
Recruiting Immunity for the Fight against Colorectal Cancer: Current Status and Challenges


 and
and
Abstract
1. Introduction
2. Monoclonal Antibody Therapy
3. Adoptive Cell Therapy
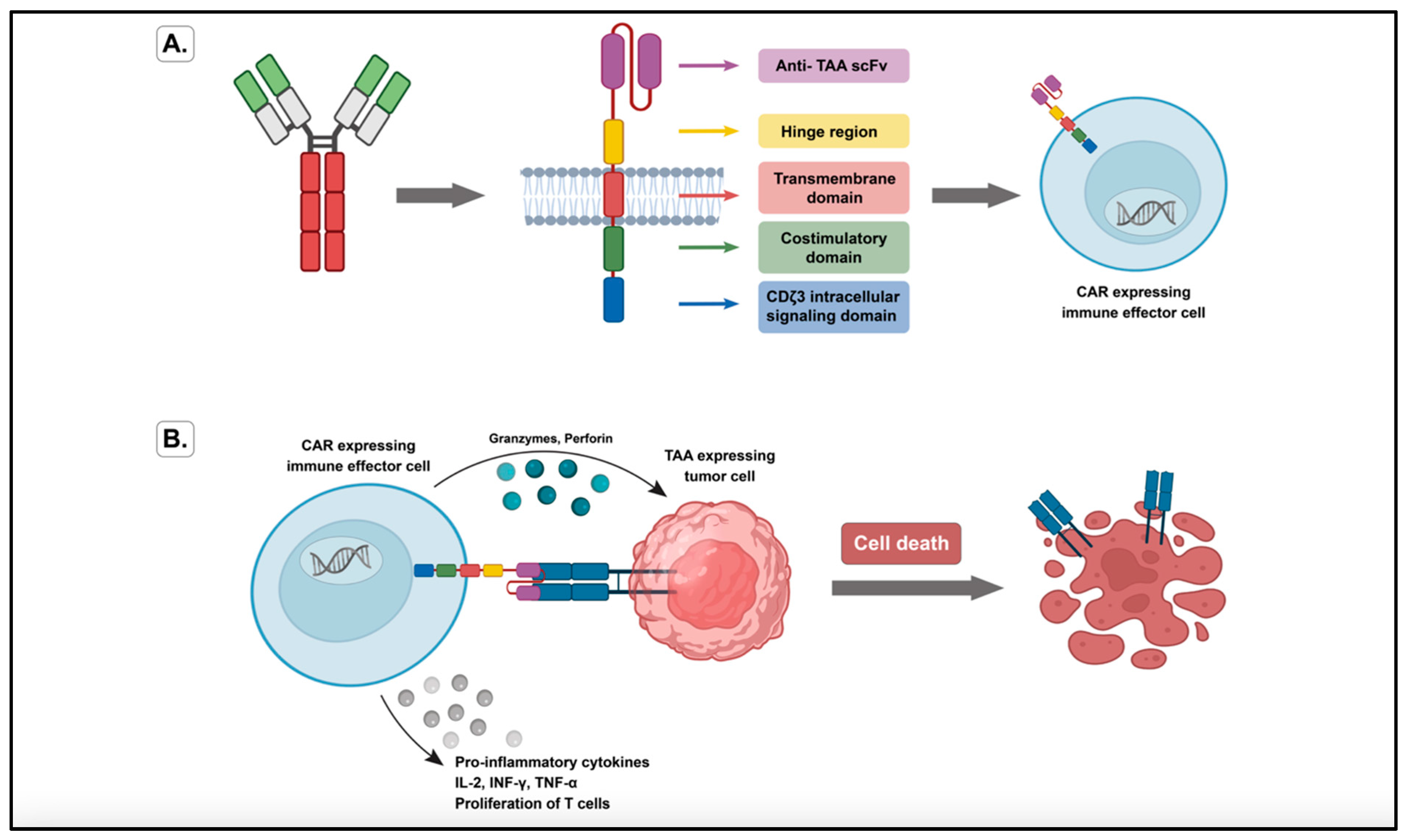
3.1. Genetically Redirected Immune Cells
3.2. Cytokine-Induced Killer Cells
3.3. Dendritic Cell-Based Vaccines
4. Targets for Immunotherapy in CRC
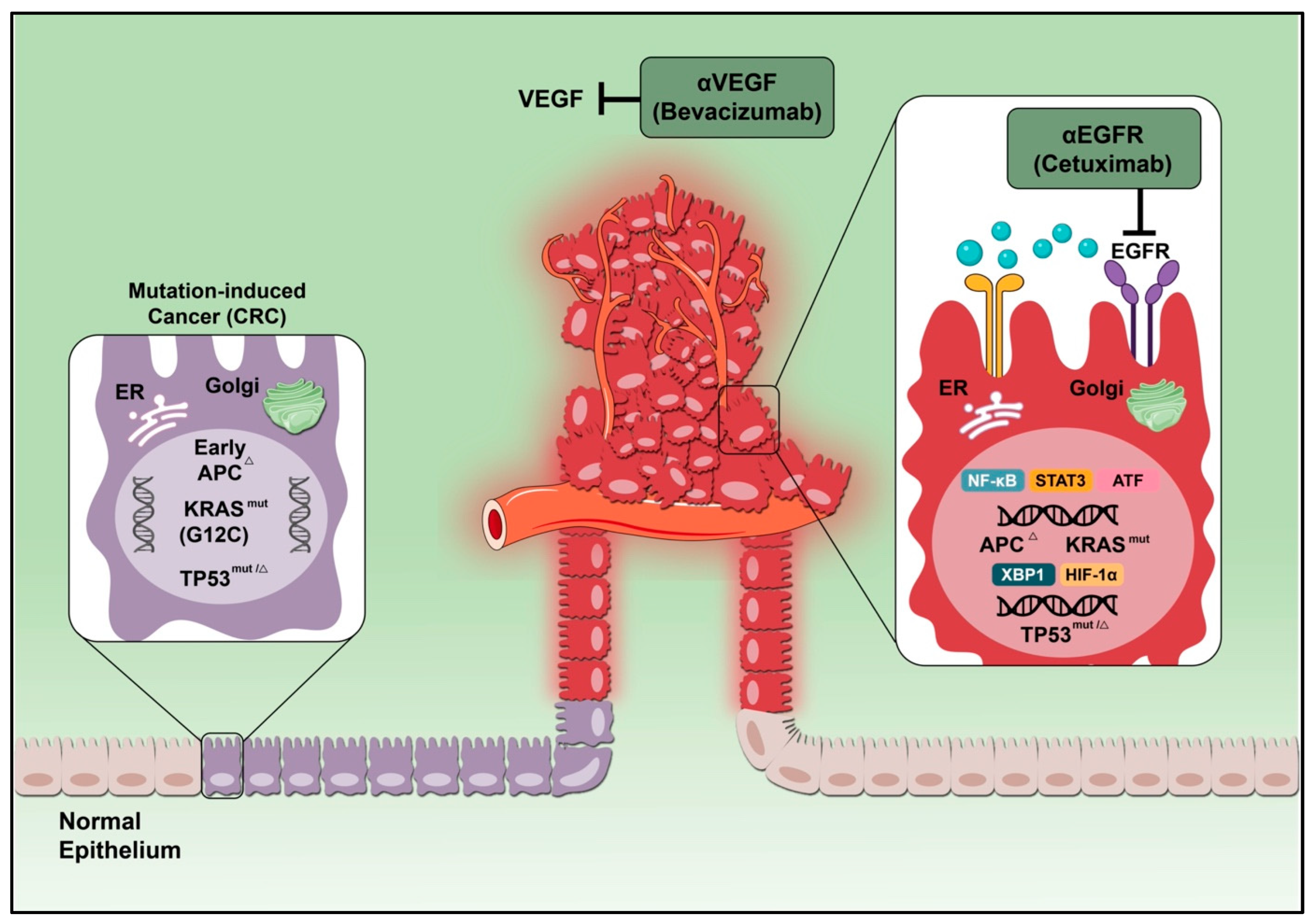
4.1. Vascular Endothelia Growth Factor
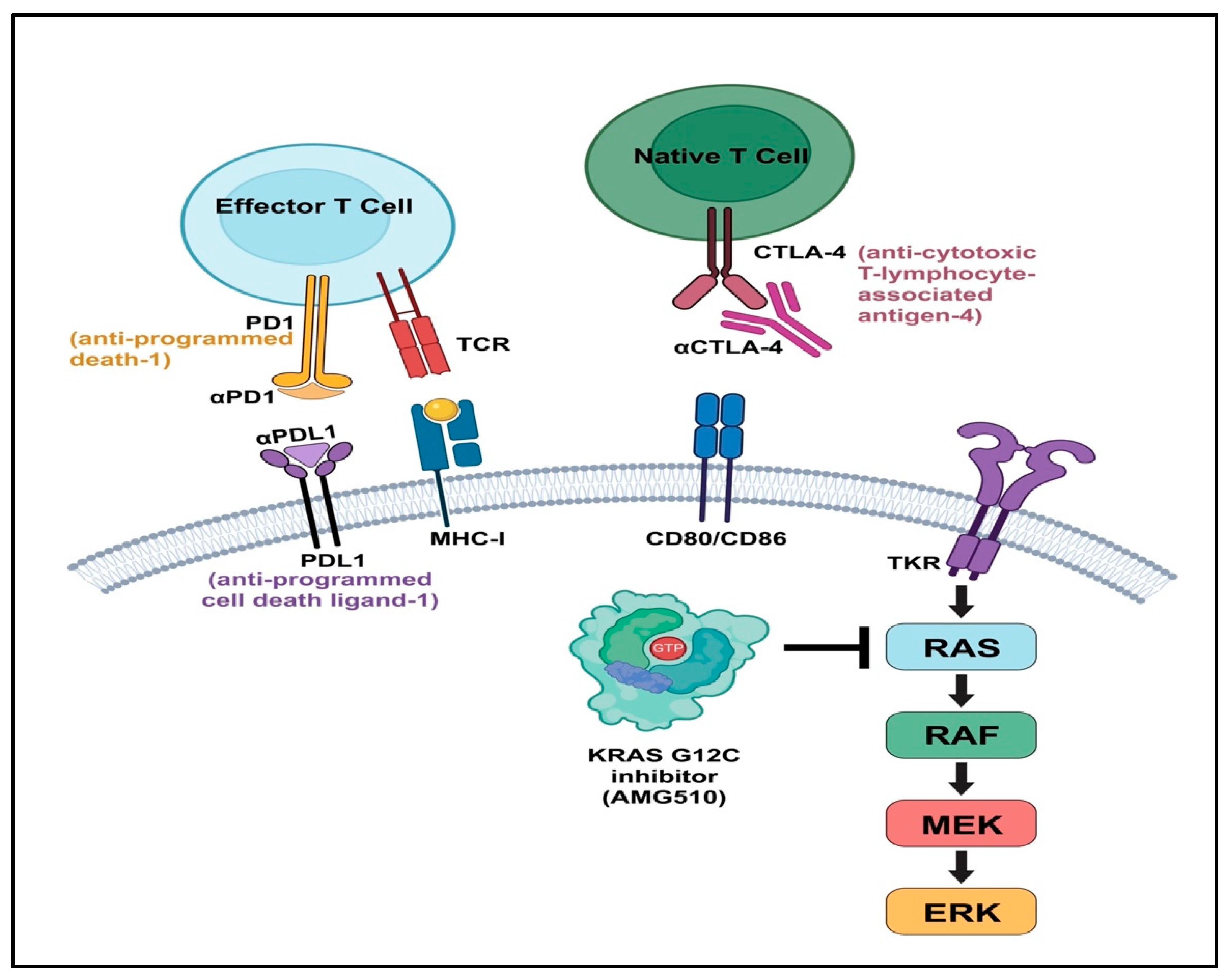
4.2. The K-RAS/B-RAF Pathway
4.3. Epidermal Growth Factor Receptor
4.4. Human Epidermal Growth Factor Receptor
4.5. Immune Checkpoint Inhibitors
4.6. Carcinoembryonic Antigen
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Biller, L.H.; Schrag, D. Diagnosis and Treatment of Metastatic Colorectal Cancer: A Review. JAMA 2021, 325, 669–685. [Google Scholar] [CrossRef] [PubMed]
- Pretzsch, E.; Bösch, F.; Neumann, J.; Ganschow, P.; Bazhin, A.; Guba, M.; Werner, J.; Angele, M. Mechanisms of Metastasis in Colorectal Cancer and Metastatic Organotropism: Hematogenous versus Peritoneal Spread. J. Oncol. 2019, 2019, 7407190. [Google Scholar] [CrossRef] [PubMed]
- Zaorsky, N.; Churilla, T.; Egleston, B.; Fisher, S.; Ridge, J.; Horwitz, E.; Meyer, J. Causes of death among cancer patients. Ann. Oncol. 2017, 28, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Burnett-Hartman, A.N.; Lee, J.K.; Demb, J.; Gupta, S. An Update on the Epidemiology, Molecular Characterization, Diagnosis, and Screening Strategies for Early-Onset Colorectal Cancer. Gastroenterology 2021, 160, 1041–1049. [Google Scholar] [CrossRef]
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef]
- Llosa, N.J.; Cruise, M.; Tam, A.; Wicks, E.C.; Hechenbleikner, E.M.; Taube, J.M.; Blosser, R.L.; Fan, H.; Wang, H.; Luber, B.S.; et al. The Vigorous Immune Microenvironment of Microsatellite Instable Colon Cancer Is Balanced by Multiple Counter-Inhibitory Checkpoints. Cancer Discov. 2015, 5, 43–51. [Google Scholar] [CrossRef]
- Giannakis, M.; Mu, X.J.; Shukla, S.A.; Qian, Z.R.; Cohen, O.; Nishihara, R.; Bahl, S.; Cao, Y.; Amin-Mansour, A.; Yamauchi, M.; et al. Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Rep. 2016, 15, 857–865. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- Vasen, H.; Tomlinson, I.; Castells, A. Clinical management of hereditary colorectal cancer syndromes. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 88–97. [Google Scholar] [CrossRef]
- Papamichael, D.; Audisio, R.A.; Glimelius, B.; de Gramont, A.; Glynne-Jones, R.; Haller, D.; Köhne, C.-H.; Rostoft, S.; Lemmens, V.; Mitry, E.; et al. Treatment of colorectal cancer in older patients: International Society of Geriatric Oncology (SIOG) consensus recommendations 2013. Ann. Oncol. 2014, 26, 463–476. [Google Scholar] [CrossRef]
- Miyake, Y.; Ikeda, K.; Osawa, H.; Nagase, H.; Hoshi, M.; Doi, T.; Makari, Y.; Oshima, S.; Iijima, S.; Kurokawa, E.; et al. Fluoropyrimidines with oxaliplatin(L-OHP) as an adjuvant chemotherapy for Stage III colon cancer. Gan Kagaku Ryoho Cancer Chemother. 2012, 39, 2161–2163. [Google Scholar]
- Kim, J.Y.; Kim, Y.J.; Lee, K.W.; Lee, J.S.; Kim, D.W.; Kang, S.B.; Lee, H.S.; Jang, N.Y.; Kim, J.S.; Kim, J.H. Practical outcome of adjuvant FOLFOX4 chemotherapy in elderly patients with stage III colon cancer: Single-center study in Korea. Jpn. J. Clin. Oncol. 2013, 43, 132–138. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Denoya, P.; Wang, H.; Sands, D.; Nogueras, J.; Weiss, E.; Wexner, S.D. Short-term outcomes of laparoscopic total mesorectal excision following neoadjuvant chemoradiotherapy. Surg. Endosc. 2009, 24, 933–938. [Google Scholar] [CrossRef] [PubMed]
- Sauer, R.; Becker, H.; Hohenberger, W.; Rodel, C.; Wittekind, C.; Fietkau, R.; Martus, P.; Tschmelitsch, J.; Hager, E.; Hess, C.F.; et al. Preoperative versus Postoperative Chemoradiotherapy for Rectal Cancer. N. Engl. J. Med. 2004, 351, 1731–1740. [Google Scholar] [CrossRef]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-Fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Giacchetti, S.; Perpoint, B.; Zidani, R.; Le Bail, N.; Faggiuolo, R.; Focan, C.; Chollet, P.; Llory, J.; Letourneau, Y.; Coudert, B.; et al. Phase III Multicenter Randomized Trial of Oxaliplatin Added to Chronomodulated Fluorouracil–Leucovorin as First-Line Treatment of Metastatic Colorectal Cancer. J. Clin. Oncol. 2000, 18, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, R.; Kikuchi, K.; Uchida, A.; Ozawa, M.; Sano, N.; Tadano, S.; Inagawa, S.; Oda, T.; Ohkohchi, N. Pathological complete response after preoperative chemotherapy including FOLFOX plus bevacizumab for locally advanced rectal cancer: A case report and literature review. Int. J. Surg. Case Rep. 2019, 62, 85–88. [Google Scholar] [CrossRef]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus Irinotecan, Fluorouracil, and Leucovorin for Metastatic Colorectal Cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef]
- Gorgulho, C.M.; Krishnamurthy, A.; Lanzi, A.; Galon, J.; Housseau, F.; Kaneno, R.; Lotze, M.T. Gutting it Out: Developing Effective Immunotherapies for Patients With Colorectal Cancer. J. Immunother. 2021, 44, 49–62. [Google Scholar] [CrossRef]
- Khalil, D.N.; Smith, E.L.; Brentjens, R.J.; Wolchok, J.D. The future of cancer treatment: Immunomodulation, CARs and combination immunotherapy. Nat. Rev. Clin. Oncol. 2016, 13, 273–290. [Google Scholar] [CrossRef]
- Abbott, M.; Ustoyev, Y. Cancer and the Immune System: The History and Background of Immunotherapy. Semin. Oncol. Nurs. 2019, 35, 150923. [Google Scholar] [CrossRef] [PubMed]
- YaYang, Y. Cancer immunotherapy: Harnessing the immune system to battle cancer. J. Clin. Investig. 2015, 125, 3335–3337. [Google Scholar] [CrossRef] [PubMed]
- Riley, R.S.; June, C.H.; Langer, R.; Mitchell, M.J. Delivery technologies for cancer immunotherapy. Nat. Rev. Drug Discov. 2019, 18, 175–196. [Google Scholar] [CrossRef] [PubMed]
- GoGoydel, R.S.; Rader, C. Antibody-based cancer therapy. Oncogene 2021, 40, 3655–3664. [Google Scholar] [CrossRef] [PubMed]
- Weiskopf, K.; Weissman, I.L. Macrophages are critical effectors of antibody therapies for cancer. MAbs 2015, 7, 303–310. [Google Scholar] [CrossRef]
- Scott, A.M.; Wolchok, J.D.; Old, L.J. Antibody therapy of cancer. Nat. Rev. Cancer 2012, 12, 278–287. [Google Scholar] [CrossRef]
- Thomas, A.; Teicher, B.A.; Hassan, R. Antibody–drug conjugates for cancer therapy. Lancet Oncol. 2016, 17, e254–e262. [Google Scholar] [CrossRef]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune Checkpoint Blockade: A Common Denominator Approach to Cancer Therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef]
- SharSharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef]
- Goulet, D.R.; Atkins, W.M. Considerations for the Design of Antibody-Based Therapeutics. J. Pharm. Sci. 2020, 109, 74–103. [Google Scholar] [CrossRef] [PubMed]
- Sveen, A.; Kopetz, S.; Lothe, R.A. Biomarker-guided therapy for colorectal cancer: Strength in complexity. Nat. Rev. Clin. Oncol. 2020, 17, 11–32. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Restifo, N.P.; Yang, J.C.; Morgan, R.A.; Dudley, M.E. Adoptive cell transfer: A clinical path to effective cancer immunotherapy. Nat. Cancer 2008, 8, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Chandran, S.S.; Klebanoff, C.A. T cell receptor-based cancer immunotherapy: Emerging efficacy and pathways of resistance. Immunol. Rev. 2019, 290, 127–147. [Google Scholar] [CrossRef]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef]
- Kerkar, S.P.; Sanchez-Perez, L.; Yang, S.; Borman, Z.A.; Muranski, P.; Ji, Y.; Chinnasamy, D.; Kaiser, A.D.; Hinrichs, C.S.; Klebanoff, C.; et al. Genetic Engineering of Murine CD8+ and CD4+ T Cells for Preclinical Adoptive Immunotherapy Studies. J. Immunother. 2011, 34, 343–352. [Google Scholar] [CrossRef]
- MoMorgan, R.A.; Dudley, M.E.; Wunderlich, J.R.; Hughes, M.S.; Yang, J.C.; Sherry, R.M.; Royal, R.E.; Topalian, S.L.; Kammula, U.S.; Restifo, N.P.; et al. Cancer Regression in Patients After Transfer of Genetically Engineered Lymphocytes. Science 2006, 314, 126–129. [Google Scholar] [CrossRef]
- BeBendle, G.M.; Linnemann, C.; Hooijkaas, A.I.; Bies, L.; De Witte, M.A.; Jorritsma, A.; Kaiser, A.D.M.; Pouw, N.; Debets, R.; Kieback, E.; et al. Lethal graft-versus-host disease in mouse models of T cell receptor gene therapy. Nat. Med. 2010, 16, 565–570. [Google Scholar] [CrossRef]
- Ma, S.; Li, X.; Wang, X.; Cheng, L.; Li, Z.; Zhang, C.; Ye, Z.; Qian, Q. Current Progress in CAR-T Cell Therapy for Solid Tumors. Int. J. Biol. Sci. 2019, 15, 2548–2560. [Google Scholar] [CrossRef]
- Retraction notice to: The Skp2 promoter integrates signaling through the NF-kB, p53, and Akt/GSK3beta pathways to regulate autophagy and apoptosis. Mol. Cell 2014, 55, 342. [CrossRef]
- Sato, T.; Neschadim, A.; Konrad, M.; Fowler, D.H.; Lavie, A.; Medin, J.A. Engineered Human tmpk/AZT As a Novel Enzyme/Prodrug Axis for Suicide Gene Therapy. Mol. Ther. 2007, 15, 962–970. [Google Scholar] [CrossRef] [PubMed]
- Neschadim, A.; Wang, J.C.; Sato, T.; Fowler, D.H.; Lavie, A.; Medin, J.A. Cell Fate Control Gene Therapy Based on Engineered Variants of Human Deoxycytidine Kinase. Mol. Ther. 2012, 20, 1002–1013. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Camino, V.; Harwood, S.L.; Alvarez-Mendez, A.; Alvarez-Vallina, L. Efficacy and toxicity management of CAR-T-cell immunotherapy: A matter of responsiveness control or tumour-specificity? Biochem. Soc. Trans. 2016, 44, 406–411. [Google Scholar] [CrossRef] [PubMed]
- Glienke, W.; Esser, R.; Priesner, C.; Suerth, J.D.; Schambach, A.; Wels, W.S.; Grez, M.; Kloess, S.; Arseniev, L.; Koehl, U. Advantages and applications of CAR-expressing natural killer cells. Front. Pharmacol. 2015, 6, 21. [Google Scholar] [CrossRef]
- Li, L.; Li, W.; Wang, C.; Yan, X.; Wang, Y.; Niu, C.; Zhang, X.; Li, M.; Tian, H.; Yao, C.; et al. Adoptive transfer of natural killer cells in combination with chemotherapy improves outcomes of patients with locally advanced colon carcinoma. Cytotherapy 2018, 20, 134–148. [Google Scholar] [CrossRef]
- Schmidt-Wolf, I.G.; Negrin, R.S.; Kiem, H.P.; Blume, K.G.; Weissman, I.L. Use of a SCID mouse/human lymphoma model to evaluate cytokine-induced killer cells with potent antitumor cell activity. J. Exp. Med. 1991, 174, 139–149. [Google Scholar] [CrossRef]
- Schmeel, L.C.; Schmeel, F.; Coch, C.; Schmidt-Wolf, I.G.H. Cytokine-induced killer (CIK) cells in cancer immunotherapy: Report of the international registry on CIK cells (IRCC). J. Cancer Res. Clin. Oncol. 2015, 141, 839–849. [Google Scholar] [CrossRef]
- Pan, K.; Wang, Q.-J.; Liu, Q.; Zheng, H.-X.; Li, Y.-Q.; Weng, D.-S.; Li, J.-J.; Huang, L.-X.; He, J.; Chen, S.-P.; et al. The phenotype of ex vivo generated cytokine-induced killer cells is associated with overall survival in patients with cancer. Tumor Biol. 2014, 35, 701–707. [Google Scholar] [CrossRef]
- Pan, Q.-Z.; Zhao, J.-J.; Yang, C.-P.; Zhou, Y.-Q.; Lin, J.-Z.; Tang, Y.; Gu, J.-M.; Wang, Q.-J.; Li, Y.-Q.; He, J.; et al. Efficacy of adjuvant cytokine-induced killer cell immunotherapy in patients with colorectal cancer after radical resection. OncoImmunology 2020, 9, 1752563. [Google Scholar] [CrossRef]
- Zhang, J.; Zhu, L.; Zhang, Q.; He, X.; Yin, Y.; Gu, Y.; Guo, R.; Lu, K.; Liu, L.; Liu, P.; et al. Effects of cytokine-induced killer cell treatment in colorectal cancer patients: A retrospective study. Biomed. Pharmacother. 2014, 68, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Wang, Y.; Yu, J.; Wei, F.; Cao, S.; Zhang, X.; Dong, N.; Li, H.; Ren, X. Autologous Cytokine-Induced Killer Cells Improves Overall Survival of Metastatic Colorectal Cancer Patients: Results From a Phase II Clinical Trial. Clin. Color. Cancer 2016, 15, 228–235. [Google Scholar] [CrossRef] [PubMed]
- DeMaria, O.; Cornen, S.; Daëron, M.; Morel, Y.; Medzhitov, R.; Vivier, E. Harnessing innate immunity in cancer therapy. Nature 2019, 574, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zeng, H.; Xu, R.-H.; Liu, B.; Li, Z. Vaccination with Human Pluripotent Stem Cells Generates a Broad Spectrum of Immunological and Clinical Responses Against Colon Cancer. Stem Cells 2009, 27, 3103–3111. [Google Scholar] [CrossRef]
- Palucka, K.; Banchereau, J. Cancer immunotherapy via dendritic cells. Nat. Rev. Cancer 2012, 12, 265–277. [Google Scholar] [CrossRef]
- Gao, D.; Li, C.; Xie, X.; Zhao, P.; Wei, X.; Sun, W.; Liu, H.-C.; Alexandrou, A.T.; Jones, J.; Zhao, R.; et al. Autologous Tumor Lysate-Pulsed Dendritic Cell Immunotherapy with Cytokine-Induced Killer Cells Improves Survival in Gastric and Colorectal Cancer Patients. PLoS ONE 2014, 9, e93886. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Huang, L.; Chen, L.; Lin, X.; Chen, L.; Zheng, Q. Effect of dendritic cell-cytokine-induced killer cells in patients with advanced colorectal cancer combined with first-line treatment. World J. Surg. Oncol. 2017, 15, 209. [Google Scholar] [CrossRef]
- Zhu, H.; Yang, X.; Li, J.; Ren, Y.; Zhang, T.; Zhang, C.; Zhang, J.; Li, J.; Pang, Y. Immune Response, Safety, and Survival and Quality of Life Outcomes for Advanced Colorectal Cancer Patients Treated with Dendritic Cell Vaccine and Cytokine-Induced Killer Cell Therapy. BioMed Res. Int. 2014, 2014, 603871. [Google Scholar] [CrossRef]
- Folkman, J. Role of angiogenesis in tumor growth and metastasis. Semin. Oncol. 2002, 29 (Suppl. 16), 15–18. [Google Scholar] [CrossRef]
- Rosen, L.S.; Jacobs, I.A.; Burkes, R.L. Bevacizumab in Colorectal Cancer: Current Role in Treatment and the Potential of Biosimilars. Target. Oncol. 2017, 12, 599–610. [Google Scholar] [CrossRef]
- Riechelmann, R.; Grothey, A. Antiangiogenic therapy for refractory colorectal cancer: Current options and future strategies. Ther. Adv. Med. Oncol. 2017, 9, 106–126. [Google Scholar] [CrossRef] [PubMed]
- FlFlorescu-Ţenea, R.; Kamal, A.; Mitruţ, P.; Mitruţ, R.; Ilie, D.; Nicolaescu, A.; Mogoantă, L. Colorectal Cancer: An Update on Treatment Options and Future Perspectives. Curr. Health Sci. J. 2019, 45, 134–141. [Google Scholar]
- JaJain, R.K. Normalization of Tumor Vasculature: An Emerging Concept in Antiangiogenic Therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef] [PubMed]
- de Aguiar, R.B.; de Moraes, J.Z. Exploring the Immunological Mechanisms Underlying the Anti-vascular Endothelial Growth Factor Activity in Tumors. Front. Immunol. 2019, 10, 1023. [Google Scholar] [CrossRef]
- Willett, C.G.; Boucher, Y.; di Tomaso, E.; Duda, D.G.; Munn, L.L.; Tong, R.T.; Chung, D.C.; Sahani, D.V.; Kalva, S.P.; Kozin, S.V.; et al. Direct evidence that the VEGF-specific antibody bevacizumab has antivascular effects in human rectal cancer. Nat. Med. 2004, 10, 145–147. [Google Scholar] [CrossRef]
- Shrimali, R.K.; Yu, Z.; Theoret, M.R.; Chinnasamy, D.; Restifo, N.P.; Rosenberg, S.A. Antiangiogenic Agents Can Increase Lymphocyte Infiltration into Tumor and Enhance the Effectiveness of Adoptive Immunotherapy of Cancer. Cancer Res. 2010, 70, 6171–6180. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, Y.; Dai, Y.; Cheng, J.-N.; Gong, Z.; Feng, Y.; Sun, C.; Jia, Q.; Zhu, B. Immune Landscape of Colorectal Cancer Tumor Microenvironment from Different Primary Tumor Location. Front. Immunol. 2018, 9, 1578. [Google Scholar] [CrossRef]
- Tada, Y.; Togashi, Y.; Kotani, D.; Kuwata, T.; Sato, E.; Kawazoe, A.; Doi, T.; Wada, H.; Nishikawa, H.; Shitara, K. Targeting VEGFR2 with Ramucirumab strongly impacts effector/activated regulatory T cells and CD8+ T cells in the tumor microenvironment. J. Immunother. Cancer 2018, 6, 106. [Google Scholar] [CrossRef]
- Wada, J.; Suzuki, H.; Fuchino, R.; Yamasaki, A.; Nagai, S.; Yanai, K.; Koga, K.; Nakamura, M.; Tanaka, M.; Morisaki, T.; et al. The contribution of vascular endothelial growth factor to the induction of regulatory T-cells in malignant effusions. Anticancer Res. 2009, 29, 881–888. [Google Scholar] [PubMed]
- Oyama, T.; Ran, S.; Ishida, T.; Nadaf, S.; Kerr, L.; Carbone, D.P.; Gabrilovich, D.I. Vascular endothelial growth factor affects dendritic cell maturation through the inhibition of nuclear factor-kappa B activation in hemopoietic progenitor cells. J. Immunol. 1998, 160, 1224–1232. [Google Scholar]
- Cassidy, J.D.; Clarke, S.; Diaz-Rubio, E.; Scheithauer, W.; Figer, A.; Wong, R.; Koski, S.; Rittweger, K.; Gilberg, F.; Saltz, L. XELOX vs FOLFOX-4 as first-line therapy for metastatic colorectal cancer: NO16966 updated results. Br. J. Cancer 2011, 105, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, P.; Grothey, A. Commentary on a Phase III Trial of Bevacizumab plus XELOX or FOLFOX4 for First-Line Treatment of Metastatic Colorectal Cancer: The NO16966 Trial. Clin. Color. Cancer 2006, 6, 261–264. [Google Scholar] [CrossRef] [PubMed]
- Giantonio, B.J.; Catalano, P.J.; Meropol, N.J.; O’Dwyer, P.J.; Mitchell, E.P.; Alberts, S.R.; Schwartz, M.A.; Benson, A.B., III. Bevacizumab in Combination With Oxaliplatin, Fluorouracil, and Leucovorin (FOLFOX4) for Previously Treated Metastatic Colorectal Cancer: Results From the Eastern Cooperative Oncology Group Study E3200. J. Clin. Oncol. 2007, 25, 1539–1544. [Google Scholar] [CrossRef]
- Yoshida, Y.; Naito, M.; Yamada, T.; Aisu, N.; Daibo, K.; Mera, T.; Tanaka, T.; Naito, K.; Yasumoto, K.; Kamigaki, T.; et al. Adoptive Chemoimmunotherapy Using Activated αβ T Cells for Stage IV Colorectal Cancer. Anticancer Res. 2016, 36, 3741–3746. [Google Scholar]
- Van Cutsem, E.; Tabernero, J.; Lakomy, R.; Prenen, H.; Prausová, J.; Macarulla, T.; Ruff, P.; van Hazel, G.A.; Moiseyenko, V.; Ferry, D.; et al. Addition of Aflibercept to Fluorouracil, Leucovorin, and Irinotecan Improves Survival in a Phase III Randomized Trial in Patients With Metastatic Colorectal Cancer Previously Treated With an Oxaliplatin-Based Regimen. J. Clin. Oncol. 2012, 30, 3499–3506. [Google Scholar] [CrossRef] [PubMed]
- Folprecht, G.; Pericay, C.; Saunders, M.; Thomas, A.; Lopez, R.L.; Roh, J.; Chistyakov, V.; Höhler, T.; Kim, J.-S.; Hofheinz, R.-D.; et al. Oxaliplatin and 5-FU/folinic acid (modified FOLFOX6) with or without aflibercept in first-line treatment of patients with metastatic colorectal cancer: The AFFIRM study. Ann. Oncol. 2016, 27, 1273–1279. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Peeters, M.; Siena, S.; Humblet, Y.; Hendlisz, A.; Neyns, B.; Canon, J.-L.; Van Laethem, J.-L.; Maurel, J.; Richardson, G.; et al. Open-Label Phase III Trial of Panitumumab Plus Best Supportive Care Compared With Best Supportive Care Alone in Patients With Chemotherapy-Refractory Metastatic Colorectal Cancer. J. Clin. Oncol. 2007, 25, 1658–1664. [Google Scholar] [CrossRef]
- Bardhan, K.; Liu, K. Epigenetics and Colorectal Cancer Pathogenesis. Cancers 2013, 5, 676–713. [Google Scholar] [CrossRef]
- Velez, A.A.; Howard, M. Tumor-suppressor genes, cell cycle regulatory checkpoints, and the skin. N. Am. J. Med Sci. 2015, 7, 176–188. [Google Scholar] [CrossRef]
- WaWang, L.-H.; Wu, C.-F.; Rajasekaran, N.; Shin, Y.K. Loss of Tumor Suppressor Gene Function in Human Cancer: An Overview. Cell. Physiol. Biochem. 2018, 51, 2647–2693. [Google Scholar] [CrossRef]
- Patelli, G.; Tosi, F.; Amatu, A.; Mauri, G.; Curaba, A.; Patanè, D.; Pani, A.; Scaglione, F.; Siena, S.; Sartore-Bianchi, A. Strategies to tackle RAS-mutated metastatic colorectal cancer. ESMO Open 2021, 6, 100156. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Nosho, K.; Kirkner, G.J.; Kawasaki, T.; Meyerhardt, J.A.; Loda, M.; Giovannucci, E.L.; Fuchs, C.S. CpG island methylator phenotype, microsatellite instability, BRAF mutation and clinical outcome in colon cancer. Gut 2009, 58, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, H.; Bardelli, A.; Lengauer, C.; Kinzler, K.W.; Vogelstein, B.; Velculescu, V.E. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature 2002, 418, 934. [Google Scholar] [CrossRef] [PubMed]
- Samowitz, W.S.; Curtin, K.; Schaffer, D.; Robertson, M.; Leppert, M.; Slattery, M.L. Relationship of Ki-ras mutations in colon cancers to tumor location, stage, and survival: A population-based study. Cancer Epidemiol. Biomark. Prev. 2000, 9, 1193–1197. [Google Scholar]
- Nosho, K.; Kawasaki, T.; Longtine, J.A.; Fuchs, C.S.; Ohnishi, M.; Suemoto, Y.; Kirkner, G.J.; Zepf, D.; Yan, L.; Ogino, S. PIK3CA Mutation in Colorectal Cancer: Relationship with Genetic and Epigenetic Alterations. Neoplasia 2008, 10, 534–541. [Google Scholar] [CrossRef]
- Barault, L.; Veyrie, N.; Jooste, V.; Lecorre, D.; Chapusot, C.; Ferraz, J.-M.; Lièvre, A.; Cortet, M.; Bouvier, A.-M.; Rat, P.; et al. Mutations in the RAS-MAPK, PI(3)K (phosphatidylinositol-3-OH kinase) signaling network correlate with poor survival in a population-based series of colon cancers. Int. J. Cancer 2008, 122, 2255–2259. [Google Scholar] [CrossRef]
- Li, X.-L.; Zhou, J.; Chen, Z.-R.; Chng, W.-J. p53mutations in colorectal cancer- molecular pathogenesis and pharmacological reactivation. World J. Gastroenterol. 2015, 21, 84–93. [Google Scholar] [CrossRef]
- Narayan, S.; Roy, D. Role of APC and DNA mismatch repair genes in the development of colorectal cancers. Mol. Cancer 2003, 2, 41. [Google Scholar] [CrossRef]
- Porru, M.; Pompili, L.; Caruso, C.; Biroccio, A.; Leonetti, C. Targeting KRAS in metastatic colorectal cancer: Current strategies and emerging opportunities. J. Exp. Clin. Cancer Res. 2018, 37, 57. [Google Scholar] [CrossRef]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef]
- Malumbres, M.; Pellicer, A. RAS pathways to cell cycle control and cell transformation. Front. Biosci. 1998, 3, d887–d912. [Google Scholar] [PubMed]
- Imamura, Y.; Morikawa, T.; Liao, X.; Lochhead, P.; Kuchiba, A.; Yamauchi, M.; Qian, Z.R.; Nishihara, R.; Meyerhardt, J.A.; Haigis, K.M.; et al. Specific mutations in KRAS codons 12 and 13, and patient prognosis in 1075 BRAF wild-type colorectal cancers. Clin. Cancer Res. 2012, 18, 4753–4763. [Google Scholar] [CrossRef] [PubMed]
- Calcagno, S.R.; Li, S.; Colon, M.; Kreinest, P.A.; Thompson, E.A.; Fields, A.P.; Murray, N.R. OncogenicK-ras promotes early carcinogenesis in the mouse proximal colon. Int. J. Cancer 2008, 122, 2462–2470. [Google Scholar] [CrossRef]
- Spandidos, D.A.; Kerr, I.B. Elevated expression of the human ras oncogene family in premalignant and malignant tumours of the colorectum. Br. J. Cancer 1984, 49, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Sansom, O.J.; Meniel, V.; Wilkins, J.A.; Cole, A.M.; Oien, K.A.; Marsh, V.; Jamieson, T.J.; Guerra, C.; Ashton, G.H.; Barbacid, M.; et al. Loss of Apc allows phenotypic manifestation of the transforming properties of an endogenous K-ras oncogene in vivo. Proc. Natl. Acad. Sci. USA 2006, 103, 14122–14127. [Google Scholar] [CrossRef] [PubMed]
- Moon, B.S.; Jeong, W.J.; Park, J.; Kim, T.I.; Min, D.S.; Choi, K.Y. Role of oncogenic K-Ras in cancer stem cell activation by aberrant Wnt/β-catenin signaling. J. Natl. Cancer Inst. 2014, 106, djt373. [Google Scholar] [CrossRef]
- Baud, V.; Karin, M. Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat. Rev. Drug Discov. 2009, 8, 33–40. [Google Scholar] [CrossRef]
- Hamarsheh, S.; Groß, O.; Brummer, T.; Zeiser, R. Immune modulatory effects of oncogenic KRAS in cancer. Nat. Commun. 2020, 11, 5439. [Google Scholar] [CrossRef]
- Prestipino, A.; Zeiser, R. Clinical implications of tumor-intrinsic mechanisms regulating PD-L1. Sci. Transl. Med. 2019, 11, eaav4810. [Google Scholar] [CrossRef]
- Liu, C.; Zheng, S.; Jin, R.; Wang, X.; Wang, F.; Zang, R.; Xu, H.; Lu, Z.; Huang, J.; Lei, Y.; et al. The superior efficacy of anti-PD-1/PD-L1 immunotherapy in KRAS-mutant non-small cell lung cancer that correlates with an inflammatory phenotype and increased immunogenicity. Cancer Lett. 2020, 470, 95–105. [Google Scholar] [CrossRef]
- Zdanov, S.; Mandapathil, M.; Abu Eid, R.; Adamson-Fadeyi, S.; Wilson, W.; Qian, J.; Carnie, A.; Tarasova, N.; Mkrtichyan, M.; Berzofsky, J.A.; et al. Mutant KRAS Conversion of Conventional T Cells into Regulatory T Cells. Cancer Immunol. Res. 2016, 4, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Testorelli, C.; Bussini, S.; De Filippi, R.; Marelli, O.; Orlando, L.; Greiner, J.W.; Grohmann, U.; Tentori, L.; Giuliani, A.; Bonmassar, E.; et al. Dacarbazine-induced immunogenicity of a murine leukemia is attenuated in cells transfected with mutated K-ras gene. J. Exp. Clin. Cancer Res. 1997, 16, 15–22. [Google Scholar] [PubMed]
- Smakman, N.; Veenendaal, L.M.; van Diest, P.; Bos, R.; Offringa, R.; Rinkes, I.H.M.B.; Kranenburg, O. Dual effect of KrasD12 knockdown on tumorigenesis: Increased immune-mediated tumor clearance and abrogation of tumor malignancy. Oncogene 2005, 24, 8338–8342. [Google Scholar] [CrossRef] [PubMed]
- Pylayeva-Gupta, Y.; Lee, K.E.; Hajdu, C.H.; Miller, G.; Bar-Sagi, D. Oncogenic Kras-Induced GM-CSF Production Promotes the Development of Pancreatic Neoplasia. Cancer Cell 2012, 21, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Bayne, L.J.; Beatty, G.L.; Jhala, N.; Clark, C.E.; Rhim, A.D.; Stanger, B.Z.; Vonderheide, R.H. Tumor-Derived Granulocyte-Macrophage Colony-Stimulating Factor Regulates Myeloid Inflammation and T Cell Immunity in Pancreatic Cancer. Cancer Cell 2012, 21, 822–835. [Google Scholar] [CrossRef] [PubMed]
- Li, W.Q.; Kawakami, K.; Ruszkiewicz, A.; Bennett, G.; Moore, J.; Iacopetta, B. BRAF mutations are associated with distinctive clinical, pathological and molecular features of colorectal cancer independently of microsatellite instability status. Mol. Cancer 2006, 5, 2. [Google Scholar] [CrossRef]
- Wan, P.T.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Cancer Genome Project; Jones, C.M.; Marshall, C.J.; Springer, C.J.; et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef]
- SaSanz-Garcia, E.; Argiles, G.; Elez, E.; Tabernero, J. BRAF mutant colorectal cancer: Prognosis, treatment, and new perspectives. Ann. Oncol. 2017, 28, 2648–2657. [Google Scholar] [CrossRef]
- Nakayama, I.; Hirota, T.; Shinozaki, E. BRAF Mutation in Colorectal Cancers: From Prognostic Marker to Targetable Mutation. Cancers 2020, 12, 3236. [Google Scholar] [CrossRef]
- Ikenoue, T.; Hikiba, Y.; Kanai, F.; Tanaka, Y.; Imamura, J.; Imamura, T.; Ohta, M.; Ijichi, H.; Tateishi, K.; Kawakami, T.; et al. Functional analysis of mutations within the kinase activation segment of B-Raf in human colorectal tumors. Cancer Res. 2003, 63, 8132–8137. [Google Scholar]
- ParPark, K.-S.; Kim, N.G.; Kim, J.J.; Kim, H.; Ahn, Y.H.; Choi, K.Y. Differential regulation of MAP kinase cascade in human colorectal tumorigenesis. Br. J. Cancer 1999, 81, 1116–1121. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nagasaka, T.; Sasamoto, H.; Notohara, K.; Cullings, H.M.; Takeda, M.; Kimura, K.; Kambara, T.; MacPhee, D.G.; Young, J.; Leggett, B.A.; et al. Colorectal Cancer With Mutation in BRAF, KRAS, and Wild-Type With Respect to Both Oncogenes Showing Different Patterns of DNA Methylation. J. Clin. Oncol. 2004, 22, 4584–4594. [Google Scholar] [CrossRef]
- Sumimoto, H.; Imabayashi, F.; Iwata, T.; Kawakami, Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J. Exp. Med. 2006, 203, 1651–1656. [Google Scholar] [CrossRef] [PubMed]
- Ilieva, K.M.; Correa, I.; Josephs, D.H.; Karagiannis, P.; Egbuniwe, I.U.; Cafferkey, M.J.; Spicer, J.F.; Harries, M.; Nestle, F.O.; Lacy, K.E.; et al. Effects of BRAF mutations and BRAF inhibition on immune responses to melanoma. Mol. Cancer Ther. 2014, 13, 2769–2783. [Google Scholar] [CrossRef] [PubMed]
- Lelliott, E.J.; McArthur, G.A.; Oliaro, J.; Sheppard, K.E. Immunomodulatory Effects of BRAF, MEK, and CDK4/6 Inhibitors: Implications for Combining Targeted Therapy and Immune Checkpoint Blockade for the Treatment of Melanoma. Front. Immunol. 2021, 12, 661737. [Google Scholar] [CrossRef]
- Wilmott, J.S.; Long, G.V.; Howle, J.R.; Haydu, L.E.; Sharma, R.N.; Thompson, J.F.; Kefford, R.F.; Hersey, P.; Scolyer, R.A. Selective BRAF Inhibitors Induce Marked T-cell Infiltration into Human Metastatic Melanoma. Clin. Cancer Res. 2012, 18, 1386–1394. [Google Scholar] [CrossRef]
- Cooper, Z.A.; Juneja, V.R.; Sage, P.T.; Frederick, D.T.; Piris, A.; Mitra, D.; Lo, J.A.; Hodi, F.S.; Freeman, G.J.; Bosenberg, M.W.; et al. Response to BRAF Inhibition in Melanoma Is Enhanced When Combined with Immune Checkpoint Blockade. Cancer Immunol. Res. 2014, 2, 643–654. [Google Scholar] [CrossRef]
- Reichardt, P.; Pink, D. Molecular targeted therapy of gastrointestinal stromal tumors. Curr. Cancer Drug Targets 2011, 11, 688–697. [Google Scholar] [CrossRef]
- ZhZhao, B.; Wang, L.; Qiu, H.; Zhang, M.; Sun, L.; Peng, P.; Yu, Q.; Yuan, X. Mechanisms of resistance to anti-EGFR therapy in colorectal cancer. Oncotarget 2017, 8, 3980–4000. [Google Scholar] [CrossRef]
- De Roock, W.; Claes, B.; Bernasconi, D.; De Schutter, J.; Biesmans, B.; Fountzilas, G.; Kalogeras, K.T.; Kotoula, V.; Papamichael, D.; Laurent-Puig, P.; et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: A retrospective consortium analysis. Lancet Oncol. 2010, 11, 753–762. [Google Scholar] [CrossRef]
- Kopetz, S.; Guthrie, K.A.; Morris, V.K.; Lenz, H.-J.; Magliocco, A.M.; Maru, D.; Yan, Y.; Lanman, R.; Manyam, G.; Hong, D.S.; et al. Randomized Trial of Irinotecan and Cetuximab With or Without Vemurafenib in BRAF-Mutant Metastatic Colorectal Cancer (SWOG S1406). J. Clin. Oncol. 2021, 39, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Huijberts, S.; Grothey, A.; Yaeger, R.; Cuyle, P.J.; Elez, E.; Fakih, M.; Montagut, C.; Peeters, M.; Yoshino, T.; et al. Binimetinib, Encorafenib, and Cetuximab Triplet Therapy for Patients With BRAF V600E-Mutant Metastatic Colorectal Cancer: Safety Lead-In Results From the Phase III BEACON Colorectal Cancer Study. J. Clin. Oncol. 2019, 37, 1460–1469. [Google Scholar] [CrossRef] [PubMed]
- Welsh, S.J.; Corrie, P.G. Management of BRAF and MEK inhibitor toxicities in patients with metastatic melanoma. Ther. Adv. Med. Oncol. 2015, 7, 122–136. [Google Scholar] [CrossRef] [PubMed]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef]
- Spano, J.-P.; Lagorce, C.; Atlan, D.; Milano, G.; Domont, J.; Benamouzig, R.; Attar, A.; Benichou, J.; Martin, A.; Morere, J.-F.; et al. Impact of EGFR expression on colorectal cancer patient prognosis and survival. Ann. Oncol. 2005, 16, 102–108. [Google Scholar] [CrossRef]
- Jin, K.-T.; Chen, B.; Liu, Y.-Y.; Lan, H.U.-R.; Yan, J.-P. Monoclonal antibodies and chimeric antigen receptor (CAR) T cells in the treatment of colorectal cancer. Cancer Cell Int. 2021, 21, 83. [Google Scholar] [CrossRef]
- Yarom, N.; Jonker, D.J. The role of the epidermal growth factor receptor in the mechanism and treatment of colorectal cancer. Discov. Med. 2011, 11, 95–105. [Google Scholar]
- Cunningham, D.; Humblet, Y.; Siena, S.; Khayat, D.; Bleiberg, H.; Santoro, A.; Bets, D.; Mueser, M.; Harstrick, A.; Verslype, C.; et al. Cetuximab Monotherapy and Cetuximab plus Irinotecan in Irinotecan-Refractory Metastatic Colorectal Cancer. N. Engl. J. Med. 2004, 351, 337–345. [Google Scholar] [CrossRef]
- Veluchamy, J.P.; Spanholtz, J.; Tordoir, M.; Thijssen, V.L.; Heideman, D.A.M.; Verheul, H.M.W.; de Gruijl, T.D.; van der Vliet, H.J. Combination of NK Cells and Cetuximab to Enhance Anti-Tumor Responses in RAS Mutant Metastatic Colorectal Cancer. PLoS ONE 2016, 11, e0157830. [Google Scholar] [CrossRef]
- Fakih, M.; Vincent, M. Adverse events associated with anti-EGFR therapies for the treatment of metastatic colorectal cancer. Curr. Oncol. 2010, 17 (Suppl. 1), S18–S30. [Google Scholar] [CrossRef]
- O’Neil, B.H.; Allen, R.; Spigel, D.R.; Stinchcombe, T.E.; Moore, D.T.; Berlin, J.D.; Goldberg, R.M. High Incidence of Cetuximab-Related Infusion Reactions in Tennessee and North Carolina and the Association With Atopic History. J. Clin. Oncol. 2007, 25, 3644–3648. [Google Scholar] [CrossRef] [PubMed]
- Misale, S.; Yaeger, R.; Hobor, S.; Scala, E.; Janakiraman, M.; Liska, D.; Valtorta, E.; Schiavo, R.; Buscarino, M.; Siravegna, G.; et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 2012, 486, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Amado, R.G.; Wolf, M.; Peeters, M.; Van Cutsem, E.; Siena, S.; Freeman, D.J.; Juan, T.; Sikorski, R.; Suggs, S.; Radinsky, R.; et al. Wild-Type KRAS Is Required for Panitumumab Efficacy in Patients With Metastatic Colorectal Cancer. J. Clin. Oncol. 2008, 26, 1626–1634. [Google Scholar] [CrossRef] [PubMed]
- Douillard, J.Y.; Oliner, K.S.; Siena, S.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N. Engl. J. Med. 2013, 369, 1023–1034. [Google Scholar] [CrossRef]
- Lièvre, A.; Bachet, J.-B.; Le Corre, D.; Boige, V.; Landi, B.; Emile, J.-F.; Côté, J.-F.; Tomasic, G.; Penna, C.; Ducreux, M.; et al. KRAS Mutation Status Is Predictive of Response to Cetuximab Therapy in Colorectal Cancer. Cancer Res. 2006, 66, 3992–3995. [Google Scholar] [CrossRef]
- Chen, S.; Li, X.; Chen, R.; Yin, M.; Zheng, Q. Cetuximab intensifies the ADCC activity of adoptive NK cells in a nude mouse colorectal cancer xenograft model. Oncol. Lett. 2016, 12, 1868–1876. [Google Scholar] [CrossRef]
- Baysal, H.; De Pauw, I.; Zaryouh, H.; Peeters, M.; Vermorken, J.B.; Lardon, F.; De Waele, J.; Wouters, A. The Right Partner in Crime: Unlocking the Potential of the Anti-EGFR Antibody Cetuximab via Combination With Natural Killer Cell Chartering Immunotherapeutic Strategies. Front. Immunol. 2021, 12, 737311. [Google Scholar] [CrossRef] [PubMed]
- Hajjo, R.; Sweidan, K. Review on Epidermal Growth Factor Receptor (EGFR) Structure, Signaling Pathways, Interactions, and Recent Updates of EGFR Inhibitors. Curr. Top. Med. Chem. 2020, 20, 815–834. [Google Scholar]
- Huang, Q.; Xia, J.; Wang, L.; Wang, X.; Ma, X.; Deng, Q.; Lu, Y.; Kumar, M.; Zhou, Z.; Li, L.; et al. miR-153 suppresses IDO1 expression and enhances CAR T cell immunotherapy. J. Hematol. Oncol. 2018, 11, 58. [Google Scholar] [CrossRef]
- Olayioye, M.A.; Neve, R.M.; Lane, H.A.; Hynes, N.E. NEW EMBO MEMBERS’ REVIEW: The ErbB signaling network: Receptor heterodimerization in development and cancer. EMBO J. 2000, 19, 3159–3167. [Google Scholar] [CrossRef]
- Ménard, S.; Pupa, S.M.; Campiglio, M.; Tagliabue, E. Biologic and therapeutic role of HER2 in cancer. Oncogene 2003, 22, 6570–6578. [Google Scholar] [CrossRef] [PubMed]
- Ushiro, H.; Cohen, S. Identification of phosphotyrosine as a product of epidermal growth factor-activated protein kinase in A-431 cell membranes. J. Biol. Chem. 1980, 255, 8363–8365. [Google Scholar] [CrossRef]
- Pohl, M.; Schmiegel, W. Therapeutic Strategies in Diseases of the Digestive Tract—2015 and Beyond Targeted Therapies in Colon Cancer Today and Tomorrow. Dig. Dis. 2016, 34, 574–579. [Google Scholar] [CrossRef] [PubMed]
- Siena, S.; Sartore-Bianchi, A.; Marsoni, S.; Hurwitz, H.; McCall, S.; Penault-Llorca, F.; Srock, S.; Bardelli, A.; Trusolino, L. Targeting the human epidermal growth factor receptor 2 (HER2) oncogene in colorectal cancer. Ann. Oncol. 2018, 29, 1108–1119. [Google Scholar] [CrossRef]
- Sartore-Bianchi, A.; Trusolino, L.; Martino, C.; Bencardino, K.; Lonardi, S.; Bergamo, F.; Zagonel, V.; Leone, F.; Depetris, I.; Martinelli, E.; et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): A proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 738–746. [Google Scholar] [CrossRef]
- Hainsworth, J.D.; Meric-Bernstam, F.; Swanton, C.; Hurwitz, H.; Spigel, D.R.; Sweeney, C.; Burris, H.A.; Bose, R.; Yoo, B.; Stein, A.; et al. Targeted Therapy for Advanced Solid Tumors on the Basis of Molecular Profiles: Results From MyPathway, an Open-Label, Phase II: A Multiple Basket Study. J. Clin. Oncol. 2018, 36, 536–542. [Google Scholar] [CrossRef]
- Pietrantonio, F.; Fucà, G.; Morano, F.; Gloghini, A.; Corso, S.; Aprile, G.; Perrone, F.; De Vita, F.; Tamborini, E.; Tomasello, G.; et al. Biomarkers of Primary Resistance to Trastuzumab in HER2-Positive Metastatic Gastric Cancer Patients: The AMNESIA Case-Control Study. Clin. Cancer Res. 2018, 24, 1082–1089. [Google Scholar] [CrossRef]
- Teng, R.; Zhao, J.; Zhao, Y.; Gao, J.; Li, H.; Zhou, S.; Wang, Y.; Sun, Q.; Lin, Z.; Yang, W.; et al. Chimeric Antigen Receptor–modified T Cells Repressed Solid Tumors and Their Relapse in an Established Patient-derived Colon Carcinoma Xenograft Model. J. Immunother. 2019, 42, 33–42. [Google Scholar] [CrossRef]
- Zhang, B.-L.; Qin, D.-Y.; Mo, Z.-M.; Li, Y.; Wei, W.; Wang, Y.-S.; Wang, W.; Wei, Y.-Q. Hurdles of CAR-T cell-based cancer immunotherapy directed against solid tumors. Sci. China Life Sci. 2016, 59, 340–348. [Google Scholar] [CrossRef]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case Report of a Serious Adverse Event Following the Administration of T Cells Transduced With a Chimeric Antigen Receptor Recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Ramakrishna, S.; Barsan, V.; Mackall, C. Prospects and challenges for use of CAR T cell therapies in solid tumors. Expert Opin. Biol. Ther. 2020, 20, 503–516. [Google Scholar] [CrossRef] [PubMed]
- CChen, D.S.; Mellman, I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.W.; Ngiow, S.F.; Ribas, A.; Smyth, M.J. Classifying Cancers Based on T-cell Infiltration and PD-L1. Cancer Res. 2015, 75, 2139–2145. [Google Scholar] [CrossRef]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef]
- Nishijima, T.F.; Shachar, S.S.; Nyrop, K.A.; Muss, H.B. Safety and Tolerability of PD-1/PD-L1 Inhibitors Compared with Chemotherapy in Patients with Advanced Cancer: A Meta-Analysis. Oncologist 2017, 22, 470–479. [Google Scholar] [CrossRef]
- Benson, A.B.; Venook, A.P.; Al-Hawary, M.M.; Arain, M.A.; Chen, Y.J.; Ciombor, K.K.; Cohen, S.; Cooper, H.S.; Deming, D.; Farkas, L.; et al. Colon Cancer, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Canc. Netw. 2021, 19, 329–359. [Google Scholar] [CrossRef]
- Le, D.T.; Kim, T.W.; Van Cutsem, E.; Geva, R.; Jäger, D.; Hara, H.; Burge, M.; O’Neil, B.; Kavan, P.; Yoshino, T.; et al. Phase II Open-Label Study of Pembrolizumab in Treatment-Refractory, Microsatellite Instability-High/Mismatch Repair-Deficient Metastatic Colorectal Cancer: KEYNOTE-164. J. Clin. Oncol. 2020, 38, 11–19. [Google Scholar] [CrossRef]
- André, T.; Shiu, K.K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef]
- Lenz, H.-J.; Van Cutsem, E.; Limon, M.L.; Wong, K.Y.M.; Hendlisz, A.; Aglietta, M.; García-Alfonso, P.; Neyns, B.; Luppi, G.; Cardin, D.B.; et al. First-Line Nivolumab Plus Low-Dose Ipilimumab for Microsatellite Instability-High/Mismatch Repair-Deficient Metastatic Colorectal Cancer: The Phase II CheckMate 142 Study. J. Clin. Oncol. 2022, 40, 161–170. [Google Scholar] [CrossRef]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.-J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair–Deficient/Microsatellite Instability–High Metastatic Colorectal Cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Abdullaev, S.; André, T.; Lei, M.; Lenz, H.J.; Novotny, J.; Paulson, A.S.; Tejpar, S.; Yamazaki, K.; Ledeine, J.M. A phase III study of nivolumab (NIVO), NIVO + ipilimumab (IPI), or chemotherapy (CT) for microsatellite instability-high (MSI-H)/mismatch repair-deficient (dMMR) metastatic colorectal cancer (mCRC): Checkmate 8HW. J. Clin. Oncol. 2020, 38 (Suppl. 4), TPS266. [Google Scholar] [CrossRef]
- Ooki, A.; Shinozaki, E.; Yamaguchi, K. Immunotherapy in Colorectal Cancer: Current and Future Strategies. J. Anus Rectum Colon 2021, 5, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Rafiq, S.; Yeku, O.O.; Jackson, H.J.; Purdon, T.J.; Van Leeuwen, D.G.; Drakes, D.J.; Song, M.; Miele, M.M.; Li, Z.; Wang, P.; et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat. Biotechnol. 2018, 36, 847–856. [Google Scholar] [CrossRef]
- Liu, X.; Ranganathan, R.; Jiang, S.; Fang, C.; Sun, J.; Kim, S.; Newick, K.; Lo, A.; June, C.H.; Zhao, Y.; et al. A Chimeric Switch-Receptor Targeting PD1 Augments the Efficacy of Second-Generation CAR T Cells in Advanced Solid Tumors. Cancer Res. 2016, 76, 1578–1590. [Google Scholar] [CrossRef]
- Huang, B.; Luo, L.; Wang, J.; He, B.; Feng, R.; Xian, N.; Zhang, Q.; Chen, L.; Huang, G. B7-H3 specific T cells with chimeric antigen receptor and decoy PD-1 receptors eradicate established solid human tumors in mouse models. OncoImmunology 2019, 9, 1684127. [Google Scholar] [CrossRef]
- Hammarström, S. The carcinoembryonic antigen (CEA) family: Structures, suggested functions and expression in normal and malignant tissues. Semin. Cancer Biol. 1999, 9, 67–81. [Google Scholar] [CrossRef]
- Nap, M.; Wilhelm, W.W. Immunohistology of carcinoembryonic antigen (CEA). Specificity of reagents, choice of control tissues and practical application in surgical pathology. Int. J. Biol. Markers 1992, 7, 148–153. [Google Scholar] [CrossRef]
- Li, Y.; Cao, H.; Jiao, Z.; Pakala, S.B.; Sirigiri, D.N.; Li, W.; Kumar, R.; Mishra, L. Carcinoembryonic antigen interacts with TGF-{beta} receptor and inhibits TGF-{beta} signaling in colorectal cancers. Cancer Res. 2010, 70, 8159–8168. [Google Scholar] [CrossRef]
- Camacholeal, P.; Stanners, C.P. The human carcinoembryonic antigen (CEA) GPI anchor mediates anoikis inhibition by inactivation of the intrinsic death pathway. Oncogene 2007, 27, 1545–1553. [Google Scholar] [CrossRef]
- Bast, R.C.; Ravdin, P.; Hayes, D.F.; Bates, S.; Fritsche, H.; Jessup, J.M.; Kemeny, N.; Locker, G.Y.; Mennel, R.G.; Somerfield, M.R. 2000 Update of Recommendations for the Use of Tumor Markers in Breast and Colorectal Cancer: Clinical Practice Guidelines of the American Society of Clinical Oncology. J. Clin. Oncol. 2001, 19, 1865–1878. [Google Scholar] [CrossRef] [PubMed]
- Vogel, I.; Francksen, H.; Soeth, E.; Henne-Bruns, D.; Kremer, B.; Juhl, H. The carcinoembryonic antigen and its prognostic impact on immunocytologically detected intraperitoneal colorectal cancer cells. Am. J. Surg. 2001, 181, 188–193. [Google Scholar] [CrossRef]
- Hostetter, R.B.; Augustus, L.B.; Mankarious, R.; Chi, K.F.; Fan, D.; Toth, C.; Thomas, P.; Jessup, J.M. Carcinoembryonic antigen as a selective enhancer of colorectal cancer metastasis. JNCI J. Natl. Cancer Inst. 1990, 82, 380–385. [Google Scholar] [CrossRef] [PubMed]
- Liersch, T.; Meller, J.; Bittrich, M.; Kulle, B.; Becker, H.; Goldenberg, D.M. Update of Carcinoembryonic Antigen Radioimmunotherapy with 131I-Labetuzumab After Salvage Resection of Colorectal Liver Metastases: Comparison of Outcome to a Contemporaneous Control Group. Ann. Surg. Oncol. 2007, 14, 2577–2590. [Google Scholar] [CrossRef]
- Sahlmann, C.O.; Homayounfar, K.; Niessner, M.; Dyczkowski, J.; Conradi, L.C.; Braulke, F.; Meller, B.; Beißbarth, T.; Ghadimi, B.M.; Meller, J.; et al. Repeated adjuvant anti-CEA radioimmunotherapy after resection of colorectal liver metastases: Safety, feasibility, and long-term efficacy results of a prospective phase 2 study. Cancer 2017, 123, 638–649. [Google Scholar] [CrossRef]
- Chi, X.; Yang, P.; Zhang, E.; Gu, J.; Xu, H.; Li, M.; Gao, X.; Li, X.; Zhang, Y.; Xu, H.; et al. Significantly increased anti-tumor activity of carcinoembryonic antigen-specific chimeric antigen receptor T cells in combination with recombinant human IL-12. Cancer Med. 2019, 8, 4753–4765. [Google Scholar] [CrossRef]
- Hombach, A.A.; Geumann, U.; Günther, C.; Hermann, F.G.; Abken, H. IL7-IL12 Engineered Mesenchymal Stem Cells (MSCs) Improve A CAR T Cell Attack Against Colorectal Cancer Cells. Cells 2020, 9, 873. [Google Scholar] [CrossRef]
- Osada, T.; Hsu, D.; Hammond, S.; Hobeika, A.; Devi, G.; Clay, T.M.; Lyerly, H.; Morse, M.A. Metastatic colorectal cancer cells from patients previously treated with chemotherapy are sensitive to T-cell killing mediated by CEA/CD3-bispecific T-cell-engaging BiTE antibody. Br. J. Cancer 2009, 102, 124–133. [Google Scholar] [CrossRef]
- Oberst, M.D.; Fuhrmann, S.; Mulgrew, K.; Amann, M.; Cheng, L.; Lutterbuese, P.; Richman, L.; Coats, S.; Baeuerle, P.A.; Hammond, S.A. CEA/CD3 bispecific antibody MEDI-565/AMG 211 activation of T cells and subsequent killing of human tumors is independent of mutations commonly found in colorectal adenocarcinomas. MAbs 2014, 6, 1571–1584. [Google Scholar] [CrossRef]
- Lutterbuese, R.; Raum, T.; Kischel, R.; Lutterbuese, P.; Schlereth, B.; Schaller, E.; Mangold, S.; Rau, D.; Meier, P.; Kiener, P.A.; et al. Potent Control of Tumor Growth by CEA/CD3-bispecific Single-chain Antibody Constructs That Are Not Competitively Inhibited by Soluble CEA. J. Immunother. 2009, 32, 341–352. [Google Scholar] [CrossRef]
- Hombach, A.A.; Rappl, G.; Abken, H. Blocking CD30 on T Cells by a Dual Specific CAR for CD30 and Colon Cancer Antigens Improves the CAR T Cell Response against CD30− Tumors. Mol. Ther. 2019, 27, 1825–1835. [Google Scholar] [CrossRef] [PubMed]
- Katz, S.; Point, G.R.; Cunetta, M.; Thorn, M.; Guha, P.; Espat, N.J.; Boutros, C.; Hanna, N.; Junghans, R.P. Regional CAR-T cell infusions for peritoneal carcinomatosis are superior to systemic delivery. Cancer Gene Ther. 2016, 23, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Parkhurst, M.R.; Joo, J.; Riley, J.P.; Yu, Z.; Li, Y.; Robbins, P.F.; Rosenberg, S.A. Characterization of Genetically Modified T-Cell Receptors that Recognize the CEA:691-699 Peptide in the Context of HLA-A2.1 on Human Colorectal Cancer Cells. Clin. Cancer Res. 2008, 15, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Parkhurst, M.R.; Yang, J.C.; Langan, R.C.; Dudley, M.E.; Nathan, D.-A.N.; Feldman, S.A.; Davis, J.L.; Morgan, R.A.; Merino, M.J.; Sherry, R.M.; et al. T Cells Targeting Carcinoembryonic Antigen Can Mediate Regression of Metastatic Colorectal Cancer but Induce Severe Transient Colitis. Mol. Ther. 2011, 19, 620–626. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, Z.; Yang, Z.; Wang, M.; Li, S.; Li, Y.; Zhang, R.; Xiong, Z.; Wei, Z.; Shen, J.; et al. Phase I Escalating-Dose Trial of CAR-T Therapy Targeting CEA + Metastatic Colorectal Cancers. Mol. Ther. 2017, 25, 1248–1258. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Target | Inhibitor | Results | References |
|---|---|---|---|
| VEGF | Bevacizumab | Improved OS, PFS and ORR | [18,71,72,73,74] |
| Aflibercept | Improve both OS and PFS | [75] | |
| Both VEGF inhibitors | Induced vascular-related complications | [77] | |
| K-RAS/B-RAF | Vemurafenib | Improved PFS | [121] |
| Encorafenib/Binimetinib | Improved ORR, was associated with toxicity | [122] | |
| EGFR | Cetuximab | Improved OS | [132] |
| HER2 | Trastuzumab/Lapatinib | Improved ORR | [145] |
| Pertuzumab/Trastuzuma | Improved ORR | [146] | |
| PD-1 | Pembrolizumab | Improved PFS | [158,159] |
| Nivolumab * | Improved OS, PFS and ORR | [160,161] | |
| CEA | 131I-labetuzumab | Promising survival advantage, associated with hepatotoxicity | [174,175] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Hujaily, E.M.; Al-Sowayan, B.S.; Alyousef, Z.; Uddin, S.; Alammari, F. Recruiting Immunity for the Fight against Colorectal Cancer: Current Status and Challenges. Int. J. Mol. Sci. 2022, 23, 13696. https://doi.org/10.3390/ijms232213696
Al-Hujaily EM, Al-Sowayan BS, Alyousef Z, Uddin S, Alammari F. Recruiting Immunity for the Fight against Colorectal Cancer: Current Status and Challenges. International Journal of Molecular Sciences. 2022; 23(22):13696. https://doi.org/10.3390/ijms232213696
Chicago/Turabian StyleAl-Hujaily, Ensaf M., Batla S. Al-Sowayan, Zeyad Alyousef, Shahab Uddin, and Farah Alammari. 2022. "Recruiting Immunity for the Fight against Colorectal Cancer: Current Status and Challenges" International Journal of Molecular Sciences 23, no. 22: 13696. https://doi.org/10.3390/ijms232213696
APA StyleAl-Hujaily, E. M., Al-Sowayan, B. S., Alyousef, Z., Uddin, S., & Alammari, F. (2022). Recruiting Immunity for the Fight against Colorectal Cancer: Current Status and Challenges. International Journal of Molecular Sciences, 23(22), 13696. https://doi.org/10.3390/ijms232213696