
Insights into the Orchestration of Gene Transcription Regulators in Helicobacter pylori

 ,
,  , , and
, , and 
Abstract
1. Introduction
2. Genome and Regulatory Functions
3. The Two-Component Systems of H. pylori
3.1. FlgRS Control the Transcription of Flagellar Genes
3.2. ArsRS Regulates the Transcription of Genes Involved in Acid Resistance
3.3. CrdRS Regulates the Transcription of Genes Important for Copper Homeostasis and Central Cellular Responses
3.4. CheY1Y2A Two-Component System
3.5. Orphan Regulators
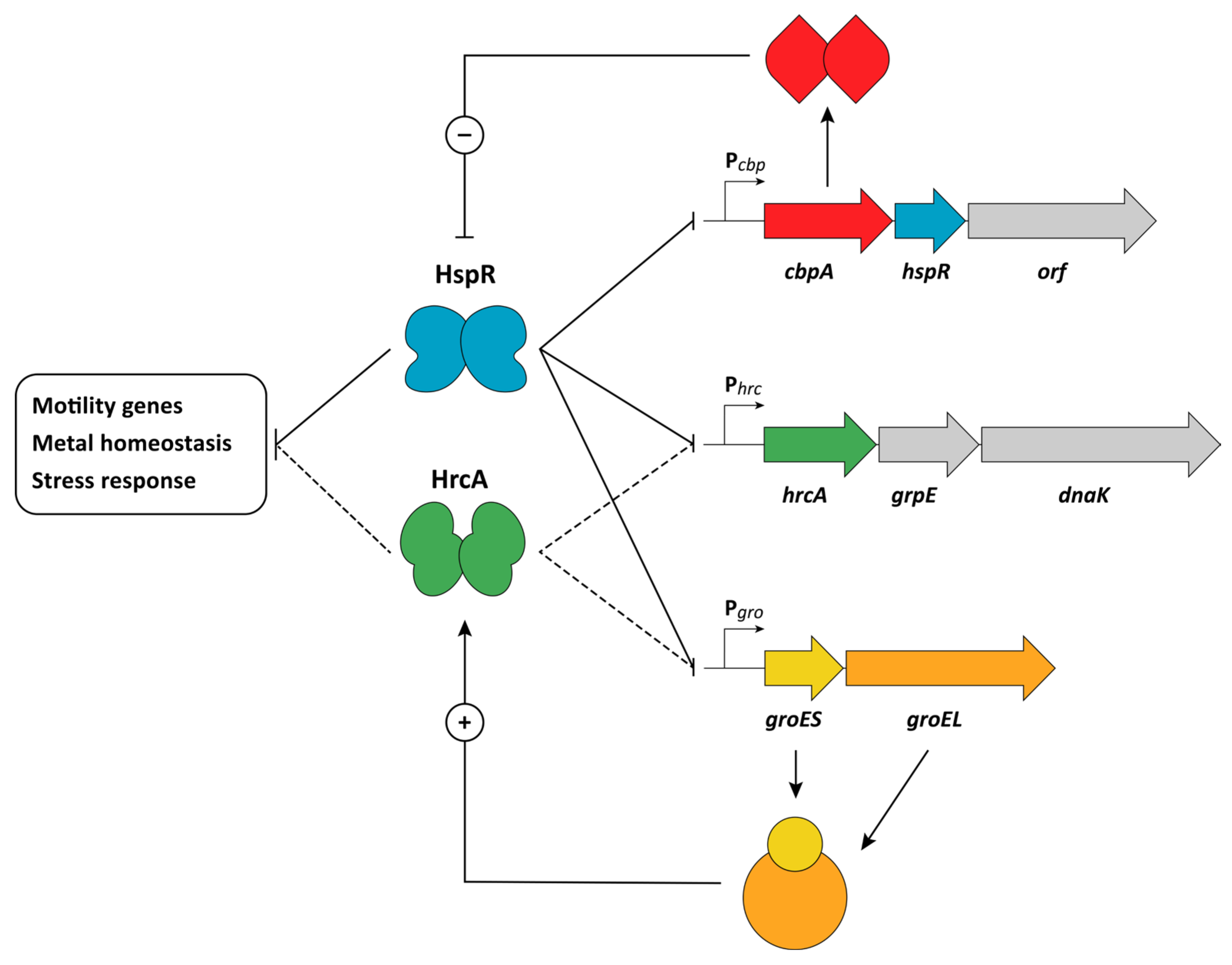
4. Heat-Shock Response
HspR and HrcA Repressors
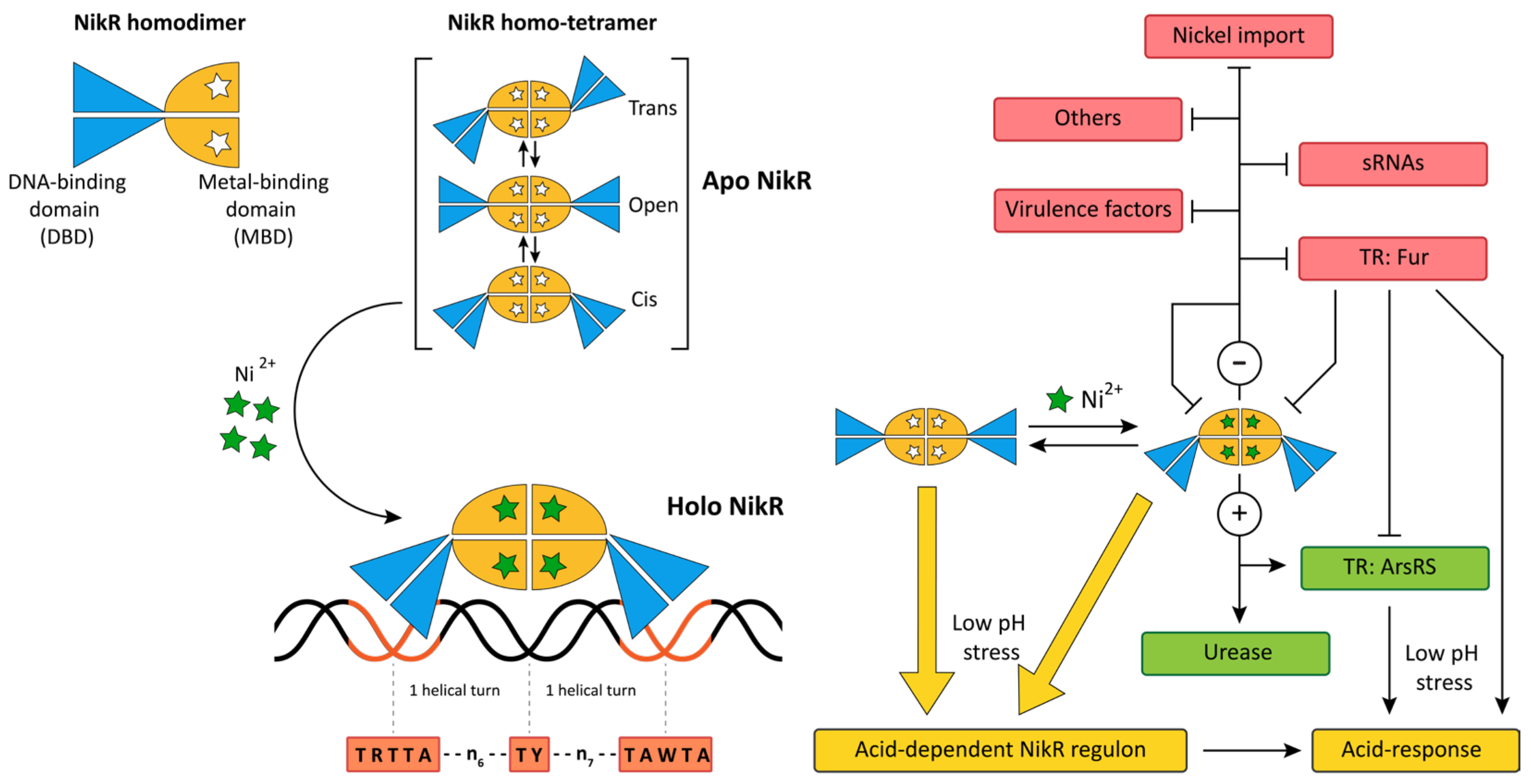
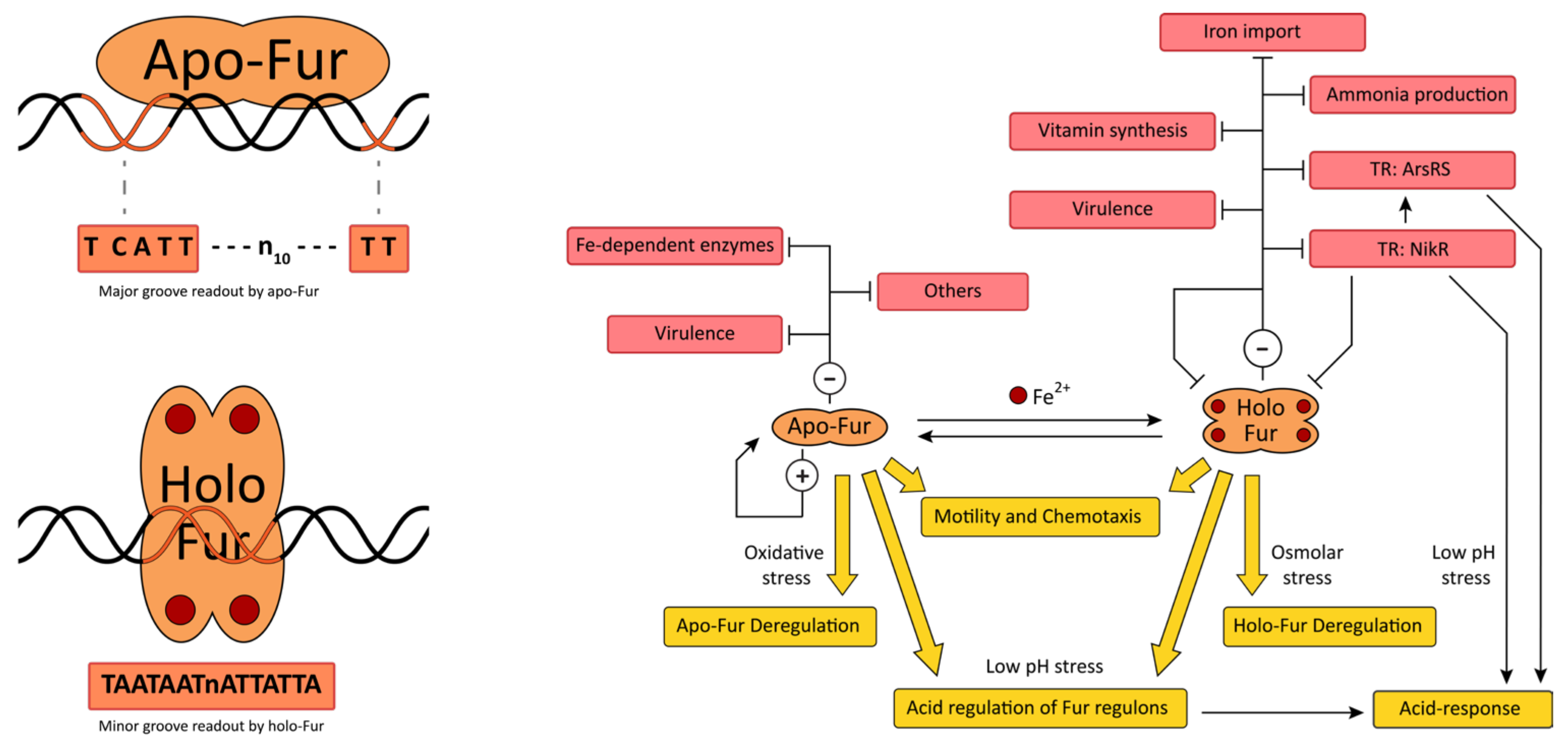
5. Metal Homeostasis
5.1. NikR
5.2. Fur
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Blaser, M.J. Helicobacter pylori and the Pathogenesis of Gastroduodenal Inflammation. J. Infect. Dis. 1990, 161, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Nomura, A.; Stemmermann, G.N.; Chyou, P.-H.; Perez, G.P.; Blaser, M.J. Helicobacter pylori Infection and the Risk for Duodenal and Gastric Ulceration. Ann. Intern. Med. 1994, 120, 977–981. [Google Scholar] [CrossRef] [PubMed]
- Parsonnet, J.; Friedman, G.D.; Vandersteen, D.P.; Chang, Y.; Vogelman, J.H.; Orentreich, N.; Sibley, R.K. Helicobacter pylori Infection and the Risk of Gastric Carcinoma. N. Engl. J. Med. 1991, 325, 1127–1131. [Google Scholar] [CrossRef] [PubMed]
- Cussac, V.; Ferrero, R.L.; Labigne, A. Expression of Helicobacter pylori urease genes in Escherichia coli grown under nitrogen-limiting conditions. J. Bacteriol. 1992, 174, 2466–2473. [Google Scholar] [CrossRef] [PubMed]
- Suerbaum, S.; Josenhans, C.; Labigne, A. Cloning and genetic characterization of the Helicobacter pylori and Helicobacter mustelae flaB flagellin genes and construction of H. pylori flaA- and flaB-negative mutants by electroporation-mediated allelic exchange. J. Bacteriol. 1993, 175, 3278–3288. [Google Scholar] [CrossRef][Green Version]
- Cover, T.; Tummuru, M.; Cao, P.; Thompson, S.; Blaser, M. Divergence of genetic sequences for the vacuolating cytotoxin among Helicobacter pylori strains. J. Biol. Chem. 1994, 269, 10566–10573. [Google Scholar] [CrossRef]
- Telford, J.L.; Ghiara, P.; Dell’Orco, M.; Comanducci, M.; Burroni, D.; Bugnoli, M.; Tecce, M.F.; Censini, S.; Covacci, A.; Xiang, Z. Gene structure of the Helicobacter pylori cytotoxin and evidence of its key role in gastric disease. J. Exp. Med. 1994, 179, 1653–1658. [Google Scholar] [CrossRef]
- Covacci, A.; Censini, S.; Bugnoli, M.; Petracca, R.; Burroni, D.; Macchia, G.; Massone, A.; Papini, E.; Xiang, Z.; Figura, N. Molecular characterization of the 128-kDa immunodominant antigen of Helicobacter pylori associated with cytotoxicity and duodenal ulcer. Proc. Natl. Acad. Sci. USA 1993, 90, 5791–5795. [Google Scholar] [CrossRef]
- Tummuru, M.K.; Cover, T.L.; Blaser, M.J. Cloning and expression of a high-molecular-mass major antigen of Helicobacter pylori: Evidence of linkage to cytotoxin production. Infect. Immun. 1993, 61, 1799–1809. [Google Scholar] [CrossRef]
- Fassbinder, F.; Vliet, V.A.H.; Gimmel, V.; Kusters, J.G.; Kist, M.; Bereswill, S. Identification of iron-regulated genes of Helicobacter pylori by a modified Fur titration assay (FURTA-Hp). FEMS Microbiol. Lett. 2000, 184, 225–229. [Google Scholar] [CrossRef][Green Version]
- Velayudhan, J.; Hughes, N.J.; McColm, A.A.; Bagshaw, J.; Clayton, C.L.; Andrews, S.; Kelly, D.J. Iron acquisition and virulence in Helicobacter pylori: A major role for FeoB, a high-affinity ferrous iron transporter. Mol. Microbiol. 2000, 37, 274–286. [Google Scholar] [CrossRef]
- Tsuda, M.; Karita, M.; Morshed, M.G.; Okita, K.; Nakazawa, T. A urease-negative mutant of Helicobacter pylori constructed by allelic exchange mutagenesis lacks the ability to colonize the nude mouse stomach. Infect. Immun. 1994, 62, 3586–3589. [Google Scholar] [CrossRef]
- Olson, J.W.; Maier, R.J. Molecular Hydrogen as an Energy Source for Helicobacter pylori. Science 2002, 298, 1788–1790. [Google Scholar] [CrossRef]
- De Reuse, H.; Vinella, D.; Cavazza, C. Common themes and unique proteins for the uptake and trafficking of nickel, a metal essential for the virulence of Helicobacter pylori. Front. Cell. Infect. Microbiol. 2013, 3, 94. [Google Scholar] [CrossRef]
- Tomb, J.-F.; White, O.; Kerlavage, A.R.; Clayton, R.A.; Sutton, G.G.; Fleischmann, R.D.; Ketchum, K.A.; Klenk, H.P.; Gill, S.; Dougherty, B.A.; et al. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 1997, 388, 539–547. [Google Scholar] [CrossRef]
- What Is Dementia? Available online: Http://Www.Alz.Org/What-Is-Dementia.Asp (accessed on 3 November 2022).
- Whitmire, J.M.; Merrell, D.S. Helicobacter pylori Genetic Polymorphisms in Gastric Disease Development. Adv. Exp. Med. Biol. 2019, 1149, 173–194. [Google Scholar] [CrossRef]
- Alm, R.A.; Ling, L.-S.L.; Moir, D.T.; King, B.L.; Brown, E.D.; Doig, P.C.; Smith, D.R.; Noonan, B.; Guild, B.C.; Dejonge, B.L.; et al. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature 1999, 397, 176–180. [Google Scholar] [CrossRef]
- Baltrus, D.A.; Amieva, M.R.; Covacci, A.; Lowe, T.M.; Merrell, D.S.; Ottemann, K.M.; Stein, M.; Salama, N.R.; Guillemin, K. The Complete Genome Sequence of Helicobacter pylori Strain G27. J. Bacteriol. 2009, 191, 447–448. [Google Scholar] [CrossRef]
- Behrens, W.; Bönig, T.; Suerbaum, S.; Josenhans, C. Genome Sequence of Helicobacter pylori hpEurope Strain N6. J. Bacteriol. 2012, 194, 3725–3726. [Google Scholar] [CrossRef]
- Zakharova, N.; Hoffman, P.S.; Berg, D.E.; Severinov, K. The Largest Subunits of RNA Polymerase from Gastric Helicobacters Are Tethered. J. Biol. Chem. 1998, 273, 19371–19374. [Google Scholar] [CrossRef]
- Zakharova, N.; Paster, B.J.; Wesley, I.; Dewhirst, F.E.; Berg, D.E.; Severinov, K.V. Fused and Overlapping rpoB and rpoC Genes in Helicobacters, Campylobacters, and Related Bacteria. J. Bacteriol. 1999, 181, 3857–3859. [Google Scholar] [CrossRef] [PubMed]
- Beier, D.; Frank, R. Molecular Characterization of Two-Component Systems of Helicobacter pylori. J. Bacteriol. 2000, 182, 2068–2076. [Google Scholar] [CrossRef] [PubMed]
- Marshall, D.G.; Dundon, W.G.; Beesley, S.M.; Smyth, C.J. Helicobacter pylori—A conundrum of genetic diversity. Microbiology 1998, 144, 2925–2939. [Google Scholar] [CrossRef] [PubMed]
- Beier, D.; Spohn, G.; Rappuoli, R.; Scarlato, V. Functional analysis of the Helicobacter pylori principal sigma subunit of RNA polymerase reveals that the spacer region is important for efficient transcription. Mol. Microbiol. 1998, 30, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Brahmachary, P.; Dashti, M.G.; Olson, J.W.; Hoover, T.R. Helicobacter pylori FlgR Is an Enhancer-Independent Activator of σ 54 -RNA Polymerase Holoenzyme. J. Bacteriol. 2004, 186, 4535–4542. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Niehus, E.; Gressmann, H.; Ye, F.; Schlapbach, R.; Dehio, M.; Dehio, C.; Stack, A.; Meyer, T.F.; Suerbaum, S.; Josenhans, C. Genome-wide analysis of transcriptional hierarchy and feedback regulation in the flagellar system of Helicobacter pylori. Mol. Microbiol. 2004, 52, 947–961. [Google Scholar] [CrossRef]
- Pflock, M.; Kennard, S.; Delany, I.; Scarlato, V.; Beier, D. Acid-Induced Activation of the Urease Promoters Is Mediated Directly by the ArsRS Two-Component System of Helicobacter pylori. Infect. Immun. 2005, 73, 6437–6445. [Google Scholar] [CrossRef]
- Waidner, B.; Melchers, K.; Stähler, F.N.; Kist, M.; Bereswill, S. The Helicobacter pylori CrdRS Two-Component Regulation System (HP1364/HP1365) Is Required for Copper-Mediated Induction of the Copper Resistance Determinant CrdA. J. Bacteriol. 2005, 187, 4683–4688. [Google Scholar] [CrossRef]
- Jiménez-Pearson, M.-A.; Delany, I.; Scarlato, V.; Beier, D. Phosphate flow in the chemotactic response system of Helicobacter pylori. Microbiology 2005, 151, 3299–3311. [Google Scholar] [CrossRef]
- Pelliciari, S.; Pinatel, E.M.; Vannini, A.; Peano, C.; Puccio, S.; De Bellis, G.; Danielli, A.; Scarlato, V.; Roncarati, D. Insight into the essential role of the Helicobacter pylori HP1043 orphan response regulator: Genome-wide identification and characterization of the DNA-binding sites. Sci. Rep. 2017, 7, srep41063. [Google Scholar] [CrossRef]
- Spohn, G.; Scarlato, V. The autoregulatory HspR repressor protein governs chaperone gene transcription in Helicobacter pylori. Mol. Microbiol. 1999, 34, 663–674. [Google Scholar] [CrossRef]
- Roncarati, D.; Scarlato, V. The Interplay between Two Transcriptional Repressors and Chaperones Orchestrates Helicobacter pylori Heat-Shock Response. Int. J. Mol. Sci. 2018, 19, 1702. [Google Scholar] [CrossRef]
- Homuth, G.; Domm, S.; Kleiner, D.; Schumann, W. Transcriptional Analysis of Major Heat Shock Genes of Helicobacter pylori. J. Bacteriol. 2000, 182, 4257–4263. [Google Scholar] [CrossRef]
- Contreras, M.; Thiberge, J.-M.; Mandrand-Berthelot, M.-A.; Labigne, A. Characterization of the roles of NikR, a nickel-responsive pleiotropic autoregulator of Helicobacter pylori. Mol. Microbiol. 2003, 49, 947–963. [Google Scholar] [CrossRef]
- Bereswill, S.; Lichte, F.; Vey, T.; Fassbinder, F.; Kist, M. Cloning and characterization of the fur gene from Helicobacter pylori. FEMS Microbiol. Lett. 1998, 159, 193–200. [Google Scholar] [CrossRef]
- Delany, I.; Pacheco, A.B.F.; Spohn, G.; Rappuoli, R.; Scarlato, V. Iron-Dependent Transcription of the frpB Gene of Helicobacter pylori Is Controlled by the Fur Repressor Protein. J. Bacteriol. 2001, 183, 4932–4937. [Google Scholar] [CrossRef]
- Chu, C.-H.; Yen, C.-Y.; Chen, B.-W.; Lin, M.-G.; Wang, L.-H.; Tang, K.-Z.; Hsiao, C.-D.; Sun, Y.-J. Crystal structures of HpSoj–DNA complexes and the nucleoid-adaptor complex formation in chromosome segregation. Nucleic Acids Res. 2019, 47, 2113–2129. [Google Scholar] [CrossRef]
- Popescu, A.; Karpay, A.; Israel, D.A.; Peek, R.M.; Krezel, A.M. Helicobacter pylori protein HP0222 belongs to Arc/MetJ family of transcriptional regulators. Proteins Struct. Funct. Bioinform. 2005, 59, 303–311. [Google Scholar] [CrossRef]
- Borin, B.N.; Krezel, A.M. Structure of HP0564 from Helicobacter pylori identifies it as a new transcriptional regulator. Proteins Struct. Funct. Bioinform. 2008, 73, 265–268. [Google Scholar] [CrossRef]
- Álvarez, A.; Toledo, H. The histone-like protein HU has a role in gene expression during the acid adaptation response in Helicobacter pylori. Helicobacter 2017, 22, e12381. [Google Scholar] [CrossRef]
- Zschiedrich, C.P.; Keidel, V.; Szurmant, H. Molecular Mechanisms of Two-Component Signal Transduction. J. Mol. Biol. 2016, 428, 3752–3775. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Shi, L. Distribution and evolution of multiple-step phosphorelay in prokaryotes: Lateral domain recruitment involved in the formation of hybrid-type histidine kinases. Microbiology 2005, 151, 2159–2173. [Google Scholar] [CrossRef] [PubMed]
- Galperin, M.Y. Diversity of structure and function of response regulator output domains. Curr. Opin. Microbiol. 2010, 13, 150–159. [Google Scholar] [CrossRef] [PubMed]
- West, A.H.; Martinez-Hackert, E.; Stock, A. Crystal Structure of the Catalytic Domain of the Chemotaxis Receptor Methylesterase, CheB. J. Mol. Biol. 1995, 250, 276–290. [Google Scholar] [CrossRef] [PubMed]
- Wassmann, P.; Chan, C.; Paul, R.; Beck, A.; Heerklotz, H.; Jenal, U.; Schirmer, T. Structure of BeF3−-Modified Response Regulator PleD: Implications for Diguanylate Cyclase Activation, Catalysis, and Feedback Inhibition. Structure 2007, 15, 915–927. [Google Scholar] [CrossRef]
- Francez-Charlot, A.; Frunzke, J.; Reichen, C.; Ebneter, J.Z.; Gourion, B.; Vorholt, J.A. Sigma factor mimicry involved in regulation of general stress response. Proc. Natl. Acad. Sci. USA 2009, 106, 3467–3472. [Google Scholar] [CrossRef]
- Mo, R.; Liu, Y.; Chen, Y.; Mao, Y.; Gao, B. Evolutionary Principles of Bacterial Signaling Capacity and Complexity. mBio 2022, 13, 1–17. [Google Scholar] [CrossRef]
- Salama, N.R.; Hartung, M.L.; Müller, A. Life in the human stomach: Persistence strategies of the bacterial pathogen Helicobacter pylori. Nat. Rev. Genet. 2013, 11, 385–399. [Google Scholar] [CrossRef]
- Scarlato, V.; Delany, I.; Spohn, G.; Beier, D. Regulation of transcription in Helicobacter pylori: Simple systems or complex circuits? Int. J. Med. Microbiol. 2001, 291, 107–117. [Google Scholar] [CrossRef]
- Spohn, G.; Scarlato, V. Motility of Helicobacter pylori Is Coordinately Regulated by the Transcriptional Activator FlgR, an NtrC Homolog. J. Bacteriol. 1999, 181, 593–599. [Google Scholar] [CrossRef]
- Alvarado, A.; Behrens, W.; Josenhans, C. Protein Activity Sensing in Bacteria in Regulating Metabolism and Motility. Front. Microbiol. 2020, 10, 3055. [Google Scholar] [CrossRef]
- Tsang, J.; Smith, T.G.; Pereira, L.E.; Hoover, T.R. Insertion mutations in Helicobacter pylori flhA reveal strain differences in RpoN-dependent gene expression. Microbiology 2013, 159, 58–67. [Google Scholar] [CrossRef]
- Tsang, J.; Hirano, T.; Hoover, T.R.; McMurry, J.L. Helicobacter pylori FlhA Binds the Sensor Kinase and Flagellar Gene Regulatory Protein FlgS with High Affinity. J. Bacteriol. 2015, 197, 1886–1892. [Google Scholar] [CrossRef]
- Tsang, J.; Hoover, T.R. Requirement of the Flagellar Protein Export Apparatus Component FliO for Optimal Expression of Flagellar Genes in Helicobacter pylori. J. Bacteriol. 2014, 196, 2709–2717. [Google Scholar] [CrossRef]
- Tsang, J.; Hoover, T.R. Basal Body Structures Differentially Affect Transcription of RpoN- and FliA-Dependent Flagellar Genes in Helicobacter pylori. J. Bacteriol. 2015, 197, 1921–1930. [Google Scholar] [CrossRef]
- Boll, J.; Hendrixson, D.R. A Regulatory Checkpoint during Flagellar Biogenesis in Campylobacter jejuni Initiates Signal Transduction to Activate Transcription of Flagellar Genes. mBio 2013, 4, e00432-13. [Google Scholar] [CrossRef]
- Wen, Y.; Feng, J.; Scott, D.R.; Marcus, E.A.; Sachs, G. The pH-Responsive Regulon of HP0244 (FlgS), the Cytoplasmic Histidine Kinase of Helicobacter pylori. J. Bacteriol. 2009, 191, 449–460. [Google Scholar] [CrossRef]
- Dietz, P.; Gerlach, G.; Beier, D. Identification of Target Genes Regulated by the Two-Component System HP166-HP165 of Helicobacter pylori. J. Bacteriol. 2002, 184, 350–362. [Google Scholar] [CrossRef]
- Pflock, M.; Finsterer, N.; Joseph, B.; Mollenkopf, H.; Meyer, T.F.; Beier, D. Characterization of the ArsRS Regulon of Helicobacter pylori, Involved in Acid Adaptation. J. Bacteriol. 2006, 188, 3449–3462. [Google Scholar] [CrossRef]
- Loh, J.T.; Shum, M.V.; Jossart, S.D.R.; Campbell, A.M.; Sawhney, N.; McDonald, W.H.; Scholz, M.B.; McClain, M.S.; Forsyth, M.H.; Cover, T.L. Delineation of the pH-Responsive Regulon Controlled by the Helicobacter pylori ArsRS Two-Component System. Infect. Immun. 2021, 89, 1–24. [Google Scholar] [CrossRef]
- Bury-Moné, S.; Thiberge, J.-M.; Contreras, M.; Maitournam, A.; Labigne, A.; De Reuse, H. Responsiveness to acidity via metal ion regulators mediates virulence in the gastric pathogen Helicobacter pylori. Mol. Microbiol. 2004, 53, 623–638. [Google Scholar] [CrossRef] [PubMed]
- Panthel, K.; Dietz, P.; Haas, R.; Beier, D. Two-Component Systems of Helicobacter pylori Contribute to Virulence in a Mouse Infection Model. Infect. Immun. 2003, 71, 5381–5385. [Google Scholar] [CrossRef] [PubMed]
- Pflock, M.; Dietz, P.; Schär, J.; Beier, D. Genetic evidence for histidine kinase HP165 being an acid sensor of Helicobacter pylori. FEMS Microbiol. Lett. 2004, 234, 51–61. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Merrell, D.S.; Goodrich, M.L.; Otto, G.; Tompkins, L.S.; Falkow, S. pH-Regulated Gene Expression of the Gastric Pathogen Helicobacter pylori. Infect. Immun. 2003, 71, 3529–3539. [Google Scholar] [CrossRef] [PubMed]
- van Vliet, A.H.M.; Kuipers, E.J.; Stoof, J.; Poppelaars, S.W.; Kusters, J.G. Acid-Responsive Gene Induction of Ammonia-Producing Enzymes in Helicobacter pylori Is Mediated via a Metal-Responsive Repressor Cascade. Infect. Immun. 2004, 72, 766–773. [Google Scholar] [CrossRef]
- Pflock, M.; Bathon, M.; Schär, J.; Müller, S.; Mollenkopf, H.; Meyer, T.F.; Beier, D. The Orphan Response Regulator HP1021 of Helicobacter pylori Regulates Transcription of a Gene Cluster Presumably Involved in Acetone Metabolism. J. Bacteriol. 2007, 189, 2339–2349. [Google Scholar] [CrossRef]
- Loh, J.T.; Gupta, S.S.; Friedman, D.B.; Krezel, A.M.; Cover, T.L. Analysis of Protein Expression Regulated by the Helicobacter pylori ArsRS Two-Component Signal Transduction System. J. Bacteriol. 2010, 192, 2034–2043. [Google Scholar] [CrossRef]
- Montefusco, S.; Esposito, R.; D’Andrea, L.; Monti, M.C.; Dunne, C.; Dolan, B.; Tosco, A.; Marzullo, L.; Clyne, M. Copper Promotes TFF1-Mediated Helicobacter pylori Colonization. PLoS ONE 2013, 8, e79455. [Google Scholar] [CrossRef]
- Sanders, L.; Andermann, T.M.; Ottemann, K.M. A supplemented soft agar chemotaxis assay demonstrates the Helicobacter pylori chemotactic response to zinc and nickel. Microbiology 2013, 159, 46–57. [Google Scholar] [CrossRef]
- Haley, K.P.; Gaddy, J.A. Metalloregulation of Helicobacter pylori physiology and pathogenesis. Front. Microbiol. 2015, 6, 911. [Google Scholar] [CrossRef]
- Waidner, B.; Melchers, K.; Ivanov, I.; Loferer, H.; Bensch, K.W.; Kist, M.; Bereswill, S. Identification by RNA Profiling and Mutational Analysis of the Novel Copper Resistance Determinants CrdA (HP1326), CrdB (HP1327), and CzcB (HP1328) in Helicobacter pylori. J. Bacteriol. 2002, 184, 6700–6708. [Google Scholar] [CrossRef]
- Hung, C.-L.; Cheng, H.-H.; Hsieh, W.-C.; Tsai, Z.T.-Y.; Tsai, H.-K.; Chu, C.-H.; Hsieh, W.-P.; Chen, Y.-F.; Tsou, Y.; Lai, C.-H.; et al. The CrdRS two-component system in Helicobacter pylori responds to nitrosative stress. Mol. Microbiol. 2015, 97, 1128–1141. [Google Scholar] [CrossRef]
- Loh, J.T.; Cover, T.L. Requirement of Histidine Kinases HP0165 and HP1364 for Acid Resistance in Helicobacter pylori. Infect. Immun. 2006, 74, 3052–3059. [Google Scholar] [CrossRef]
- Danielli, A.; Scarlato, V. Regulatory circuits in Helicobacter pylori: Network motifs and regulators involved in metal-dependent responses. FEMS Microbiol. Rev. 2010, 34, 738–752. [Google Scholar] [CrossRef]
- Foynes, S.; Dorrell, N.; Ward, S.J.; Stabler, R.A.; McColm, A.A.; Rycroft, A.N.; Wren, B.W. Helicobacter pylori Possesses Two CheY Response Regulators and a Histidine Kinase Sensor, CheA, Which Are Essential for Chemotaxis and Colonization of the Gastric Mucosa. Infect. Immun. 2000, 68, 2016–2023. [Google Scholar] [CrossRef]
- Wadhams, G.H.; Armitage, J.P. Making sense of it all: Bacterial chemotaxis. Nat. Rev. Mol. Cell Biol. 2004, 5, 1024–1037. [Google Scholar] [CrossRef]
- Keilberg, D.; Ottemann, K.M. How Helicobacter pylori senses, targets and interacts with the gastric epithelium. Environ. Microbiol. 2016, 18, 791–806. [Google Scholar] [CrossRef]
- Johnson, K.S.; Ottemann, K.M. Colonization, localization, and inflammation: The roles of H. pylori chemotaxis in vivo. Curr. Opin. Microbiol. 2017, 41, 51–57. [Google Scholar] [CrossRef]
- Terry, K.; Go, A.C.; Ottemann, K.M. Proteomic mapping of a suppressor of non-chemotactic cheW mutants reveals that Helicobacter pylori contains a new chemotaxis protein. Mol. Microbiol. 2006, 61, 871–882. [Google Scholar] [CrossRef]
- Schär, J.; Sickmann, A.; Beier, D. Phosphorylation-Independent Activity of Atypical Response Regulators of Helicobacter pylori. J. Bacteriol. 2005, 187, 3100–3109. [Google Scholar] [CrossRef]
- Müller, S.; Pflock, M.; Schär, J.; Kennard, S.; Beier, D. Regulation of expression of atypical orphan response regulators of Helicobacter pylori. Microbiol. Res. 2007, 162, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Delany, I.; Spohn, G.; Rappuoli, R.; Scarlato, V. Growth Phase-Dependent Regulation of Target Gene Promoters for Binding of the Essential Orphan Response Regulator HP1043 of Helicobacter pylori. J. Bacteriol. 2002, 184, 4800–4810. [Google Scholar] [CrossRef] [PubMed]
- De la Cruz, M.A.; Ares, M.; Von Bargen, K.; Panunzi, L.G.; Martínez-Cruz, J.; Valdez-Salazar, H.-A.; Jiménez-Galicia, C.; Torres, J. Gene Expression Profiling of Transcription Factors of Helicobacter pylori under Different Environmental Conditions. Front. Microbiol. 2017, 8, 615. [Google Scholar] [CrossRef]
- Olekhnovich, I.N.; Vitko, S.; Valliere, M.; Hoffman, P.S. Response to Metronidazole and Oxidative Stress Is Mediated through Homeostatic Regulator HsrA (HP1043) in Helicobacter pylori. J. Bacteriol. 2014, 196, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Donczew, R.; Makowski, L.; Jaworski, P.; Bezulska, M.; Nowaczyk, M.; Zakrzewska-Czerwińska, J.; Zawilak-Pawlik, A. The atypical response regulator HP1021 controls formation of the Helicobacter pylori replication initiation complex. Mol. Microbiol. 2014, 95, 297–312. [Google Scholar] [CrossRef]
- Szczepanowski, P.; Noszka, M.; Żyła-Uklejewicz, D.; Pikuła, F.; Nowaczyk-Cieszewska, M.; Krężel, A.; Stingl, K.; Zawilak-Pawlik, A. HP1021 is a redox switch protein identified in Helicobacter pylori. Nucleic Acids Res. 2021, 49, 6863–6879. [Google Scholar] [CrossRef]
- Roncarati, D.; Scarlato, V. Regulation of heat-shock genes in bacteria: From signal sensing to gene expression output. FEMS Microbiol. Rev. 2017, 41, 549–574. [Google Scholar] [CrossRef]
- Spohn, G.; Delany, I.; Rappuoli, R.; Scarlato, V. Characterization of the HspR-Mediated Stress Response in Helicobacter pylori. J. Bacteriol. 2002, 184, 2925–2930. [Google Scholar] [CrossRef]
- Spohn, G.; Danielli, A.; Roncarati, D.; Delany, I.; Rappuoli, R.; Scarlato, V. Dual Control of Helicobacter pylori Heat Shock Gene Transcription by HspR and HrcA. J. Bacteriol. 2004, 186, 2956–2965. [Google Scholar] [CrossRef]
- Roncarati, D.; Danielli, A.; Spohn, G.; Delany, I.; Scarlato, V. Transcriptional Regulation of Stress Response and Motility Functions in Helicobacter pylori Is Mediated by HspR and HrcA. J. Bacteriol. 2007, 189, 7234–7243. [Google Scholar] [CrossRef]
- Palombo, M.; Scarlato, V.; Roncarati, D. Cooperative Regulation of Campylobacter jejuni Heat-Shock Genes by HspR and HrcA. Microorganisms 2020, 8, 1161. [Google Scholar] [CrossRef]
- Roncarati, D.; Danielli, A.; Scarlato, V. The HrcA repressor is the thermosensor of the heat-shock regulatory circuit in the human pathogen Helicobacter pylori. Mol. Microbiol. 2014, 92, 910–920. [Google Scholar] [CrossRef]
- Hurme, R.; Berndt, K.; Normark, S.J.; Rhen, M. A Proteinaceous Gene Regulatory Thermometer in Salmonella. Cell 1997, 90, 55–64. [Google Scholar] [CrossRef]
- Servant, P.; Grandvalet, C.; Mazodier, P. The RheA repressor is the thermosensor of the HSP18 heat shock response in Streptomyces albus. Proc. Natl. Acad. Sci. USA 2000, 97, 3538–3543. [Google Scholar] [CrossRef]
- Hitomi, M.; Nishimura, H.; Tsujimoto, Y.; Matsui, H.; Watanabe, K. Identification of a Helix-Turn-Helix Motif of Bacillus thermoglucosidasius HrcA Essential for Binding to the CIRCE Element and Thermostability of the HrcA-CIRCE Complex, Indicating a Role as a Thermosensor. J. Bacteriol. 2003, 185, 381–385. [Google Scholar] [CrossRef]
- Herbst, K.; Bujara, M.; Heroven, A.K.; Opitz, W.; Weichert, M.; Zimmermann, A.; Dersch, P. Intrinsic Thermal Sensing Controls Proteolysis of Yersinia Virulence Regulator RovA. PLoS Pathog. 2009, 5, e1000435. [Google Scholar] [CrossRef]
- Elsholz, A.K.W.; Michalik, S.; Zühlke, D.; Hecker, M.; Gerth, U. CtsR, the Gram-positive master regulator of protein quality control, feels the heat. EMBO J. 2010, 29, 3621–3629. [Google Scholar] [CrossRef]
- Versace, G.; Palombo, M.; Menon, A.; Scarlato, V.; Roncarati, D. Feeling the Heat: The Campylobacter jejuni HrcA Transcriptional Repressor Is an Intrinsic Protein Thermosensor. Biomolecules 2021, 11, 1413. [Google Scholar] [CrossRef]
- Mogk, A.; Völker, A.; Engelmann, S.; Hecker, M.; Schumann, W.; Völker, U. Nonnative Proteins Induce Expression of the Bacillus subtilis CIRCE Regulon. J. Bacteriol. 1998, 180, 2895–2900. [Google Scholar] [CrossRef]
- Wilson, A.C.; Wu, C.C.; Yates, J.R.; Tan, M. Chlamydial GroEL Autoregulates Its Own Expression through Direct Interactions with the HrcA Repressor Protein. J. Bacteriol. 2005, 187, 7535–7542. [Google Scholar] [CrossRef]
- Roncarati, D.; Danielli, A.; Scarlato, V. CbpA Acts as a Modulator of HspR Repressor DNA Binding Activity in Helicobacter pylori. J. Bacteriol. 2011, 193, 5629–5636. [Google Scholar] [CrossRef] [PubMed]
- Bucca, G.; Brassington, A.M.E.; Schönfeld, H.; Smith, C.P. The HspR regulon of Streptomyces coelicolor: A role for the DnaK chaperone as a transcriptional co-repressor. Mol. Microbiol. 2002, 38, 1093–1103. [Google Scholar] [CrossRef] [PubMed]
- Parijat, P.; Batra, J.K. Role of DnaK in HspR-HAIR interaction ofMycobacterium tuberculosis. IUBMB Life 2015, 67, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Pepe, S.; Scarlato, V.; Roncarati, D. The Helicobacter pylori HspR-Modulator CbpA Is a Multifunctional Heat-Shock Protein. Microorganisms 2020, 8, 251. [Google Scholar] [CrossRef] [PubMed]
- Holmes, C.W.; Penn, C.W.; Lund, P.A. The hrcA and hspR regulons of Campylobacter jejuni. Microbiology 2010, 156, 158–166. [Google Scholar] [CrossRef]
- Pepe, S.; Pinatel, E.M.; Fiore, E.; Puccio, S.; Peano, C.; Brignoli, T.; Vannini, A.; Danielli, A.; Scarlato, V.; Roncarati, D. The Helicobacter pylori Heat-Shock Repressor HspR: Definition of Its Direct Regulon and Characterization of the Cooperative DNA-Binding Mechanism on Its Own Promoter. Front. Microbiol. 2018, 9, 1887. [Google Scholar] [CrossRef]
- Roncarati, D.; Pinatel, E.; Fiore, E.; Peano, C.; Loibman, S.; Scarlato, V. Helicobacter pylori Stress-Response: Definition of the HrcA Regulon. Microorganisms 2019, 7, 436. [Google Scholar] [CrossRef]
- Stewart, G.R.; Wernisch, L.; Stabler, R.; Mangan, J.A.; Hinds, J.; Laing, K.G.; Young, D.B.; Butcher, P.D. Dissection of the heat-shock response in Mycobacterium tuberculosis using mutants and microarrays a aA list of the 100 ORFs most highly induced by heat shock is provided as supplementary data with the online version of this paper (http://mic.sgmjournals.org). Microbiology 2002, 148, 3129–3138. [Google Scholar] [CrossRef]
- Andersen, M.T.; Brøndsted, L.; Pearson, B.M.; Mulholland, F.; Parker, M.; Pin, C.; Wells, J.M.; Ingmer, H. Diverse roles for HspR in Campylobacter jejuni revealed by the proteome, transcriptome and phenotypic characterization of an hspR mutant. Microbiology 2005, 151, 905–915. [Google Scholar] [CrossRef]
- Hu, Y.; Oliver, H.F.; Raengpradub, S.; Palmer, M.E.; Orsi, R.H.; Wiedmann, M.; Boor, K.J. Transcriptomic and Phenotypic Analyses Suggest a Network between the Transcriptional Regulators HrcA and σ B in Listeria monocytogenes. Appl. Environ. Microbiol. 2007, 73, 7981–7991. [Google Scholar] [CrossRef]
- Bucca, G.; Laing, E.; Mersinias, V.; Allenby, N.; Hurd, D.; Holdstock, J.; Brenner, V.; Harrison, M.; Smith, C.P. Development and application of versatile high density microarrays for genome-wide analysis of Streptomyces coelicolor: Characterization of the HspR regulon. Genome Biol. 2009, 10, R5–R14. [Google Scholar] [CrossRef]
- Veen, H.V.B.-V.D.; Bongers, R.S.; Wels, M.; Bron, P.A.; Kleerebezem, M. Transcriptome signatures of class I and III stress response deregulation in Lactobacillus plantarum reveal pleiotropic adaptation. Microb. Cell Factories 2013, 12, 112. [Google Scholar] [CrossRef]
- Macomber, L.; Hausinger, R.P. Mechanisms of nickel toxicity in microorganisms. Metallomics 2011, 3, 1153–1162. [Google Scholar] [CrossRef]
- Giedroc, D.P.; Arunkumar, A.I. Metal sensor proteins: Nature’s metalloregulated allosteric switches. Dalton Trans. 2007, 29, 3107–3120. [Google Scholar] [CrossRef]
- Benoit, S.L.; Maier, R.J.; Sawers, R.G.; Greening, C. Molecular Hydrogen Metabolism: A Widespread Trait of Pathogenic Bacteria and Protists. Microbiol. Mol. Biol. Rev. 2020, 84, e00092-19. [Google Scholar] [CrossRef]
- Scott, D.R.; Marcus, E.A.; Weeks, D.L.; Sachs, G. Mechanisms of acid resistance due to the urease system of Helicobacter pylori. Gastroenterology 2002, 123, 187–195. [Google Scholar] [CrossRef]
- Schreiter, E.; Sintchak, M.D.; Guo, Y.; Chivers, P.; Sauer, R.; Drennan, C.L. Crystal structure of the nickel-responsive transcription factor NikR. Nat. Struct. Mol. Biol. 2003, 10, 794–799. [Google Scholar] [CrossRef]
- Rodionov, D.A.; Hebbeln, P.; Gelfand, M.; Eitinger, T. Comparative and Functional Genomic Analysis of Prokaryotic Nickel and Cobalt Uptake Transporters: Evidence for a Novel Group of ATP-Binding Cassette Transporters. J. Bacteriol. 2006, 188, 317–327. [Google Scholar] [CrossRef]
- Zambelli, B.; Danielli, A.; Romagnoli, S.; Neyroz, P.; Ciurli, S.; Scarlato, V. High-Affinity Ni2+ Binding Selectively Promotes Binding of Helicobacter pylori NikR to Its Target Urease Promoter. J. Mol. Biol. 2008, 383, 1129–1143. [Google Scholar] [CrossRef]
- Higgins, K.A.; Carr, C.E.; Maroney, M.J. Specific Metal Recognition in Nickel Trafficking. Biochemistry 2012, 51, 7816–7832. [Google Scholar] [CrossRef]
- Danielli, A.; Amore, G.; Scarlato, V. Built Shallow to Maintain Homeostasis and Persistent Infection: Insight into the Transcriptional Regulatory Network of the Gastric Human Pathogen Helicobacter pylori. PLoS Pathog. 2010, 6, e1000938. [Google Scholar] [CrossRef]
- Carpenter, B.M.; West, A.L.; Gancz, H.; Servetas, S.L.; Pich, O.Q.; Gilbreath, J.J.; Hallinger, D.R.; Forsyth, M.H.; Merrell, D.S.; Michel, S.L.J. Crosstalk between the HpArsRS two-component system and HpNikR is necessary for maximal activation of urease transcription. Front. Microbiol. 2015, 6, 558. [Google Scholar] [CrossRef] [PubMed]
- Bahlawane, C.; Dian, C.; Muller, C.; Round, A.; Fauquant, C.; Schauer, K.; De Reuse, H.; Terradot, L.; Michaud-Soret, I. Structural and mechanistic insights into Helicobacter pylori NikR activation. Nucleic Acids Res. 2010, 38, 3106–3118. [Google Scholar] [CrossRef] [PubMed]
- West, A.L.; John, F.S.; Lopes, P.E.M.; MacKerell, J.A.D.; Pozharski, E.; Michel, S.L.J. Holo-Ni(II)HpNikR Is an Asymmetric Tetramer Containing Two Different Nickel-Binding Sites. J. Am. Chem. Soc. 2010, 132, 14447–14456. [Google Scholar] [CrossRef] [PubMed]
- Benini, S.; Cianci, M.; Ciurli, S. Holo-Ni2+Helicobacter pylori NikR contains four square-planar nickel-binding sites at physiological pH. Dalton Trans. 2011, 40, 7831–7833. [Google Scholar] [CrossRef]
- Delany, I.; Ieva, R.; Soragni, A.; Hilleringmann, M.; Rappuoli, R.; Scarlato, V. In Vitro Analysis of Protein-Operator Interactions of the NikR and Fur Metal-Responsive Regulators of Coregulated Genes in Helicobacter pylori. J. Bacteriol. 2005, 187, 7703–7715. [Google Scholar] [CrossRef]
- Dosanjh, N.S.; West, A.L.; Michel, S.L.J. Helicobacter pylori NikR’s Interaction with DNA: A Two-Tiered Mode of Recognition. Biochemistry 2009, 48, 527–536. [Google Scholar] [CrossRef]
- Stoof, J.; Kuipers, E.J.; van Vliet, A. Characterization of NikR-responsive promoters of urease and metal transport genes of Helicobacter mustelae. BioMetals 2009, 23, 145–159. [Google Scholar] [CrossRef]
- Vannini, A.; Pinatel, E.; Costantini, P.E.; Pelliciari, S.; Roncarati, D.; Puccio, S.; De Bellis, G.; Peano, C.; Danielli, A. Comprehensive mapping of the Helicobacter pylori NikR regulon provides new insights in bacterial nickel responses. Sci. Rep. 2017, 7, srep45458. [Google Scholar] [CrossRef]
- Benanti, E.L.; Chivers, P.T. The N-terminal Arm of the Helicobacter pylori Ni2+-dependent Transcription Factor NikR Is Required for Specific DNA Binding. J. Biol. Chem. 2007, 282, 20365–20375. [Google Scholar] [CrossRef]
- Musiani, F.; Ciurli, S.; Dikiy, A. Interaction of Selenoprotein W with 14-3-3 Proteins: A Computational Approach. J. Proteome Res. 2011, 10, 968–976. [Google Scholar] [CrossRef]
- Mazzei, L.; Dobrovolska, O.; Musiani, F.; Zambelli, B.; Ciurli, S. On the interaction of Helicobacter pylori NikR, a Ni(II)-responsive transcription factor, with the urease operator: In solution and in silico studies. J. Biol. Inorg. Chem. 2015, 20, 1021–1037. [Google Scholar] [CrossRef]
- Baksh, K.A.; Pichugin, D.; Prosser, R.S.; Zamble, D.B. Allosteric regulation of the nickel-responsive NikR transcription factor from Helicobacter pylori. J. Biol. Chem. 2021, 296, 100069. [Google Scholar] [CrossRef]
- West, A.L.; Evans, S.E.; González, J.M.; Carter, L.G.; Tsuruta, H.; Pozharski, E.; Michel, S.L.J. Ni(II) coordination to mixed sites modulates DNA binding of Hp NikR via a long-range effect. Proc. Natl. Acad. Sci. USA 2012, 109, 5633–5638. [Google Scholar] [CrossRef]
- Evans, S.E.; Michel, S.L.J. Dissecting the role of DNA sequence in Helicobacter pylori NikR/DNA recognition. Dalton Trans. 2012, 41, 7946–7951. [Google Scholar] [CrossRef]
- Ernst, F.D.; Kuipers, E.J.; Heijens, A.; Sarwari, R.; Stoof, J.; Penn, C.W.; Kusters, J.G.; van Vliet, A.H.M. The Nickel-Responsive Regulator NikR Controls Activation and Repression of Gene Transcription in Helicobacter pylori. Infect. Immun. 2005, 73, 7252–7258. [Google Scholar] [CrossRef]
- Ernst, F.D.; Stoof, J.; Horrevoets, W.M.; Kuipers, E.J.; Kusters, J.G.; van Vliet, A.H.M. NikR Mediates Nickel-Responsive Transcriptional Repression of the Helicobacter pylori Outer Membrane Proteins FecA3 (HP1400) and FrpB4 (HP1512). Infect. Immun. 2006, 74, 6821–6828. [Google Scholar] [CrossRef]
- Davis, G.S.; Flannery, E.L.; Mobley, H.L.T. Helicobacter pylori HP1512 Is a Nickel-Responsive NikR-Regulated Outer Membrane Protein. Infect. Immun. 2006, 74, 6811–6820. [Google Scholar] [CrossRef]
- Wolfram, L.; Haas, E.; Bauerfeind, P. Nickel Represses the Synthesis of the Nickel Permease NixA of Helicobacter pylori. J. Bacteriol. 2006, 188, 1245–1250. [Google Scholar] [CrossRef]
- Danielli, A.; Romagnoli, S.; Roncarati, D.; Costantino, L.; Delany, I.; Scarlato, V. Growth Phase and Metal-Dependent Transcriptional Regulation of the fecA Genes in Helicobacter pylori. J. Bacteriol. 2009, 191, 3717–3725. [Google Scholar] [CrossRef]
- Romagnoli, S.; Agriesti, F.; Scarlato, V. In Vivo Recognition of the fecA3 Target Promoter by Helicobacter pylori NikR. J. Bacteriol. 2011, 193, 1131–1141. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jones, M.D.; Ademi, I.; Yin, X.; Gong, Y.; Zamble, D.B. Nickel-responsive regulation of two novel Helicobacter pylori NikR-targeted genes. Metallomics 2015, 7, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Muller, C.; Bahlawane, C.; Aubert, S.; DeLay, C.M.; Schauer, K.; Michaud-Soret, I.; De Reuse, H. Hierarchical regulation of the NikR-mediated nickel response in Helicobacter pylori. Nucleic Acids Res. 2011, 39, 7564–7575. [Google Scholar] [CrossRef]
- van Vliet, A.H.M.; Poppelaars, S.W.; Davies, B.J.; Stoof, J.; Bereswill, S.; Kist, M.; Penn, C.W.; Kuipers, E.J.; Kusters, J.G. NikR Mediates Nickel-Responsive Transcriptional Induction of Urease Expression in Helicobacter pylori. Infect. Immun. 2002, 70, 2846–2852. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.D.; Li, Y.; Zamble, D.B. Acid-responsive activity of the Helicobacter pylori metalloregulator NikR. Proc. Natl. Acad. Sci. USA 2018, 115, 8966–8971. [Google Scholar] [CrossRef] [PubMed]
- Abraham, L.O.; Li, Y.; Zamble, D.B. The metal- and DNA-binding activities of Helicobacter pylori NikR. J. Inorg. Biochem. 2006, 100, 1005–1014. [Google Scholar] [CrossRef]
- Eisenbart, S.K.; Alzheimer, M.; Pernitzsch, S.R.; Dietrich, S.; Stahl, S.; Sharma, C.M. A Repeat-Associated Small RNA Controls the Major Virulence Factors of Helicobacter pylori. Mol. Cell 2020, 80, 210–226.e7. [Google Scholar] [CrossRef]
- Kinoshita-Daitoku, R.; Kiga, K.; Miyakoshi, M.; Otsubo, R.; Ogura, Y.; Sanada, T.; Bo, Z.; Phuoc, T.V.; Okano, T.; Iida, T.; et al. A bacterial small RNA regulates the adaptation of Helicobacter pylori to the host environment. Nat. Commun. 2021, 12, 2085. [Google Scholar] [CrossRef]
- Roncarati, D.; Pelliciari, S.; Doniselli, N.; Maggi, S.; Vannini, A.; Valzania, L.; Mazzei, L.; Zambelli, B.; Rivetti, C.; Danielli, A. Metal-responsive promoter DNA compaction by the ferric uptake regulator. Nat. Commun. 2016, 7, 12593. [Google Scholar] [CrossRef]
- Borin, B.N.; Tang, W.; Krezel, A.M. Helicobacter pylori RNA polymerase α-subunit C-terminal domain shows features unique to ɛ-proteobacteria and binds NikR/DNA complexes. Protein Sci. 2014, 23, 454–463. [Google Scholar] [CrossRef]
- Visweswariah, S.S.; Busby, S.J. Evolution of bacterial transcription factors: How proteins take on new tasks, but do not always stop doing the old ones. Trends Microbiol. 2015, 23, 463–467. [Google Scholar] [CrossRef]
- van Vliet, A.H.; Ernst, F.D.; Kusters, J.G. NikR-mediated regulation of Helicobacter pylori acid adaptation. Trends Microbiol. 2004, 12, 489–494. [Google Scholar] [CrossRef]
- Li, Y.; Zamble, D.B. pH-Responsive DNA-Binding Activity of Helicobacter pylori NikR. Biochemistry 2009, 48, 2486–2496. [Google Scholar] [CrossRef]
- Pich, O.Q.; Merrell, D.S. The ferric uptake regulator of Helicobacter pylori: A critical player in the battle for iron and colonization of the stomach. Futur. Microbiol. 2013, 8, 725–738. [Google Scholar] [CrossRef]
- Agriesti, F.; Roncarati, D.; Musiani, F.; Del Campo, C.; Iurlaro, M.; Sparla, F.; Ciurli, S.; Danielli, A.; Scarlato, V. FeON-FeOFF: The Helicobacter pylori Fur regulator commutates iron-responsive transcription by discriminative readout of opposed DNA grooves. Nucleic Acids Res. 2014, 42, 3138–3151. [Google Scholar] [CrossRef]
- Butcher, J.; Sarvan, S.; Brunzelle, J.S.; Couture, J.-F.; Stintzi, A. Structure and regulon of Campylobacter jejuni ferric uptake regulator Fur define apo-Fur regulation. Proc. Natl. Acad. Sci. USA 2012, 109, 10047–10052. [Google Scholar] [CrossRef]
- Pohl, E.; Haller, J.C.; Mijovilovich, A.; Meyer-Klaucke, W.; Garman, E.; Vasil, M.L. Architecture of a protein central to iron homeostasis: Crystal structure and spectroscopic analysis of the ferric uptake regulator. Mol. Microbiol. 2003, 47, 903–915. [Google Scholar] [CrossRef]
- Vitale, S.; Fauquant, C.; Lascoux, D.; Schauer, K.; Saint-Pierre, C.; Michaud-Soret, I. A ZnS4 Structural Zinc Site in the Helicobacter pylori Ferric Uptake Regulator. Biochemistry 2009, 48, 5582–5591. [Google Scholar] [CrossRef]
- Dian, C.; Vitale, S.; Leonard, G.A.; Bahlawane, C.; Fauquant, C.; Leduc, D.; Muller, C.; de Reuse, H.; Michaud-Soret, I.; Terradot, L. The structure of the Helicobacter pylori ferric uptake regulator Fur reveals three functional metal binding sites. Mol. Microbiol. 2011, 79, 1260–1275. [Google Scholar] [CrossRef]
- Gilbreath, J.J.; Pich, O.Q.; Benoit, S.L.; Besold, A.N.; Cha, J.-H.; Maier, R.J.; Michel, S.L.J.; Maynard, E.L.; Merrell, D.S. Random and site-specific mutagenesis of the Helicobacter pyloriferric uptake regulator provides insight into Fur structure-function relationships. Mol. Microbiol. 2013, 89, 304–323. [Google Scholar] [CrossRef]
- Danielli, A.; Roncarati, D.; Delany, I.; Chiarini, V.; Rappuoli, R.; Scarlato, V. In Vivo Dissection of the Helicobacter pylori Fur Regulatory Circuit by Genome-Wide Location Analysis. J. Bacteriol. 2006, 188, 4654–4662. [Google Scholar] [CrossRef] [PubMed]
- Bereswill, S.; Greiner, S.; van Vliet, A.H.M.; Waidner, B.; Fassbinder, F.; Schiltz, E.; Kusters, J.G.; Kist, M. Regulation of Ferritin-Mediated Cytoplasmic Iron Storage by the Ferric Uptake Regulator Homolog (Fur) of Helicobacter pylori. J. Bacteriol. 2000, 182, 5948–5953. [Google Scholar] [CrossRef]
- Delany, I.; Spohn, G.; Rappuoli, R.; Scarlato, V. The Fur repressor controls transcription of iron-activated and -repressed genes in Helicobacter pylori. Mol. Microbiol. 2002, 42, 1297–1309. [Google Scholar] [CrossRef] [PubMed]
- Ernst, F.D.; Homuth, G.; Stoof, J.; Mäder, U.; Waidner, B.; Kuipers, E.J.; Kist, M.; Kusters, J.G.; Bereswill, S.; van Vliet, A.H.M. Iron-Responsive Regulation of the Helicobacter pylori Iron-Cofactored Superoxide Dismutase SodB Is Mediated by Fur. J. Bacteriol. 2005, 187, 3687–3692. [Google Scholar] [CrossRef]
- Gancz, H.; Censini, S.; Merrell, D.S. Iron and pH Homeostasis Intersect at the Level of Fur Regulation in the Gastric Pathogen Helicobacter pylori. Infect. Immun. 2006, 74, 602–614. [Google Scholar] [CrossRef]
- Gilbreath, J.J.; West, A.L.; Pich, O.Q.; Carpenter, B.M.; Michel, S.; Merrell, D.S. Fur Activates Expression of the 2-Oxoglutarate Oxidoreductase Genes (oorDABC) in Helicobacter pylori. J. Bacteriol. 2012, 194, 6490–6497. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, B.M.; Gilbreath, J.J.; Pich, O.Q.; McKelvey, A.M.; Maynard, E.L.; Li, Z.-Z.; Merrell, D.S. Identification and Characterization of Novel Helicobacter pylori apo-Fur-Regulated Target Genes. J. Bacteriol. 2013, 195, 5526–5539. [Google Scholar] [CrossRef]
- Pich, O.Q.; Carpenter, B.M.; Gilbreath, J.J.; Merrell, D.S. Detailed analysis of Helicobacter pylori Fur-regulated promoters reveals a Fur box core sequence and novel Fur-regulated genes. Mol. Microbiol. 2012, 84, 921–941. [Google Scholar] [CrossRef]
- Vannini, A.; Roncarati, D.; Spinsanti, M.; Scarlato, V.; Danielli, A. In Depth Analysis of the Helicobacter pylori cag Pathogenicity Island Transcriptional Responses. PLoS ONE 2014, 9, e98416. [Google Scholar] [CrossRef]
- Merrell, D.S.; Thompson, L.J.; Kim, C.C.; Mitchell, H.; Tompkins, L.S.; Lee, A.; Falkow, S. Growth Phase-Dependent Response of Helicobacter pylori to Iron Starvation. Infect. Immun. 2003, 71, 6510–6525. [Google Scholar] [CrossRef]
- van Vliet, A.H.M.; Stoof, J.; Poppelaars, S.W.; Bereswill, S.; Homuth, G.; Kist, M.; Kuipers, E.J.; Kusters, J.G. Differential Regulation of Amidase- and Formamidase-mediated Ammonia Production by the Helicobacter pylori Fur Repressor. J. Biol. Chem. 2003, 278, 9052–9057. [Google Scholar] [CrossRef]
- Ernst, F.D.; Bereswill, S.; Waidner, B.; Stoof, J.; Mäder, U.; Kusters, J.G.; Kuipers, E.J.; Kist, M.; Van Vliet, A.H.M.; Homuth, G. Transcriptional profiling of Helicobacter pylori Fur- and iron-regulated gene expression. Microbiology 2005, 151, 533–546. [Google Scholar] [CrossRef]
- Delany, I.; Spohn, G.; Pacheco, A.-B.F.; Ieva, R.; Alaimo, C.; Rappuoli, R.; Scarlato, V. Autoregulation of Helicobacter pylori Fur revealed by functional analysis of the iron-binding site. Mol. Microbiol. 2002, 46, 1107–1122. [Google Scholar] [CrossRef]
- Delany, I.; Ieva, R.; Alaimo, C.; Rappuoli, R.; Scarlato, V. The Iron-Responsive Regulator Fur Is Transcriptionally Autoregulated and Not Essential in Neisseria meningitidis. J. Bacteriol. 2003, 185, 6032–6041. [Google Scholar] [CrossRef]
- Delany, I.; Spohn, G.; Rappuoli, R.; Scarlato, V. An anti-repression Fur operator upstream of the promoter is required for iron-mediated transcriptional autoregulation in Helicobacter pylori. Mol. Microbiol. 2003, 50, 1329–1338. [Google Scholar] [CrossRef]
- Sharma, C.M.; Hoffmann, S.; Darfeuille, F.; Reignier, J.; Findeiß, S.; Sittka, A.; Chabas, S.; Reiche, K.; Hackermüller, J.; Reinhardt, R.; et al. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 2010, 464, 250–255. [Google Scholar] [CrossRef]
- Pelliciari, S.; Vannini, A.; Roncarati, D.; Edanielli, A. The allosteric behavior of Fur mediates oxidative stress signal transduction in Helicobacter pylori. Front. Microbiol. 2015, 6, 840. [Google Scholar] [CrossRef]
- Bijlsma, J.J.E.; Waidner, B.; van Vliet, A.H.M.; Hughes, N.J.; Häg, S.; Bereswill, S.; Kelly, D.J.; Vandenbroucke-Grauls, C.M.J.E.; Kist, M.; Kusters, J.G. The Helicobacter pylori Homologue of the Ferric Uptake Regulator Is Involved in Acid Resistance. Infect. Immun. 2002, 70, 606–611. [Google Scholar] [CrossRef]
- Gancz, H.; Merrell, D.S. The Helicobacter pylori Ferric Uptake Regulator (Fur) is essential for growth under sodium chloride stress. J. Microbiol. 2011, 49, 294–298. [Google Scholar] [CrossRef]
- Loh, J.T.; Gaddy, J.A.; Algood, H.M.S.; Gaudieri, S.; Mallal, S.; Cover, T.L. Helicobacter pylori Adaptation In Vivo in Response to a High-Salt Diet. Infect. Immun. 2015, 83, 4871–4883. [Google Scholar] [CrossRef]
- Lee, A.-Y.; Kao, C.-Y.; Wang, Y.-K.; Lin, S.-Y.; Lai, T.-Y.; Sheu, B.-S.; Lo, C.-J.; Wu, J.-J. Inactivation of ferric uptake regulator (Fur) attenuates Helicobacter pylori J99 motility by disturbing the flagellar motor switch and autoinducer-2 production. Helicobacter 2017, 22, e12388. [Google Scholar] [CrossRef] [PubMed]
- Miles, S.; Piazuelo, M.B.; Semino-Mora, C.; Washington, M.K.; Dubois, A.; Peek, R.M.; Correa, P.; Merrell, D.S. Detailed In Vivo Analysis of the Role of Helicobacter pylori Fur in Colonization and Disease. Infect. Immun. 2010, 78, 3073–3082. [Google Scholar] [CrossRef] [PubMed]
- Roncarati, D.; Scarlato, V.; Vannini, A. Targeting of Regulators as a Promising Approach in the Search for Novel Antimicrobial Agents. Microorganisms 2022, 10, 185. [Google Scholar] [CrossRef] [PubMed]
- Antoniciello, F.; Roncarati, D.; Zannoni, A.; Chiti, E.; Scarlato, V.; Chiappori, F. Targeting the Essential Transcription Factor HP1043 of Helicobacter pylori: A Drug Repositioning Study. Front. Mol. Biosci. 2022, 9, 887564. [Google Scholar] [CrossRef]
- González, A.; Casado, J.; Chueca, E.; Salillas, S.; Velázquez-Campoy, A.; Angarica, V.E.; Bénejat, L.; Guignard, J.; Giese, A.; Sancho, J.; et al. Repurposing Dihydropyridines for Treatment of Helicobacter pylori Infection. Pharmaceutics 2019, 11, 681. [Google Scholar] [CrossRef]
- Casado, J.; Lanas, Á.; González, A. Two-component regulatory systems in Helicobacter pylori and Campylobacter jejuni: Attractive targets for novel antibacterial drugs. Front. Cell. Infect. Microbiol. 2022, 12, 977944. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Locus No. in Strain 26695 | Putative Function (Gene) | References |
|---|---|---|
| Sigma factors | ||
| HP0088 | RNA polymerase sigma-80 factor (rpoD) | [25] |
| HP0714 | RNA polymerase sigma-54 factor (rpoN) | [26] |
| HP1032 | RNA polymerase sigma-28 factor (fliA) | [27] |
| Histidine kinases and response regulators | ||
| HP0166/0165 | Signal-transducing system (arsRS) | [23,28] |
| HP0703/0244 | Signal-transducing system (flgRS) | [23,26] |
| HP1365/1364 | Signal-transducing system (crdRS) | [23,29] |
| HP1067/0392 | Signal-transducing system (cheY1Y2A) | [30] |
| HP1021 | Response regulator | [23] |
| HP1043 | Response regulator (hp1043, hsrA) | [23,31] |
| Other regulators | ||
| HP1025 | Chaperone gene repressor (hspR) | [32,33] |
| HP0111 | Transcriptional repressor (hrcA) | [33,34] |
| HP1338 | Nickel response transcriptional regulator (nikR) | [35] |
| HP1027 | Ferric uptake regulator (fur) | [36,37] |
| HP1139 | Putative repressor, (soj) | [38] |
| HP0222 | Putative transcriptional regulator | [39] |
| HP0564 | Putative transcriptional repressor | [40] |
| HP0835 | Putative transcriptional regulator (hup) | [41] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vannini, A.; Roncarati, D.; D’Agostino, F.; Antoniciello, F.; Scarlato, V. Insights into the Orchestration of Gene Transcription Regulators in Helicobacter pylori. Int. J. Mol. Sci. 2022, 23, 13688. https://doi.org/10.3390/ijms232213688
Vannini A, Roncarati D, D’Agostino F, Antoniciello F, Scarlato V. Insights into the Orchestration of Gene Transcription Regulators in Helicobacter pylori. International Journal of Molecular Sciences. 2022; 23(22):13688. https://doi.org/10.3390/ijms232213688
Chicago/Turabian StyleVannini, Andrea, Davide Roncarati, Federico D’Agostino, Federico Antoniciello, and Vincenzo Scarlato. 2022. "Insights into the Orchestration of Gene Transcription Regulators in Helicobacter pylori" International Journal of Molecular Sciences 23, no. 22: 13688. https://doi.org/10.3390/ijms232213688
APA StyleVannini, A., Roncarati, D., D’Agostino, F., Antoniciello, F., & Scarlato, V. (2022). Insights into the Orchestration of Gene Transcription Regulators in Helicobacter pylori. International Journal of Molecular Sciences, 23(22), 13688. https://doi.org/10.3390/ijms232213688