, J.B. Helicobacter pylori and mitogen-activated protein kinases regulate the cell cycle, proliferation and apoptosis in gastric epithelial cells. J. Gastroenterol. Hepatol. 2008, 23, e67–e78. [Google Scholar] [CrossRef]
- Kusano, M.; Toyota, M.; Suzuki, H.; Akino, K.; Aoki, F.; Fujita, M.; Hosokawa, M.; Shinomura, Y.; Imai, K.; Tokino, T. Genetic, epigenetic, and clinicopathologic features of gastric carcinomas with the CpG island methylator phenotype and an association with Epstein–Barr virus. Cancer 2006, 106, 1467–1479. [Google Scholar] [CrossRef]
- Loh, M.; Liem, N.; Vaithilingam, A.; Lim, P.L.; Sapari, N.S.; Elahi, E.; Mok, Z.Y.; Cheng, C.L.; Yan, B.; Pang, B. DNA methylation subgroups and the CpG island methylator phenotype in gastric cancer: A comprehensive profiling approach. BMC Gastroenterol. 2014, 14, 55. [Google Scholar] [CrossRef]
- Wang, H.; Ding, Y.; Chen, Y.; Jiang, J.; Chen, Y.; Lu, J.; Kong, M.; Mo, F.; Huang, Y.; Zhao, W. A novel genomic classification system of gastric cancer via integrating multidimensional genomic characteristics. Gastric Cancer 2021, 24, 1227–1241. [Google Scholar] [CrossRef]
- Zhu, Z.; Qin, J.; Dong, C.; Yang, J.; Yang, M.; Tian, J.; Zhong, X. Identification of four gastric cancer subtypes based on genetic analysis of cholesterogenic and glycolytic pathways. Bioengineered 2021, 12, 4780–4793. [Google Scholar] [CrossRef]
- Yusefi, A.R.; Lankarani, K.B.; Bastani, P.; Radinmanesh, M.; Kavosi, Z. Risk factors for gastric cancer: A systematic review. Asian Pac. J. Cancer Prev. APJCP 2018, 19, 591. [Google Scholar] [PubMed]
- Hooi, J.K.; Lai, W.Y.; Ng, W.K.; Suen, M.M.; Underwood, F.E.; Tanyingoh, D.; Malfertheiner, P.; Graham, D.Y.; Wong, V.W.; Wu, J.C. Global prevalence of Helicobacter pylori infection: Systematic review and meta-analysis. Gastroenterology 2017, 153, 420–429. [Google Scholar] [CrossRef]
- Li, F.; Du, H.; Li, S.; Liu, J. The association between metabolic syndrome and gastric cancer in Chinese. Front. Oncol. 2018, 8, 326. [Google Scholar] [CrossRef] [PubMed]
- Tsugane, S.; Sasazuki, S. Diet and the risk of gastric cancer: Review of epidemiological evidence. Gastric Cancer 2007, 10, 75–83. [Google Scholar] [CrossRef]
- Arnold, M.; Park, J.Y.; Camargo, M.C.; Lunet, N.; Forman, D.; Soerjomataram, I. Is gastric cancer becoming a rare disease? A global assessment of predicted incidence trends to 2035. Gut 2020, 69, 823–829. [Google Scholar] [CrossRef]
- Heer, E.V.; Harper, A.S.; Sung, H.; Jemal, A.; Fidler-Benaoudia, M.M. Emerging cancer incidence trends in Canada: The growing burden of young adult cancers. Cancer 2020, 126, 4553–4562. [Google Scholar] [CrossRef] [PubMed]
- Alegría-Torres, J.A.; Baccarelli, A.; Bollati, V. Epigenetics and lifestyle. Epigenomics 2011, 3, 267–277. [Google Scholar] [CrossRef]
- Lauby-Secretan, B.; Scoccianti, C.; Loomis, D.; Grosse, Y.; Bianchini, F.; Straif, K. Body fatness and cancer—Viewpoint of the IARC Working Group. N. Engl. J. Med. 2016, 375, 794–798. [Google Scholar] [CrossRef]
- Haydon, A.M.; MacInnis, R.J.; English, D.R.; Giles, G.G. Effect of physical activity and body size on survival after diagnosis with colorectal cancer. Gut 2006, 55, 62–67. [Google Scholar] [CrossRef]
- Renehan, A.G.; Soerjomataram, I.; Tyson, M.; Egger, M.; Zwahlen, M.; Coebergh, J.W.; Buchan, I. Incident cancer burden attributable to excess body mass index in 30 European countries. Int. J. Cancer 2010, 126, 692–702. [Google Scholar] [CrossRef]
- Ley, R.E.; Bäckhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef]
- Wei, M.-Y.; Shi, S.; Liang, C.; Meng, Q.-C.; Hua, J.; Zhang, Y.-Y.; Liu, J.; Zhang, B.; Xu, J.; Yu, X.-J. The microbiota and microbiome in pancreatic cancer: More influential than expected. Mol. Cancer 2019, 18, 97. [Google Scholar] [CrossRef]
- Kim, M.-H.; Yun, K.E.; Kim, J.; Park, E.; Chang, Y.; Ryu, S.; Kim, H.-L.; Kim, H.-N. Gut microbiota and metabolic health among overweight and obese individuals. Sci. Rep. 2020, 10, 19417. [Google Scholar] [CrossRef]
- Tseng, C.-H.; Tseng, F.-H. Diabetes and gastric cancer: The potential links. World J. Gastroenterol. WJG 2014, 20, 1701. [Google Scholar] [CrossRef]
- Dabo, B.; Pelucchi, C.; Rota, M.; Jain, H.; Bertuccio, P.; Bonzi, R.; Palli, D.; Ferraroni, M.; Zhang, Z.-F.; Sanchez-Anguiano, A. The association between diabetes and gastric cancer: Results from the Stomach Cancer Pooling Project Consortium. Eur. J. Cancer Prev. 2022, 31, 260. [Google Scholar] [CrossRef] [PubMed]
- Derakhshan, M.H.; Malekzadeh, R.; Watabe, H.; Yazdanbod, A.; Fyfe, V.; Kazemi, A.; Rakhshani, N.; Didevar, R.; Sotoudeh, M.; Zolfeghari, A. Combination of gastric atrophy, reflux symptoms and histological subtype indicates two distinct aetiologies of gastric cardia cancer. Gut 2008, 57, 298–305. [Google Scholar] [CrossRef]
- Rubenstein, J.H.; Taylor, J. Meta-analysis: The association of oesophageal adenocarcinoma with symptoms of gastro-oesophageal reflux. Aliment. Pharmacol. Ther. 2010, 32, 1222–1227. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.H.; Tseng, C.C.; Bernstein, L. Hiatal hernia, reflux symptoms, body size, and risk of esophageal and gastric adenocarcinoma. Cancer Interdiscip. Int. J. Am. Cancer Soc. 2003, 98, 940–948. [Google Scholar] [CrossRef] [PubMed]
- Ye, W.; Chow, W.-H.; Lagergren, J.; Yin, L.; Nyrén, O. Risk of adenocarcinomas of the esophagus and gastric cardia in patients with gastroesophageal reflux diseases and after antireflux surgery. Gastroenterology 2001, 121, 1286–1293. [Google Scholar] [CrossRef]
- Li, Y.; Feng, A.; Zheng, S.; Chen, C.; Lyu, J. Recent Estimates and Predictions of 5-Year Survival in Patients with Gastric Cancer: A Model-Based Period Analysis. Cancer Control 2022, 29, 10732748221099227. [Google Scholar] [CrossRef]
- Mukaisho, K.-i.; Nakayama, T.; Hagiwara, T.; Hattori, T.; Sugihara, H. Two distinct etiologies of gastric cardia adenocarcinoma: Interactions among pH, Helicobacter pylori, and bile acids. Front. Microbiol. 2015, 6, 412. [Google Scholar] [CrossRef] [PubMed]
- Misumi, A.; Murakami, A.; Harada, K.; Baba, K.; Akagi, M. Definition of carcinoma of the gastric cardia. Langenbecks Arch. Chir. 1989, 374, 221–226. [Google Scholar] [CrossRef]
- Alkhathami, A.M.; Alzahrani, A.A.; Alzhrani, M.A.; Alsuwat, O.B.; Mahfouz, M.E.M. Risk factors for gastroesophageal reflux disease in Saudi Arabia. Gastroenterol. Res. 2017, 10, 294–300. [Google Scholar] [CrossRef]
- Naito, Y.; Kashiwagi, K.; Takagi, T.; Andoh, A.; Inoue, R. Intestinal dysbiosis secondary to proton-pump inhibitor use. Digestion 2018, 97, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Polat, F.R.; Polat, S. The effect of Helicobacter pylori on gastroesophageal reflux disease. JSLS J. Soc. Laparoendosc. Surg. 2012, 16, 260. [Google Scholar] [CrossRef]
- Argilés, J.M.; López-Soriano, F.J. The role of cytokines in cancer cachexia. Med. Res. Rev. 1999, 19, 223–248. [Google Scholar] [CrossRef]
- Germano, G.; Allavena, P.; Mantovani, A. Cytokines as a key component of cancer-related inflammation. Cytokine 2008, 43, 374–379. [Google Scholar] [CrossRef]
- Nicolini, A.; Carpi, A.; Rossi, G. Cytokines in breast cancer. Cytokine Growth Factor Rev. 2006, 17, 325–337. [Google Scholar] [CrossRef]
- Baj, J.; Forma, A.; Sitarz, M.; Portincasa, P.; Garruti, G.; Krasowska, D.; Maciejewski, R. Helicobacter pylori virulence factors—mechanisms of bacterial pathogenicity in the gastric microenvironment. Cells 2020, 10, 27. [Google Scholar] [CrossRef]
- Ding, S.-Z.; Goldberg, J.B.; Hatakeyama, M. Helicobacter pylori infection, oncogenic pathways and epigenetic mechanisms in gastric carcinogenesis. Future Oncol. 2010, 6, 851–862. [Google Scholar] [CrossRef]
- Pai, S.G.; Carneiro, B.A.; Mota, J.M.; Costa, R.; Leite, C.A.; Barroso-Sousa, R.; Kaplan, J.B.; Chae, Y.K.; Giles, F.J. Wnt/beta-catenin pathway: Modulating anticancer immune response. J. Hematol. Oncol. 2017, 10, 101. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Jiang, W.; Wang, S.; Wang, L.; Xie, K. Role of Wnt/β-catenin signaling in drug resistance of pancreatic cancer. Curr. Pharm. Des. 2012, 18, 2464–2471. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, D.R.; Derakhshan, M.H.; Wirz, A.A.; Orange, C.; Ballantyne, S.A.; Going, J.J.; McColl, K.E. The gastric acid pocket is attenuated in H. pylori infected subjects. Gut 2017, 66, 1555–1562. [Google Scholar] [CrossRef] [PubMed]
- Hansson, L.-E.; Nyrén, O.; Hsing, A.W.; Bergström, R.; Josefsson, S.; Chow, W.-H.; Fraumeni, J.F., Jr.; Adami, H.-O. The risk of stomach cancer in patients with gastric or duodenal ulcer disease. N. Engl. J. Med. 1996, 335, 242–249. [Google Scholar] [CrossRef]
- Mohammadi, S.O.; Yadegar, A.; Kargar, M.; Mirjalali, H.; Kafilzadeh, F. The impact of Helicobacter pylori infection on gut microbiota-endocrine system axis; modulation of metabolic hormone levels and energy homeostasis. J. Diabetes Metab. Disord. 2020, 19, 1855–1861. [Google Scholar] [CrossRef]
- Bauerfeind, P.; Garner, R.; Dunn, B.; Mobley, H. Synthesis and activity of Helicobacter pylori urease and catalase at low pH. Gut 1997, 40, 25–30. [Google Scholar] [CrossRef]
- Ahmed, A.; Smoot, D.; Littleton, G.; Tackey, R.; Walters, C.S.; Kashanchi, F.; Allen, C.R.; Ashktorab, H. Helicobacter pylori inhibits gastric cell cycle progression. Microbes Infect. 2000, 2, 1159–1169. [Google Scholar] [CrossRef]
- Koeppel, M.; Garcia-Alcalde, F.; Glowinski, F.; Schlaermann, P.; Meyer, T.F. Helicobacter pylori infection causes characteristic DNA damage patterns in human cells. Cell Rep. 2015, 11, 1703–1713. [Google Scholar] [CrossRef]
- Rossi, C.; Cicalini, I.; Cufaro, M.C.; Consalvo, A.; Upadhyaya, P.; Sala, G.; Antonucci, I.; Del Boccio, P.; Stuppia, L.; De Laurenzi, V. Breast cancer in the era of integrating “Omics” approaches. Oncogenesis 2022, 11, 17. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.M.; Holden, K.A.; Jeong, A.-R.; Kim, L.; Fitzgerald, K.D.; Almasri, E.; McLennan, G.; Eisenberg, M.; Jahromi, A.H.; Hoh, C. Response to CAR-T Therapy Can be Monitored Using Genome-Wide Sequencing of Cell-Free DNA in Patients with DLBCL. Blood 2020, 136, 17. [Google Scholar] [CrossRef]
- Goonetilleke, K.; Siriwardena, A. Systematic review of carbohydrate antigen (CA 19-9) as a biochemical marker in the diagnosis of pancreatic cancer. Eur. J. Surg. Oncol. (EJSO) 2007, 33, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Ray, K. Biomarkers for the early detection of PDAC. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 505. [Google Scholar] [CrossRef]
- Breijyeh, Z.; Jubeh, B.; Karaman, R. Resistance of gram-negative bacteria to current antibacterial agents and approaches to resolve it. Molecules 2020, 25, 1340. [Google Scholar] [CrossRef]
- Choi, I.J.; Kook, M.-C.; Kim, Y.-I.; Cho, S.-J.; Lee, J.Y.; Kim, C.G.; Park, B.; Nam, B.-H. Helicobacter pylori therapy for the prevention of metachronous gastric cancer. N. Engl. J. Med. 2018, 378, 1085–1095. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.J.; Kim, C.G.; Lee, J.Y.; Kim, Y.-I.; Kook, M.-C.; Park, B.; Joo, J. Family history of gastric cancer and Helicobacter pylori treatment. N. Engl. J. Med. 2020, 382, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Pappas-Gogos, G.; Tepelenis, K.; Fousekis, F.; Katsanos, K.; Pitiakoudis, M.; Vlachos, K. The Implication of Gastric Microbiome in the Treatment of Gastric Cancer. Cancers 2022, 14, 2039. [Google Scholar] [CrossRef]
- Guo, X.; Ma, N.; Wang, J.; Song, J.; Bu, X.; Cheng, Y.; Sun, K.; Xiong, H.; Jiang, G.; Zhang, B. Increased p38-MAPK is responsible for chemotherapy resistance in human gastric cancer cells. BMC Cancer 2008, 8, 375. [Google Scholar] [CrossRef]
- Zhao, Y.; Shen, S.; Guo, J.; Chen, H.; Greenblatt, D.Y.; Kleeff, J.; Liao, Q.; Chen, G.; Friess, H.; Leung, P.S. Mitogen-activated protein kinases and chemoresistance in pancreatic cancer cells. J. Surg. Res. 2006, 136, 325–335. [Google Scholar] [CrossRef]
- Tohidpour, A.; Gorrell, R.J.; Roujeinikova, A.; Kwok, T. The middle fragment of Helicobacter pylori CagA induces actin rearrangement and triggers its own uptake into gastric epithelial cells. Toxins 2017, 9, 237. [Google Scholar] [CrossRef]
- Krisch, L.M.; Posselt, G.; Hammerl, P.; Wessler, S. CagA phosphorylation in Helicobacter pylori-infected B cells is mediated by the nonreceptor tyrosine kinases of the Src and Abl families. Infect. Immun. 2016, 84, 2671–2680. [Google Scholar] [CrossRef]
- Selbach, M.; Paul, F.E.; Brandt, S.; Guye, P.; Daumke, O.; Backert, S.; Dehio, C.; Mann, M. Host cell interactome of tyrosine-phosphorylated bacterial proteins. Cell Host Microbe 2009, 5, 397–403. [Google Scholar] [CrossRef]
- Shimizu, T.; Tolcher, A.W.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Smith, L.S.; Gunn, S.; Smetzer, L.; Mays, T.A.; Kaiser, B. The Clinical Effect of the Dual-Targeting Strategy Involving PI3K/AKT/mTOR and RAS/MEK/ERK Pathways in Patients with Advanced CancerClinical Effect of Dual PI3K and MAPK Pathways Inhibitions. Clin. Cancer Res. 2012, 18, 2316–2325. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Ma, S.; Che, K.; Pobbati, A.V.; Rubin, B.P. Inhibition of PI3K and MAPK pathways along with KIT inhibitors as a strategy to overcome drug resistance in gastrointestinal stromal tumors. PLoS ONE 2021, 16, e0252689. [Google Scholar] [CrossRef] [PubMed]
- Roberts, P.J.; Usary, J.E.; Darr, D.B.; Dillon, P.M.; Pfefferle, A.D.; Whittle, M.C.; Duncan, J.S.; Johnson, S.M.; Combest, A.J.; Jin, J. Combined PI3K/mTOR and MEK Inhibition Provides Broad Antitumor Activity in Faithful Murine Cancer ModelsCombined PI3K/mTOR and MEK Inhibition. Clin. Cancer Res. 2012, 18, 5290–5303. [Google Scholar] [CrossRef] [PubMed]
- Khwanraj, K.; Madlah, S.; Grataitong, K.; Dharmasaroja, P. Comparative mRNA expression of eEF1A isoforms and a PI3K/Akt/mTOR pathway in a cellular model of Parkinson’s disease. Parkinson’s Dis. 2016, 2016, 8716016. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Tao, H.; Carloni, E.; Leung, W.K.; Graham, D.Y.; Sepulveda, A.R. Helicobacter pylori impairs DNA mismatch repair in gastric epithelial cells. Gastroenterology 2002, 123, 542–553. [Google Scholar] [CrossRef]
- Weir, C.B.; Le, J.K. Metronidazole. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Adachi, T.; Matsui, S.; Watanabe, T.; Okamoto, K.; Okamoto, A.; Kono, M.; Yamada, M.; Nagai, T.; Komeda, Y.; Minaga, K. Comparative study of clarithromycin-versus metronidazole-based triple therapy as first-line eradication for Helicobacter pylori. Oncology 2017, 93, 15–19. [Google Scholar] [CrossRef]
- Jenks, P.J.; Edwards, D.I. Metronidazole resistance in Helicobacter pylori. Int. J. Antimicrob. Agents 2002, 19, 1–7. [Google Scholar] [CrossRef]
- Boehm, E.T.; Thon, C.; Kupcinskas, J.; Steponaitiene, R.; Skieceviciene, J.; Canbay, A.; Malfertheiner, P.; Link, A. Fusobacterium nucleatum is associated with worse prognosis in Lauren’s diffuse type gastric cancer patients. Sci. Rep. 2020, 10, 16240. [Google Scholar] [CrossRef]
- Mima, K.; Nishihara, R.; Qian, Z.R.; Cao, Y.; Sukawa, Y.; Nowak, J.A.; Yang, J.; Dou, R.; Masugi, Y.; Song, M. Fusobacterium nucleatum in colorectal carcinoma tissue and patient prognosis. Gut 2016, 65, 1973–1980. [Google Scholar] [CrossRef]
- Oh, H.J.; Kim, J.H.; Bae, J.M.; Kim, H.J.; Cho, N.-Y.; Kang, G.H. Prognostic impact of Fusobacterium nucleatum depends on combined tumor location and microsatellite instability status in stage II/III colorectal cancers treated with adjuvant chemotherapy. J. Pathol. Transl. Med. 2019, 53, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Yun, S.Y.; Lee, Y.; Lee, H.; Yong, D.; Lee, K. Clinical differences in patients infected with fusobacterium and antimicrobial susceptibility of fusobacterium isolates recovered at a tertiary-care hospital in korea. Ann. Lab. Med. 2022, 42, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Tavakoli, A.; Monavari, S.H.; Solaymani Mohammadi, F.; Kiani, S.J.; Armat, S.; Farahmand, M. Association between Epstein-Barr virus infection and gastric cancer: A systematic review and meta-analysis. BMC Cancer 2020, 20, 493. [Google Scholar] [CrossRef] [PubMed]
- Iizasa, H.; Nanbo, A.; Nishikawa, J.; Jinushi, M.; Yoshiyama, H. Epstein-Barr Virus (EBV)-associated gastric carcinoma. Viruses 2012, 4, 3420–3439. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, X.-L.; Liu, K.; Bai, D.; Zhang, W.-H.; Chen, X.-Z.; Hu, J.-K. Associations between gastric cancer risk and virus infection other than Epstein-Barr virus: A systematic review and meta-analysis based on epidemiological studies. Clin. Transl. Gastroenterol. 2020, 11, e00201. [Google Scholar] [CrossRef] [PubMed]
- Malarkey, D.E.; Hoenerhoff, M.J.; Maronpot, R.R. Carcinogenesis: Manifestation and mechanisms. Fundam. Toxicol. Pathol. 2018, 83–104. [Google Scholar] [CrossRef]
- Pitot, H.C. The molecular biology of carcinogenesis. Cancer 1993, 72, 962–970. [Google Scholar] [CrossRef]
- Elaskandrany, M.; Patel, R.; Patel, M.; Miller, G.; Saxena, D.; Saxena, A. Fungi, host immune response, and tumorigenesis. Am. J. Physiol. -Gastrointest. Liver Physiol. 2021, 321, G213–G222. [Google Scholar] [CrossRef]
- Kaźmierczak-Siedlecka, K.; Dvořák, A.; Folwarski, M.; Daca, A.; Przewłócka, K.; Makarewicz, W. Fungal gut microbiota dysbiosis and its role in colorectal, oral, and pancreatic carcinogenesis. Cancers 2020, 12, 1326. [Google Scholar] [CrossRef]
- Zackular, J.P.; Baxter, N.T.; Iverson, K.D.; Sadler, W.D.; Petrosino, J.F.; Chen, G.Y.; Schloss, P.D. The gut microbiome modulates colon tumorigenesis. MBio 2013, 4, e00692-13. [Google Scholar] [CrossRef]
- Geng, F.; Zhang, Y.; Lu, Z.; Zhang, S.; Pan, Y. Fusobacterium nucleatum caused DNA damage and promoted cell proliferation by the Ku70/p53 pathway in oral cancer cells. DNA Cell Biol. 2020, 39, 144–151. [Google Scholar] [CrossRef]
- Cani, P.D. Human gut microbiome: Hopes, threats and promises. Gut 2018, 67, 1716–1725. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H. Harness the functions of gut microbiome in tumorigenesis for cancer treatment. Cancer Commun. 2021, 41, 937–967. [Google Scholar] [CrossRef]
- Knight, R.; Callewaert, C.; Marotz, C.; Hyde, E.R.; Debelius, J.W.; McDonald, D.; Sogin, M.L. The microbiome and human biology. Annu. Rev. Genom. Hum. Genet. 2017, 18, 65–86. [Google Scholar] [CrossRef]
- Oppler, B. Zur Kenntniss des Mageninhalts beim Carcinoma ventriculi1. DMW-Dtsch. Med. Wochenschr. 1895, 21, 73–75. [Google Scholar] [CrossRef][Green Version]
- Galt, H.; Iles, C. A study of the Boas-Oppler bacillus. J. Pathol. Bacteriol. 1915, 19, 239–244. [Google Scholar] [CrossRef]
- Lertpiriyapong, K.; Whary, M.T.; Muthupalani, S.; Lofgren, J.L.; Gamazon, E.R.; Feng, Y.; Ge, Z.; Wang, T.C.; Fox, J.G. Gastric colonisation with a restricted commensal microbiota replicates the promotion of neoplastic lesions by diverse intestinal microbiota in the Helicobacter pylori INS-GAS mouse model of gastric carcinogenesis. Gut 2014, 63, 54–63. [Google Scholar] [CrossRef]
- Li, Z.-P.; Liu, J.-X.; Lu, L.-L.; Wang, L.-L.; Xu, L.; Guo, Z.-H.; Dong, Q.-J. Overgrowth of Lactobacillus in gastric cancer. World J. Gastrointest. Oncol. 2021, 13, 1099. [Google Scholar] [CrossRef]
- Xu, L.; Qu, Y.-H.; Chu, X.-D.; Wang, R.; Nelson, H.H.; Gao, Y.-T.; Yuan, J.-M. Urinary levels of N-nitroso compounds in relation to risk of gastric cancer: Findings from the shanghai cohort study. PLoS ONE 2015, 10, e0117326. [Google Scholar] [CrossRef]
- Butler, M.; Leach, R. 1116 Proceedings ofthe Royal Society ofMedicine 40. J. Bact 1956, 71, 362. [Google Scholar]
- Waites, K.B.; Katz, B.; Schelonka, R.L. Mycoplasmas and ureaplasmas as neonatal pathogens. Clin. Microbiol. Rev. 2005, 18, 757–789. [Google Scholar] [CrossRef]
- Lanao, A.E.; Chakraborty, R.K.; Pearson-Shaver, A.L. Mycoplasma infections. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Duan, H.; Qu, L.; Shou, C. Activation of EGFR-PI3K-AKT signaling is required for Mycoplasma hyorhinis-promoted gastric cancer cell migration. Cancer Cell Int. 2014, 14, 135. [Google Scholar] [CrossRef]
- Ketcham, C.M.; Anai, S.; Reutzel, R.; Sheng, S.; Schuster, S.M.; Brenes, R.B.; Agbandje-McKenna, M.; McKenna, R.; Rosser, C.J.; Boehlein, S.K. p37 induces tumor invasiveness. Mol. Cancer Ther. 2005, 4, 1031–1038. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Qu, L.; Ma, H.; Chen, L.; Liu, W.; Liu, C.; Meng, L.; Wu, J.; Shou, C. Mycoplasma hyorhinisinfection in gastric carcinoma and its effects on the malignant phenotypes of gastric cancer cells. BMC Gastroenterol. 2010, 10, 132. [Google Scholar] [CrossRef] [PubMed]
- Nascimento Araujo, C.d.; Amorim, A.T.; Barbosa, M.S.; Alexandre, J.C.P.L.; Campos, G.B.; Macedo, C.L.; Marques, L.M.; Timenetsky, J. Evaluating the presence of Mycoplasma hyorhinis, Fusobacterium nucleatum, and Helicobacter pylori in biopsies of patients with gastric cancer. Infect. Agents Cancer 2021, 16, 70. [Google Scholar] [CrossRef] [PubMed]
- Gomersall, A.C.; Phan, H.A.; Iacuone, S.; Li, S.F.; Parish, R.W. The Mycoplasma hyorhinis p37 protein rapidly induces genes in fibroblasts associated with inflammation and cancer. PLoS ONE 2015, 10, e0140753. [Google Scholar] [CrossRef]
- Patil, S.; Rao, R.S.; Raj, A.T. Role of Mycoplasma in the initiation and progression of oral cancer. J. Int. Oral Health JIOH 2015, 7, i–ii. [Google Scholar]
- Liu, W.-B.; Zhang, J.-Z.; Jiang, B.-H.; Ren, T.-T.; Gong, M.-M.; Meng, L.; Shou, C.-C. Lipoprotein p37 from Mycoplasma hyorhinis inhibiting mammalian cell adhesion. J. Biomed. Sci. 2006, 13, 323–331. [Google Scholar] [CrossRef][Green Version]
- Schmidhauser, C.; Dudler, R.; Schmidt, T.; Parish, R. A mycoplasmal protein influences tumour cell invasiveness and contact inhibition in vitro. J. Cell Sci. 1990, 95, 499–506. [Google Scholar] [CrossRef]
- Takahashi-Yanaga, F.; Kahn, M. Targeting Wnt signaling: Can we safely eradicate cancer stem cells? Clin. Cancer Res. 2010, 16, 3153–3162. [Google Scholar] [CrossRef]
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef]
- Zholobenko, V.; Rutten, F.; Zholobenko, A.; Holmes, A. In situ spectroscopic identification of the six types of asbestos. J. Hazard. Mater. 2021, 403, 123951. [Google Scholar] [CrossRef] [PubMed]
- Landrigan, P.J.; Nicholson, W.J.; Suzuki, Y.; LaDou, J. The hazards of chrysotile asbestos: A critical review. Ind. Health 1999, 37, 271–280. [Google Scholar] [CrossRef] [PubMed]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Arsenic, Metals, Fibres, and Dusts. IARC Monogr. Eval. Carcinog. Risks Hum. 2012, 100, 11. [Google Scholar]
- Kim, S.J.; Williams, D.; Cheresh, P.; Kamp, D.W. Asbestos-induced gastrointestinal cancer: An update. J. Gastrointest. Dig. Syst. 2013, 3, 135. [Google Scholar] [CrossRef]
- Selikoff, I.J.; Hammond, E.C.; Churg, J. Asbestos exposure, smoking, and neoplasia. JAMA 1968, 204, 106–112. [Google Scholar] [CrossRef]
- Oksa, P.; Wolff, H.; Vehmas, T.; Pallasaho, P.; Frilander, H. Asbestos, Asbestosis, and Cancer: Helsinki Criteria for Diagnosis and Attribution 2014; Finnish Institute of Occupational Health: Helsinki, Finland, 2014. [Google Scholar]
- Patel-Mandlik, K.; Millette, J. Evidence of migration of ingested asbestos into various baboon organs. Scan. Electron Microsc. 1980, 1, 347–354. [Google Scholar]
- Luus, K. Asbestos: Mining exposure, health effects and policy implications. McGill J. Med. MJM 2007, 10, 121. [Google Scholar] [CrossRef]
- Manning, C.B.; Vallyathan, V.; Mossman, B.T. Diseases caused by asbestos: Mechanisms of injury and disease development. Int. Immunopharmacol. 2002, 2, 191–200. [Google Scholar] [CrossRef]
- Stanik, I.A.; Cedzynska, K.; Zakowska, Z. Destruction of the chrysotile asbestos structure with a population of bacteria Lactobacillus casei and Lactobacillus plantarum. Fresenius Environ. Bull. 2006, 15, 640. [Google Scholar]
- Seshan, K. How are the physical and chemical properties of chrysotile asbestos altered by a 10-year residence in water and up to 5 days in simulated stomach acid? Environ. Health Perspect. 1983, 53, 143–148. [Google Scholar] [CrossRef]
- Pan, M.; Wan, C.; Xie, Q.; Huang, R.; Tao, X.; Shah, N.P.; Wei, H. Changes in gastric microbiota induced by Helicobacter pylori infection and preventive effects of Lactobacillus plantarum ZDY 2013 against such infection. J. Dairy Sci. 2016, 99, 970–981. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.W.; Baek, Y.-M.; Yang, K.E.; Yoo, H.-S.; Cho, C.-K.; Lee, Y.-W.; Park, J.; Eom, C.-Y.; Lee, Z.-W.; Choi, J.-S. Lactobacillus casei extract induces apoptosis in gastric cancer by inhibiting NF-κB and mTOR-mediated signaling. Integr. Cancer Ther. 2013, 12, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Scanlan, R. Formation and occurrence of nitrosamines in food. Cancer Res. 1983, 43, 2435s–2440s. [Google Scholar] [PubMed]
- Kankanamage, R.N.; Ghosh, A.B.; Jiang, D.; Gkika, K.; Keyes, T.; Achola, L.A.; Suib, S.; Rusling, J.F. Metabolites of tobacco-and e-cigarette-related nitrosamines can drive Cu2+-mediated DNA oxidation. Chem. Res. Toxicol. 2020, 33, 2072–2086. [Google Scholar] [CrossRef]
- de la Monte, S.M.; Neusner, A.; Chu, J.; Lawton, M. Epidemiological trends strongly suggest exposures as etiologic agents in the pathogenesis of sporadic Alzheimer’s disease, diabetes mellitus, and non-alcoholic steatohepatitis. J. Alzheimer’s Dis. 2009, 17, 519–529. [Google Scholar] [CrossRef]
- Gankhuyag, N.; Lee, K.-H.; Cho, J.-Y. The role of nitrosamine (NNK) in breast cancer carcinogenesis. J. Mammary Gland. Biol. Neoplasia 2017, 22, 159–170. [Google Scholar] [CrossRef]
- Tong, M.; Neusner, A.; Longato, L.; Lawton, M.; Wands, J.R.; de la Monte, S.M. Nitrosamine exposure causes insulin resistance diseases: Relevance to type 2 diabetes mellitus, non-alcoholic steatohepatitis, and Alzheimer’s disease. J. Alzheimer’s Dis. 2009, 17, 827–844. [Google Scholar]
- Lee, C.-H.; Chang, Y.-C.; Chen, C.-S.; Tu, S.-H.; Wang, Y.-J.; Chen, L.-C.; Chang, Y.-J.; Wei, P.-L.; Chang, H.-W.; Chang, C.-H. Crosstalk between nicotine and estrogen-induced estrogen receptor activation induces α9-nicotinic acetylcholine receptor expression in human breast cancer cells. Breast Cancer Res. Treat. 2011, 129, 331–345. [Google Scholar] [CrossRef]
- Wen, J.; Fu, J.-H.; Zhang, W.; Guo, M. Lung carcinoma signaling pathways activated by smoking. Chin. J. Cancer 2011, 30, 551. [Google Scholar] [CrossRef]
- Song, P.; Wu, L.; Guan, W. Dietary nitrates, nitrites, and nitrosamines intake and the risk of gastric cancer: A meta-analysis. Nutrients 2015, 7, 9872–9895. [Google Scholar] [CrossRef] [PubMed]
- La Vecchia, C.; D’Avanzo, B.; Airoldi, L.; Braga, C.; Decarli, A. Nitrosamine intake and gastric cancer risk. Eur. J. Cancer Prev. 1995, 4, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Taneja, P.; Labhasetwar, P.; Nagarnaik, P.; Ensink, J.H. The risk of cancer as a result of elevated levels of nitrate in drinking water and vegetables in Central India. J. Water Health 2017, 15, 602–614. [Google Scholar] [CrossRef] [PubMed]
- Stewart, V. Regulation of nitrate and nitrite reductase synthesis in enterobacteria. Antonie Van Leeuwenhoek 1994, 66, 37–45. [Google Scholar] [CrossRef]
- Sarhadi, V.; Mathew, B.; Kokkola, A.; Karla, T.; Tikkanen, M.; Rautelin, H.; Lahti, L.; Puolakkainen, P.; Knuutila, S. Gut microbiota of patients with different subtypes of gastric cancer and gastrointestinal stromal tumors. Gut Pathog. 2021, 13, 11. [Google Scholar] [CrossRef]
- Liu, S.; Dai, J.; Lan, X.; Fan, B.; Dong, T.; Zhang, Y.; Han, M. Intestinal bacteria are potential biomarkers and therapeutic targets for gastric cancer. Microb. Pathog. 2021, 151, 104747. [Google Scholar] [CrossRef]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef]
- Rooks, M.G.; Garrett, W.S. Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol. 2016, 16, 341–352. [Google Scholar] [CrossRef]
- Johnson, C.; Warmoes, M.O.; Shen, X.; Locasale, J.W. Epigenetics and cancer metabolism. Cancer Lett. 2015, 356, 309–314. [Google Scholar] [CrossRef]
- Prasher, D.; Greenway, S.C.; Singh, R.B. The impact of epigenetics on cardiovascular disease. Biochem. Cell Biol. 2020, 98, 12–22. [Google Scholar] [CrossRef]
- Yoda, Y.; Takeshima, H.; Niwa, T.; Kim, J.G.; Ando, T.; Kushima, R.; Sugiyama, T.; Katai, H.; Noshiro, H.; Ushijima, T. Integrated analysis of cancer-related pathways affected by genetic and epigenetic alterations in gastric cancer. Gastric Cancer 2015, 18, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Canale, M.; Casadei-Gardini, A.; Ulivi, P.; Arechederra, M.; Berasain, C.; Lollini, P.-L.; Fernández-Barrena, M.G.; Avila, M.A. Epigenetic mechanisms in gastric cancer: Potential new therapeutic opportunities. Int. J. Mol. Sci. 2020, 21, 5500. [Google Scholar] [CrossRef]
- Kanherkar, R.R.; Bhatia-Dey, N.; Csoka, A.B. Epigenetics across the human lifespan. Front. Cell Dev. Biol. 2014, 2, 49. [Google Scholar] [CrossRef]
- Muhammad, J.S.; Eladl, M.A.; Khoder, G. Helicobacter pylori-induced DNA methylation as an epigenetic modulator of gastric cancer: Recent outcomes and future direction. Pathogens 2019, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Ushijima, T. Detection and interpretation of altered methylation patterns in cancer cells. Nat. Rev. Cancer 2005, 5, 223–231. [Google Scholar] [CrossRef]
- Kang, G.H.; Lee, S.; Kim, W.H.; Lee, H.W.; Kim, J.C.; Rhyu, M.-G.; Ro, J.Y. Epstein-barr virus-positive gastric carcinoma demonstrates frequent aberrant methylation of multiple genes and constitutes CpG island methylator phenotype-positive gastric carcinoma. Am. J. Pathol. 2002, 160, 787–794. [Google Scholar] [CrossRef]
- Niwa, T.; Toyoda, T.; Tsukamoto, T.; Mori, A.; Tatematsu, M.; Ushijima, T. Prevention of Helicobacter pylori–Induced Gastric Cancers in Gerbils by a DNA Demethylating Agent. Cancer Prev. Res. 2013, 6, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Abrahami, D.; McDonald, E.G.; Schnitzer, M.E.; Barkun, A.N.; Suissa, S.; Azoulay, L. Proton pump inhibitors and risk of gastric cancer: Population-based cohort study. Gut 2022, 71, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.S.; Leung, W.K. Long-term use of proton-pump inhibitors and risk of gastric cancer: A review of the current evidence. Ther. Adv. Gastroenterol. 2019, 12, 1756284819834511. [Google Scholar] [CrossRef]
- Poulsen, A.; Christensen, S.; McLaughlin, J.; Thomsen, R.; Sørensen, H.; Olsen, J.; Friis, S. Proton pump inhibitors and risk of gastric cancer: A population-based cohort study. Br. J. Cancer 2009, 100, 1503–1507. [Google Scholar] [CrossRef]
- Ahmed, A.; Clarke, J.O. Proton pump inhibitors (PPI). Drug 2020, 56, 307–335. [Google Scholar]
- Cheng, T.; Liu, B.-F.; Han, T.-Y.; Gu, Z.-H.; Pan, P.; Yu, H. Effectiveness and safety of proton pump inhibitors for treating acute pancreatitis: A protocol for systematic review and meta analysis. Medicine 2021, 100, e24808. [Google Scholar] [CrossRef] [PubMed]
- Nieto, J.M.; Pisegna, J.R. The role of proton pump inhibitors in the treatment of Zollinger–Ellison syndrome. Expert Opin. Pharmacother. 2006, 7, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.M.; Kim, N. Pharmacokinetics and pharmacodynamics of the proton pump inhibitors. J. Neurogastroenterol. Motil. 2013, 19, 25. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Cao, L.; Tian, Y.; Zhang, P.; Ding, C.; Lu, W.; Jia, C.; Shao, C.; Liu, W.; Wang, D. Butyrate suppresses the proliferation of colorectal cancer cells via targeting pyruvate kinase M2 and metabolic reprogramming. Mol. Cell. Proteom. 2018, 17, 1531–1545. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef]
- Phan, L.M.; Yeung, S.-C.J.; Lee, M.-H. Cancer metabolic reprogramming: Importance, main features, and potentials for precise targeted anti-cancer therapies. Cancer Biol. Med. 2014, 11, 1. [Google Scholar] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519. [Google Scholar] [CrossRef]
- Pfeiffer, T.; Schuster, S.; Bonhoeffer, S. Cooperation and competition in the evolution of ATP-producing pathways. Science 2001, 292, 504–507. [Google Scholar] [CrossRef]
- Marçais, A.; Cherfils-Vicini, J.; Viant, C.; Degouve, S.; Viel, S.; Fenis, A.; Rabilloud, J.; Mayol, K.; Tavares, A.; Bienvenu, J. The metabolic checkpoint kinase mTOR is essential for IL-15 signaling during the development and activation of NK cells. Nat. Immunol. 2014, 15, 749–757. [Google Scholar] [CrossRef]
- Zahra, K.; Dey, T.; Mishra, S.P.; Pandey, U. Pyruvate kinase M2 and cancer: The role of PKM2 in promoting tumorigenesis. Front. Oncol. 2020, 10, 159. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.-B.; Zhang, Y.-C.; Huang, H.-H.; Lin, J. Prospects for clinical applications of butyrate-producing bacteria. World J. Clin. Pediatrics 2021, 10, 84. [Google Scholar] [CrossRef] [PubMed]
- Bouwens, T.; Trouw, L.; Veerhuis, R.; Dirven, C.; Lamfers, M.; Al-Khawaja, H. Complement activation in Glioblastoma multiforme pathophysiology: Evidence from serum levels and presence of complement activation products in tumor tissue. J. Neuroimmunol. 2015, 278, 271–276. [Google Scholar] [CrossRef]
- Geng, H.-W.; Yin, F.-Y.; Zhang, Z.-F.; Gong, X.; Yang, Y. Butyrate suppresses glucose metabolism of colorectal cancer cells via GPR109a-AKT signaling pathway and enhances chemotherapy. Front. Mol. Biosci. 2021, 8, 634874. [Google Scholar] [CrossRef]
- Donohoe, D.R.; Collins, L.B.; Wali, A.; Bigler, R.; Sun, W.; Bultman, S.J. The Warburg effect dictates the mechanism of butyrate-mediated histone acetylation and cell proliferation. Mol. Cell 2012, 48, 612–626. [Google Scholar] [CrossRef] [PubMed]
- Stanley, J.; Linton, D.; Burnens, A.P.; Dewhirst, F.E.; On, S.L.; Porter, A.; Owen, R.J.; Costas, M. Helicobacter pullorumsp. nov.-genotype and phenotype of a new species isolated from poultry and from human patients with gastroenteritis. Microbiology 1994, 140, 3441–3449. [Google Scholar] [CrossRef]
- De Vos, D.; Lim, A., Jr.; Pirnay, J.-P.; Struelens, M.; Vandenvelde, C.; Duinslaeger, L.; Vanderkelen, A.; Cornelis, P. Direct detection and identification of Pseudomonas aeruginosa in clinical samples such as skin biopsy specimens and expectorations by multiplex PCR based on two outer membrane lipoprotein genes, oprI and oprL. J. Clin. Microbiol. 1997, 35, 1295–1299. [Google Scholar] [CrossRef]
- Soleimanpour, S.; Hasanian, S.M.; Avan, A.; Yaghoubi, A.; Khazaei, M. Bacteriotherapy in gastrointestinal cancer. Life Sci. 2020, 254, 117754. [Google Scholar] [CrossRef]
- Cotter, P.D.; Ross, R.P.; Hill, C. Bacteriocins—A viable alternative to antibiotics? Nat. Rev. Microbiol. 2013, 11, 95–105. [Google Scholar] [CrossRef]
- Dicks, L.M.; Dreyer, L.; Smith, C.; Van Staden, A.D. A review: The fate of bacteriocins in the human gastro-intestinal tract: Do they cross the gut–blood barrier? Front. Microbiol. 2018, 9, 2297. [Google Scholar] [CrossRef]
- Ou, J.; Carbonero, F.; Zoetendal, E.G.; DeLany, J.P.; Wang, M.; Newton, K.; Gaskins, H.R.; O’Keefe, S.J. Diet, microbiota, and microbial metabolites in colon cancer risk in rural Africans and African Americans. Am. J. Clin. Nutr. 2013, 98, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.; Lee, Y.S.; Lee, Y.C. Sodium butyrate-induced DAPK-mediated apoptosis in human gastric cancer cells. Oncol. Rep. 2012, 27, 1111–1115. [Google Scholar] [CrossRef] [PubMed]
- Panebianco, C.; Villani, A.; Pisati, F.; Orsenigo, F.; Ulaszewska, M.; Latiano, T.P.; Potenza, A.; Andolfo, A.; Terracciano, F.; Tripodo, C. Butyrate, a postbiotic of intestinal bacteria, affects pancreatic cancer and gemcitabine response in in vitro and in vivo models. Biomed. Pharmacother. 2022, 151, 113163. [Google Scholar] [CrossRef]
- Huang, L.; Yu, Z.; Zhang, T.; Zhao, X.; Huang, G. HSP40 interacts with pyruvate kinase M2 and regulates glycolysis and cell proliferation in tumor cells. PLoS ONE 2014, 9, e92949. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Jiang, J.; Ji, J.; Cai, Q.; Chen, X.; Yu, Y.; Zhu, Z.; Zhang, J. PKM2 promotes cell migration and inhibits autophagy by mediating PI3K/AKT activation and contributes to the malignant development of gastric cancer. Sci. Rep. 2017, 7, 2886. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.S.; Li, D.; Zhang, J.; Wang, Y.S.; Yang, L.; Zhang, H.L.; Wang, X.H.; Mu, B.; Wang, W.; Ma, Y. Silencing of pkm2 increases the efficacy of docetaxel in human lung cancer xenografts in mice. Cancer Sci. 2010, 101, 1447–1453. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Qiao, T.; Zhuang, X.; Chen, W.; Xing, N.; Zhang, Q. Knockdown of the M2 isoform of pyruvate kinase (PKM2) with shRNA enhances the effect of docetaxel in human NSCLC cell lines in vitro. Yonsei Med. J. 2016, 57, 1312–1323. [Google Scholar] [CrossRef]
- Yu, T.; Guo, F.; Yu, Y.; Sun, T.; Ma, D.; Han, J.; Qian, Y.; Kryczek, I.; Sun, D.; Nagarsheth, N. Fusobacterium nucleatum promotes chemoresistance to colorectal cancer by modulating autophagy. Cell 2017, 170, 548–563.e16. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Su, G.; Sun, J.; Zhang, Y. Activation of the TLR4/MyD88 signaling pathway contributes to the development of human hepatocellular carcinoma via upregulation of IL-23 and IL-17A. Oncol. Lett. 2018, 15, 9647–9654. [Google Scholar] [CrossRef]
- Xia, L.; Tan, S.; Zhou, Y.; Lin, J.; Wang, H.; Oyang, L.; Tian, Y.; Liu, L.; Su, M.; Wang, H. Role of the NFκB-signaling pathway in cancer. OncoTargets Ther. 2018, 11, 2063. [Google Scholar] [CrossRef]
- Division of Gastroenterology and Hepatology, Ren-Ji Hospital, Shanghai Jiao-Tong University School of Medicine, Shanghai Institute of Digestive Disease; Key Laboratory of Gastroenterology & Hepatology, Ministry of Health; Ruijin Hospital affiliated to Shanghai Jiao Tong University School of Medicine; Fudan University Shanghai Cancer Center; Zhongshan Hospital affiliated to Fudan University. Study of Oral Metronidazole on Postoperative Chemotherapy in Colorectal Cancer. 2020. Available online: https://ClinicalTrials.gov/show/NCT04264676 (accessed on 22 July 2022).
- Preoperative Endoscopic Treatment with Fosfomycin and Metronidazole in Patients with Right-Sided Colon Cancer and Colon Adenoma (MEFO-Trial). Available online: https://ClinicalTrials.gov/show/NCT04312360 (accessed on 22 July 2022).
- Bullman, S.; Pedamallu, C.S.; Sicinska, E.; Clancy, T.E.; Zhang, X.; Cai, D.; Neuberg, D.; Huang, K.; Guevara, F.; Nelson, T. Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science 2017, 358, 1443–1448. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Kanno, S.; Nosho, K.; Sukawa, Y.; Mitsuhashi, K.; Kurihara, H.; Igarashi, H.; Takahashi, T.; Tachibana, M.; Takahashi, H. Association of Fusobacterium nucleatum with clinical and molecular features in colorectal serrated pathway. Int. J. Cancer 2015, 137, 1258–1268. [Google Scholar] [CrossRef]
- Tomkovich, S. The Interplay between Inflammation and Microbial Activities in Colorectal Cancer. Ph.D. Thesis, University of North Carolina at Chapel Hill, Chapel Hill, NC, USA, 2016. Available online: https://doi.org/10.17615/ffds-3m05 (accessed on 22 July 2022).
- Evers, B.; Jastrzebski, K.; Heijmans, J.P.; Grernrum, W.; Beijersbergen, R.L.; Bernards, R. CRISPR knockout screening outperforms shRNA and CRISPRi in identifying essential genes. Nat. Biotechnol. 2016, 34, 631–633. [Google Scholar] [CrossRef]
- Platt, R.J.; Chen, S.; Zhou, Y.; Yim, M.J.; Swiech, L.; Kempton, H.R.; Dahlman, J.E.; Parnas, O.; Eisenhaure, T.M.; Jovanovic, M. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 2014, 159, 440–455. [Google Scholar] [CrossRef] [PubMed]
- Ishino, Y.; Shinagawa, H.; Makino, K.; Amemura, M.; Nakata, A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J. Bacteriol. 1987, 169, 5429–5433. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.; Embden, J.D.v.; Gaastra, W.; Schouls, L.M. Identification of genes that are associated with DNA repeats in prokaryotes. Mol. Microbiol. 2002, 43, 1565–1575. [Google Scholar] [CrossRef]
- Mojica, F.J.; Díez-Villaseñor, C.; Soria, E.; Juez, G. Biological significance of a family of regularly spaced repeats in the genomes of Archaea, Bacteria and mitochondria. Mol. Microbiol. 2000, 36, 244–246. [Google Scholar] [CrossRef]
- Han, W.; She, Q. CRISPR history: Discovery, characterization, and prosperity. Prog. Mol. Biol. Transl. Sci. 2017, 152, 1–21. [Google Scholar]
- Liu, Z.; Dong, H.; Cui, Y.; Cong, L.; Zhang, D. Application of different types of CRISPR/Cas-based systems in bacteria. Microb. Cell Factories 2020, 19, 172. [Google Scholar] [CrossRef]
- Mei, Y.; Wang, Y.; Chen, H.; Sun, Z.S.; Ju, X.-D. Recent progress in CRISPR/Cas9 technology. J. Genet. Genom. 2016, 43, 63–75. [Google Scholar] [CrossRef]
- Lu, Y.; Xue, J.; Deng, T.; Zhou, X.; Yu, K.; Deng, L.; Huang, M.; Yi, X.; Liang, M.; Wang, Y. Safety and feasibility of CRISPR-edited T cells in patients with refractory non-small-cell lung cancer. Nat. Med. 2020, 26, 732–740. [Google Scholar] [CrossRef]
- Su, S.; Zou, Z.; Chen, F.; Ding, N.; Du, J.; Shao, J.; Li, L.; Fu, Y.; Hu, B.; Yang, Y. CRISPR-Cas9-mediated disruption of PD-1 on human T cells for adoptive cellular therapies of EBV positive gastric cancer. Oncoimmunology 2017, 6, e1249558. [Google Scholar] [CrossRef]
- Schubert, M.G.; Goodman, D.B.; Wannier, T.M.; Kaur, D.; Farzadfard, F.; Lu, T.K.; Shipman, S.L.; Church, G.M. High-throughput functional variant screens via in vivo production of single-stranded DNA. Proc. Natl. Acad. Sci. USA 2021, 118, e2018181118. [Google Scholar] [CrossRef]
- Meng, D.; He, W.; Zhang, Y.; Liang, Z.; Zheng, J.; Zhang, X.; Zheng, X.; Zhan, P.; Chen, H.; Li, W. Development of PI3K inhibitors: Advances in clinical trials and new strategies. Pharmacol. Res. 2021, 173, 105900. [Google Scholar] [CrossRef]
- Willems, L.; Tamburini, J.; Chapuis, N.; Lacombe, C.; Mayeux, P.; Bouscary, D. PI3K and mTOR signaling pathways in cancer: New data on targeted therapies. Curr. Oncol. Rep. 2012, 14, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Terracciano, L.M.; Piscuoglio, S.; Ng, C.K. Hepatocellular carcinoma: Pathology and genetics. In Encyclopedia of Cancer, 3rd ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 198–210. [Google Scholar]
- Katso, R.; Okkenhaug, K.; Ahmadi, K.; White, S.; Timms, J.; Waterfield, M.D. Cellular function of phosphoinositide 3-kinases: Implications for development, immunity, homeostasis, and cancer. Annu. Rev. Cell Dev. Biol. 2001, 17, 615–675. [Google Scholar] [CrossRef]
- Pedrero, J.M.G.; Carracedo, D.G.; Pinto, C.M.; Zapatero, A.H.; Rodrigo, J.P.; Nieto, C.S.; Gonzalez, M.V. Retracted: Frequent genetic and biochemical alterations of the PI 3-K/AKT/PTEN pathway in head and neck squamous cell carcinoma. Int. J. Cancer 2005, 114, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Samuels, Y.; Diaz, L.A., Jr.; Schmidt-Kittler, O.; Cummins, J.M.; DeLong, L.; Cheong, I.; Rago, C.; Huso, D.L.; Lengauer, C.; Kinzler, K.W. Mutant PIK3CA promotes cell growth and invasion of human cancer cells. Cancer Cell 2005, 7, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, K.; Matsuzaki, T.; Farnworth, E. Handbook of Fermented Functional Foods; CRC Press: Boca Raton, FL, USA, 2008. [Google Scholar]
- Husni, R.N.; Gordon, S.M.; Washington, J.A.; Longworth, D.L. Lactobacillus bacteremia and endocarditis: Review of 45 cases. Clin. Infect. Dis. 1997, 25, 1048–1055. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Sharma, A.; Gupta, M.; Padwad, Y.; Sharma, R. Cell-free culture supernatant of probiotic Lactobacillus fermentum protects against H2O2-induced premature senescence by suppressing ROS-Akt-mTOR axis in murine preadipocytes. Probiotics Antimicrob. Proteins 2020, 12, 563–576. [Google Scholar] [CrossRef]
- Gopalakrishnan, V.; Helmink, B.A.; Spencer, C.N.; Reuben, A.; Wargo, J.A. The Influence of the Gut Microbiome on Cancer, Immunity, and Cancer Immunotherapy. Cancer Cell 2018, 33, 570–580. [Google Scholar] [CrossRef] [PubMed]
- Martín-Núñez, G.M.; Cornejo-Pareja, I.; Clemente-Postigo, M.; Tinahones, F.J.; Moreno-Indias, I. Helicobacter pylori Eradication Therapy Affect the Gut Microbiota and Ghrelin Levels. Front. Med. 2021, 8, 712908. [Google Scholar] [CrossRef] [PubMed]
- Aykut, B.; Pushalkar, S.; Chen, R.; Li, Q.; Abengozar, R.; Kim, J.I.; Shadaloey, S.A.; Wu, D.; Preiss, P.; Verma, N. The fungal mycobiome promotes pancreatic oncogenesis via activation of MBL. Nature 2019, 574, 264–267. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classification | Subtypes | Prognosis | Associated Genes | Ref. |
|---|---|---|---|---|
| Intrinsic subtypes | G- INT | Better overall survival | FUT, LGALS4, CDH17 | [20] |
| G-DIF | Poor | AURKB, ELOVL5 | ||
| Lei subtypes | Proliferative | Short disease-free survival |  TP53 TP53 | [22] |
| Metabolic |  TP53 TP53 | |||
| Mesenchymal | TP53 | |||
| TCGA | EBV- positive | Best | PIK3CA, JAK2, PD-L1/2, BCOR | [23] |
| MSI | Moderate with no adjuvant chemotherapy response | PIK3CA, ERBB2/3, EGFR, PD-L1, MLH1, TP53 | ||
| GS | Worse | CDH1, RHOA | ||
| CIN | Moderate | SMAD4, APC, TP53 | ||
| ACGA | MSI- high | Best with lowest recurrence frequency | ARID1A, MTOR, KRAS, PIK3CA, ALK, PTEN | [24] |
| MSS/EMT | Worse with highest recurrence frequency | CDH1 | ||
| MSS/TP53+ | Moderate | APC, ARID1A, KRAS, PIK3CA, SMAD4 | ||
| MSS/TP53- | Moderate | ERBB2, EGFR, CCNE1, CCND1, MDM2, ROBO2, GATA6, MYC | ||
| Combined TCGA and ACRG | EBV- positive | Best | PIK3CA, JAK2, PD-L1/2, BCOR | [25,26] |
| MSI- high | Best with lowest recurrence frequency | ARID1A, MTOR, KRAS, PIK3CA, ALK, PTEN | ||
| GC with aberrant E-cadherin | * | * | ||
| GC with aberrant p53 expression | * | * | ||
| GC with normal p53 expression | * | * | ||
| CIMP | CIMP-H | * | EBV-associated | [27,28,29,30,31] |
| CIMP-L | * | * | ||
| CIMP-N | Worse survival | * |
) and decreased () mutations.Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mathebela, P.; Damane, B.P.; Mulaudzi, T.V.; Mkhize-Khwitshana, Z.L.; Gaudji, G.R.; Dlamini, Z. Influence of the Microbiome Metagenomics and Epigenomics on Gastric Cancer. Int. J. Mol. Sci. 2022, 23, 13750. https://doi.org/10.3390/ijms232213750
Mathebela P, Damane BP, Mulaudzi TV, Mkhize-Khwitshana ZL, Gaudji GR, Dlamini Z. Influence of the Microbiome Metagenomics and Epigenomics on Gastric Cancer. International Journal of Molecular Sciences. 2022; 23(22):13750. https://doi.org/10.3390/ijms232213750
Chicago/Turabian StyleMathebela, Precious, Botle Precious Damane, Thanyani Victor Mulaudzi, Zilungile Lynette Mkhize-Khwitshana, Guy Roger Gaudji, and Zodwa Dlamini. 2022. "Influence of the Microbiome Metagenomics and Epigenomics on Gastric Cancer" International Journal of Molecular Sciences 23, no. 22: 13750. https://doi.org/10.3390/ijms232213750
APA StyleMathebela, P., Damane, B. P., Mulaudzi, T. V., Mkhize-Khwitshana, Z. L., Gaudji, G. R., & Dlamini, Z. (2022). Influence of the Microbiome Metagenomics and Epigenomics on Gastric Cancer. International Journal of Molecular Sciences, 23(22), 13750. https://doi.org/10.3390/ijms232213750