
Buffy Coat Transcriptomic Analysis Reveals Alterations in Host Cell Protein Synthesis and Cell Cycle in Severe COVID-19 Patients

 , , , , and
, , , , and
Abstract
1. Introduction
2. Results
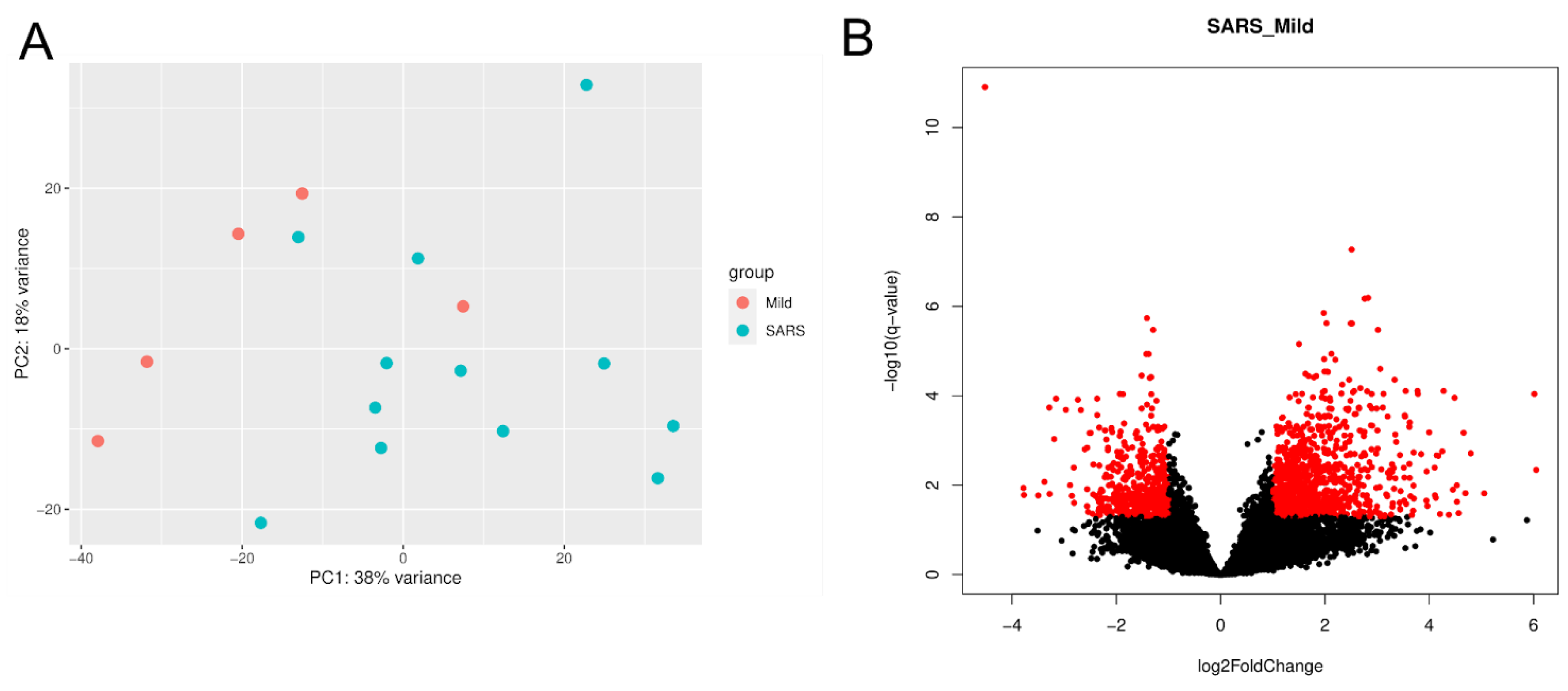
2.1. Cohort Data
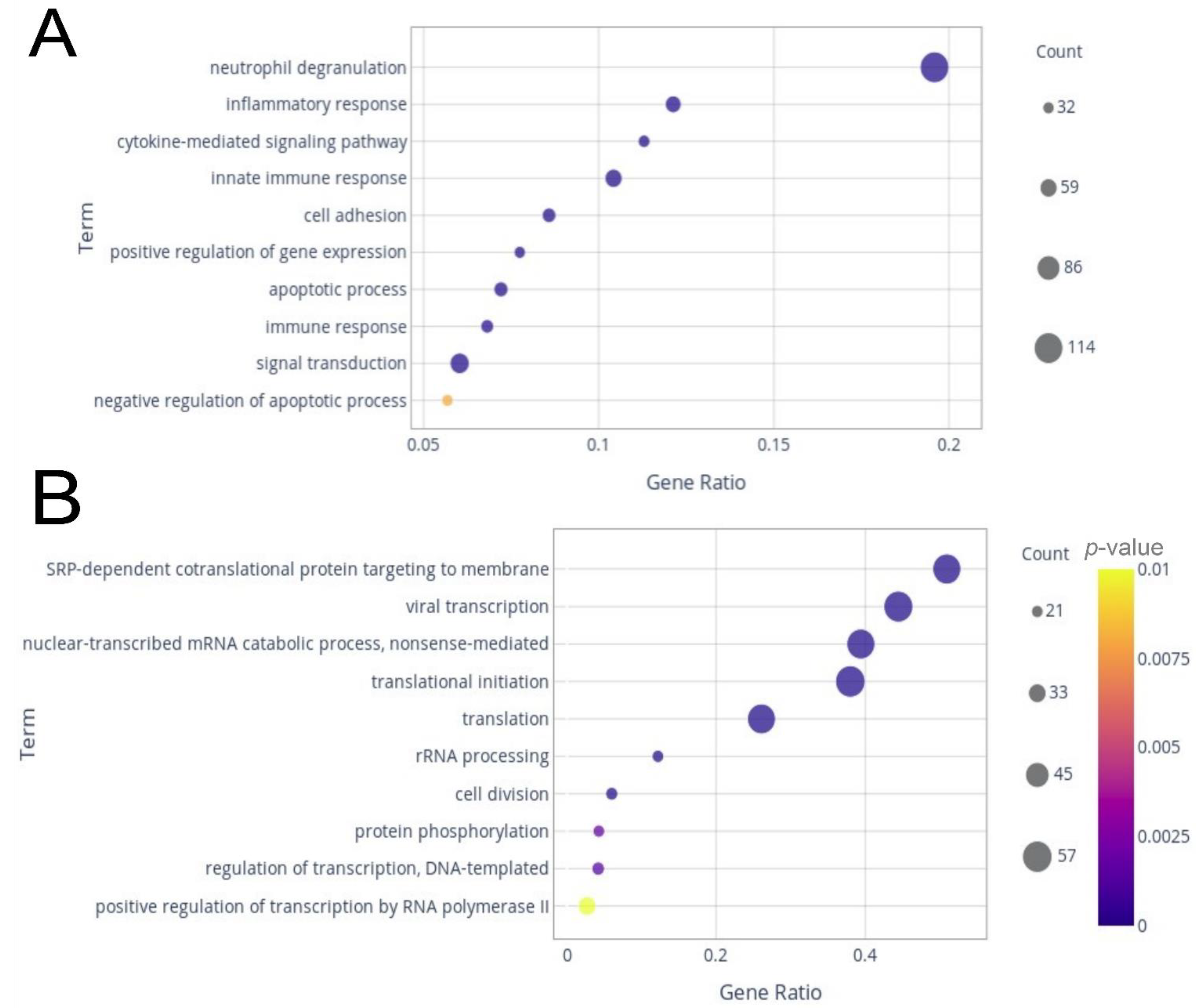
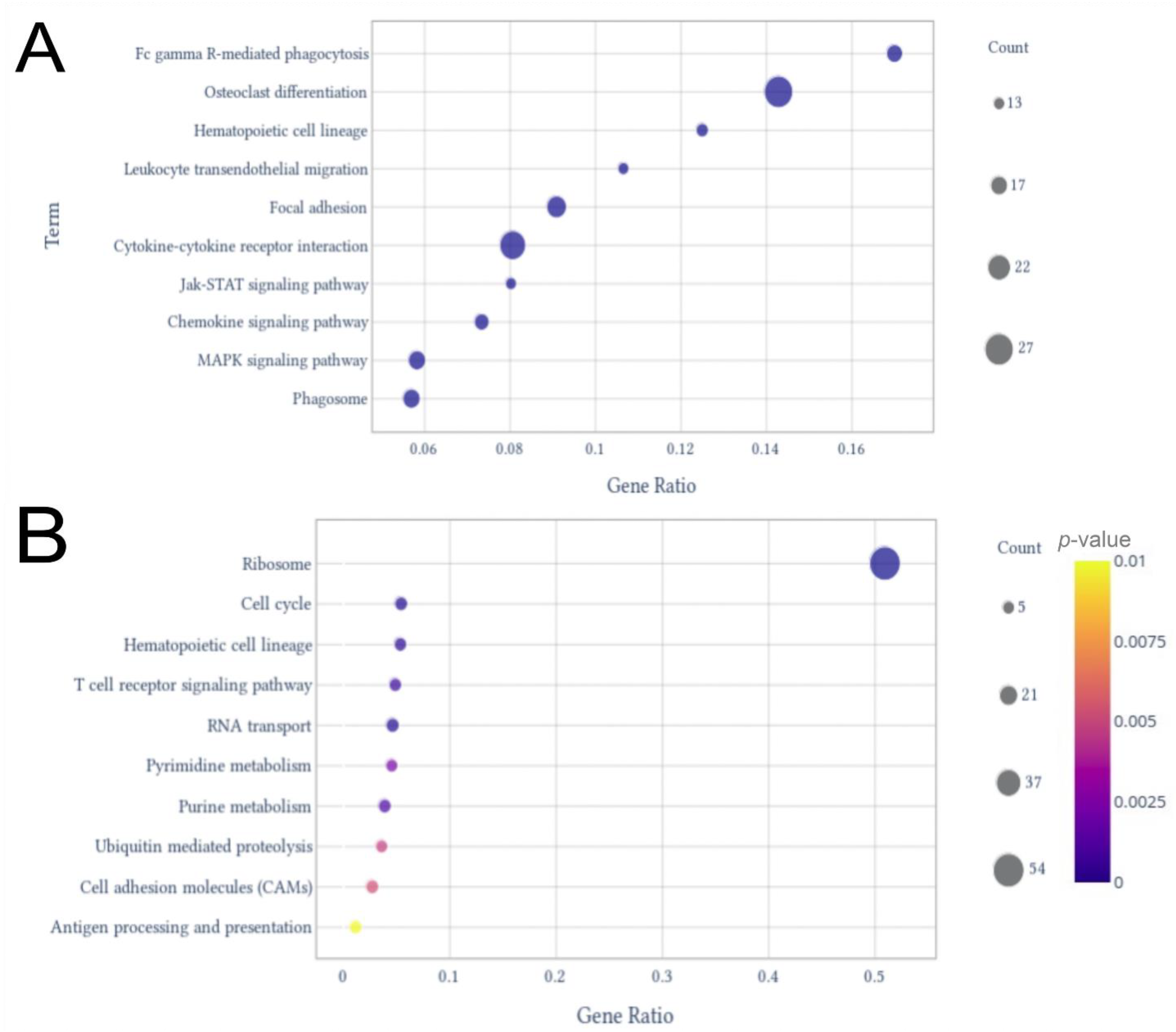
2.2. Differentially Expressed Genes (DEGs) and Enrichment Analysis
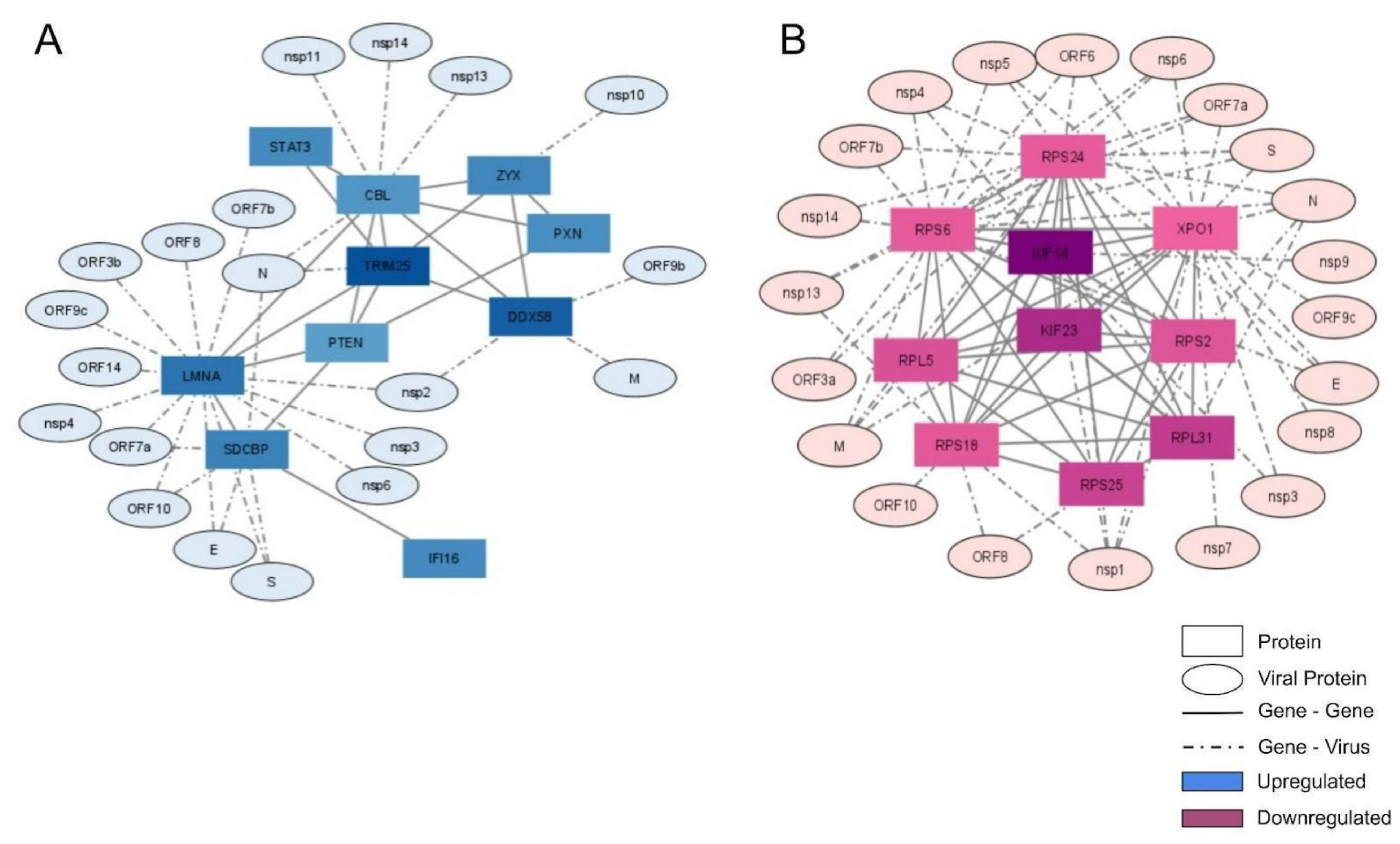
2.3. Interactome
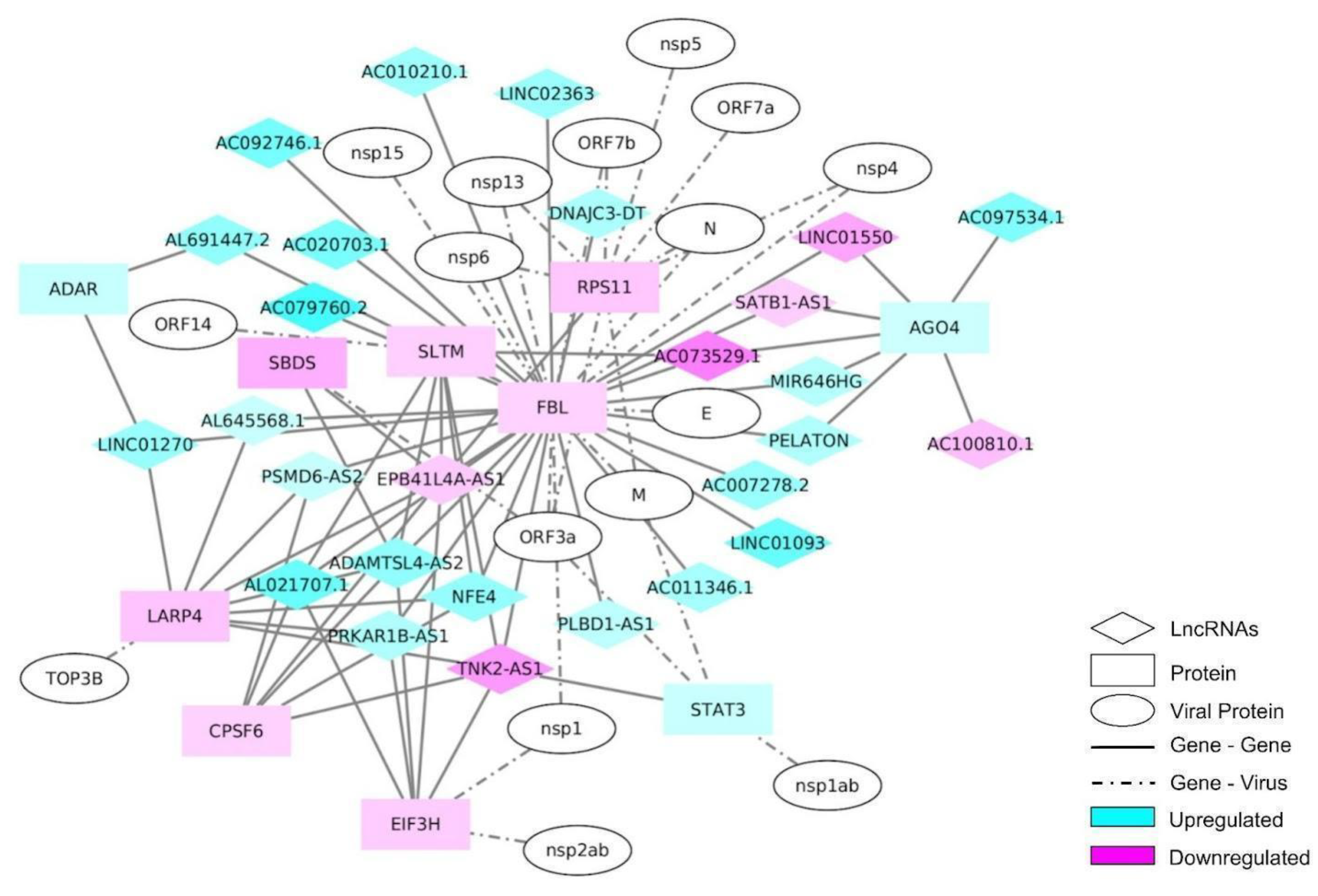
2.4. lncRNA Interactome
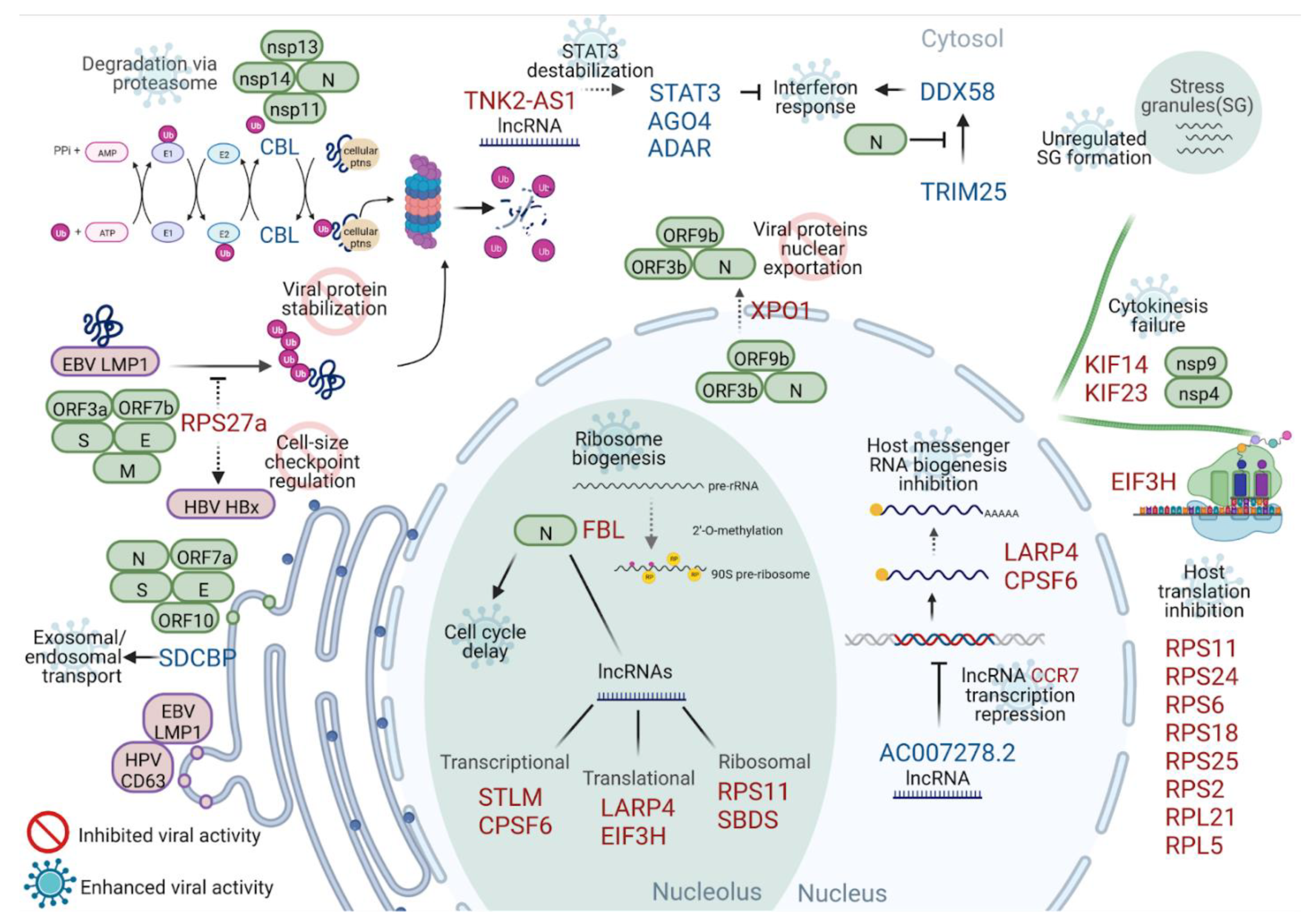
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Patient Cohort and Sample Processing
5.2. SARS-CoV-2 Detection and Quantification
5.3. Severity Biomarker Investigation
5.4. RNA Extraction and Library Preparation
5.5. RNA Sequencing (RNA-seq) Analysis
5.6. Enrichment Analysis
5.7. Interactome Analysis
5.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cascella, M.; Rajnik, M.; Aleem, A.; Dulebohn, S.C.; Di Napoli, R. Features, Evaluation, and Treatment of Coronavirus (COVID-19); StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Li, J.; Huang, D.Q.; Zou, B.; Yang, H.; Hui, W.Z.; Rui, F.; Yee, N.T.S.; Liu, C.; Nerurkar, S.N.; Kai, J.C.Y.; et al. Epidemiology of COVID-19: A systematic review and meta-analysis of clinical characteristics, risk factors, and outcomes. J. Med. Virol. 2021, 93, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, L.; Sang, L.; Ye, F.; Ruan, S.; Zhong, B.; Song, T.; Alshukairi, A.N.; Chen, R.; Zhang, Z.; et al. Kinetics of viral load and antibody response in relation to COVID-19 severity. J. Clin. Investig. 2020, 130, 5235–5244. [Google Scholar] [CrossRef] [PubMed]
- De Lorenzo, R.; Loré, N.I.; Finardi, A.; Mandelli, A.; Cirillo, D.M.; Tresoldi, C.; Benedetti, F.; Ciceri, F.; Rovere-Querini, P.; Comi, G.; et al. Blood neurofilament light chain and total tau levels at admission predict death in COVID-19 patients. J. Neurol. 2021, 268, 4436–4442. [Google Scholar] [CrossRef] [PubMed]
- Fällmar, D.; Rostami, E.; Kumlien, E.; Ashton, N.J.; Jackmann, S.; Pavel, R.; Blennow, K.; Hultström, M.; Lipcsey, M.; Frithiof, R.; et al. The extent of neuroradiological findings in COVID-19 shows correlation with blood biomarkers, Glasgow coma scale score and days in intensive care. J. Neuroradiol. 2021, 49, 421–427. [Google Scholar] [CrossRef]
- Passos, F.R.S.; Heimfarth, L.; Monteiro, B.S.; Corrêa, C.B.; de Moura, T.R.; de Souza Araújo, A.A.; Martins-Filho, P.R.; Quintans-Júnior, L.J.; de Souza Siqueira Quintans, J. Oxidative stress and inflammatory markers in patients with COVID-19: Potential role of RAGE, HMGB1, GFAP and COX-2 in disease severity. Int. Immunopharmacol. 2022, 104, 108502. [Google Scholar] [CrossRef]
- Overmyer, K.A.; Shishkova, E.; Miller, I.J.; Balnis, J.; Bernstein, M.N.; Peters-Clarke, T.M.; Meyer, J.G.; Quan, Q.; Muehlbauer, L.K.; Trujillo, E.A.; et al. Large-Scale Multi-omic Analysis of COVID-19 Severity. Cell Syst. 2021, 12, 23–40.e7. [Google Scholar] [CrossRef]
- Xiong, Y.; Liu, Y.; Cao, L.; Wang, D.; Guo, M.; Jiang, A.; Guo, D.; Hu, W.; Yang, J.; Tang, Z.; et al. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerg. Microbes Infect. 2020, 9, 761–770. [Google Scholar] [CrossRef]
- Gill, S.E.; Dos Santos, C.C.; O’Gorman, D.B.; Carter, D.E.; Patterson, E.K.; Slessarev, M.; Martin, C.; Daley, M.; Miller, M.R.; Cepinskas, G.; et al. Transcriptional profiling of leukocytes in critically ill COVID19 patients: Implications for interferon response and coagulation. Intensive Care Med. Exp. 2020, 8, 75. [Google Scholar] [CrossRef]
- Banerjee, A.K.; Blanco, M.R.; Bruce, E.A.; Honson, D.D.; Chen, L.M.; Chow, A.; Bhat, P.; Ollikainen, N.; Quinodoz, S.A.; Loney, C.; et al. SARS-CoV-2 Disrupts Splicing, Translation, and Protein Trafficking to Suppress Host Defenses. Cell 2020, 183, 1325–1339.e21. [Google Scholar] [CrossRef]
- Lee, J.-G.; Huang, W.; Lee, H.; van de Leemput, J.; Kane, M.A.; Han, Z. Characterization of SARS-CoV-2 proteins reveals Orf6 pathogenicity, subcellular localization, host interactions and attenuation by Selinexor. Cell Biosci. 2021, 11, 58. [Google Scholar] [CrossRef]
- Lock, P.; Stacey, T.T.; Straffon, A.F.L.; Schieb, H.; Hovens, C.M.; Stylli, S.S. Spred-2 steady-state levels are regulated by phosphorylation and Cbl-mediated ubiquitination. Biochem. Biophys. Res. Commun. 2006, 351, 1018–1023. [Google Scholar] [CrossRef] [PubMed]
- Roca Suarez, A.A.; Van Renne, N.; Baumert, T.F.; Lupberger, J. Viral manipulation of STAT3: Evade, exploit, and injure. PLoS Pathog. 2018, 14, e1006839. [Google Scholar] [CrossRef]
- Barral, P.M.; Sarkar, D.; Su, Z.-Z.; Barber, G.N.; DeSalle, R.; Racaniello, V.R.; Fisher, P.B. Functions of the cytoplasmic RNA sensors RIG-I and MDA-5: Key regulators of innate immunity. Pharmacol. Ther. 2009, 124, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Gack, M.U.; Shin, Y.C.; Joo, C.-H.; Urano, T.; Liang, C.; Sun, L.; Takeuchi, O.; Akira, S.; Chen, Z.; Inoue, S.; et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 2007, 446, 916–920. [Google Scholar] [CrossRef]
- Carty, M.; Guy, C.; Bowie, A.G. Detection of Viral Infections by Innate Immunity. Biochem. Pharmacol. 2021, 183, 114316. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Zhang, X.; Zhang, J.; Chen, S.; Ma, Y.; Hu, J.; Yue, T.; Wang, J.; Zhu, J.; Wu, T.; et al. Paxillin knockdown suppresses metastasis and epithelial-mesenchymal transition in colorectal cancer via the ERK signalling pathway. Oncol. Rep. 2020, 44, 1105–1115. [Google Scholar] [CrossRef] [PubMed]
- Yoshigi, M.; Hoffman, L.M.; Jensen, C.C.; Yost, H.J.; Beckerle, M.C. Mechanical force mobilizes zyxin from focal adhesions to actin filaments and regulates cytoskeletal reinforcement. J. Cell Biol. 2005, 171, 209–215. [Google Scholar] [CrossRef]
- Chen, C.-Y.; Chen, J.; He, L.; Stiles, B.L. PTEN: Tumor Suppressor and Metabolic Regulator. Front. Endocrinol. 2018, 9, 338. [Google Scholar] [CrossRef]
- Philley, J.V.; Kannan, A.; Dasgupta, S. MDA-9/Syntenin Control. J. Cell Physiol. 2016, 231, 545–550. [Google Scholar] [CrossRef]
- Granneman, S.; Baserga, S.J. Ribosome biogenesis: Of knobs and RNA processing. Exp. Cell Res. 2004, 296, 43–50. [Google Scholar] [CrossRef]
- Landry, D.M.; Hertz, M.I.; Thompson, S.R. RPS25 is essential for translation initiation by the Dicistroviridae and hepatitis C viral IRESs. Genes Dev. 2009, 23, 2753–2764. [Google Scholar] [CrossRef]
- Carleton, M.; Mao, M.; Biery, M.; Warrener, P.; Kim, S.; Buser, C.C.; Marshall, C.G.; Fernandes, C.; Annis, J.; Linsley, P.S. RNA interference-mediated silencing of mitotic kinesin KIF14 disrupts cell cycle progression and induces cytokinesis failure. Mol. Cell Biol. 2006, 26, 3853–3863. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Wang, J.; Zhang, D.; Chen, W.; Hou, L.; Wu, X.; Lu, Y. Inhibition of KIF14 Suppresses Tumor Cell Growth and Promotes Apoptosis in Human Glioblastoma. Cell Physiol. Biochem. 2015, 37, 1659–1670. [Google Scholar] [CrossRef]
- Glotzer, M. The molecular requirements for cytokinesis. Science 2005, 307, 1735–1739. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Han, M.-E.; Oh, S.-O. The molecular mechanism for nuclear transport and its application. Anat. Cell Biol. 2017, 50, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Tabe, Y.; Kojima, K.; Yamamoto, S.; Sekihara, K.; Matsushita, H.; Davis, R.E.; Wang, Z.; Ma, W.; Ishizawa, J.; Kazuno, S.; et al. Ribosomal Biogenesis and Translational Flux Inhibition by the Selective Inhibitor of Nuclear Export (SINE) XPO1 Antagonist KPT-185. PLoS ONE 2015, 10, e0137210. [Google Scholar] [CrossRef] [PubMed]
- Dimitrova, D.G.; Teysset, L.; Carré, C. RNA 2′-O-Methylation (Nm) Modification in Human Diseases. Genes 2019, 10, 117. [Google Scholar] [CrossRef] [PubMed]
- Karijolich, J.; Kantartzis, A.; Yu, Y.-T. RNA modifications: A mechanism that modulates gene expression. Methods Mol. Biol. 2010, 629, 1–19. [Google Scholar]
- Jain, S.; Wheeler, J.R.; Walters, R.W.; Agrawal, A.; Barsic, A.; Parker, R. ATPase-Modulated Stress Granules Contain a Diverse Proteome and Substructure. Cell 2016, 164, 487–498. [Google Scholar] [CrossRef]
- Zheng, Z.-Q.; Wang, S.-Y.; Xu, Z.-S.; Fu, Y.-Z.; Wang, Y.-Y. SARS-CoV-2 nucleocapsid protein impairs stress granule formation to promote viral replication. Cell Discov. 2021, 7, 38. [Google Scholar] [CrossRef]
- Prasad, K.; Alasmari, A.F.; Ali, N.; Khan, R.; Alghamdi, A.; Kumar, V. Insights into the SARS-CoV-2-Mediated Alteration in the Stress Granule Protein Regulatory Networks in Humans. Pathogens 2021, 10, 1459. [Google Scholar] [CrossRef] [PubMed]
- Jafarzadeh, A.; Nemati, M.; Jafarzadeh, S. Contribution of STAT3 to the Pathogenesis of COVID-19. Microb Pathog. 2021, 154, 104836. [Google Scholar] [CrossRef] [PubMed]
- Ringlander, J.; Fingal, J.; Kann, H.; Prakash, K.; Rydell, G.; Andersson, M.; Martner, A.; Lindh, M.; Horal, P.; Hellstrand, K.; et al. Impact of ADAR-induced editing of minor viral RNA populations on replication and transmission of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2022, 119, e2112663119. [Google Scholar] [CrossRef] [PubMed]
- Plassart, L.; Shayan, R.; Montellese, C.; Rinaldi, D.; Larburu, N.; Pichereaux, C.; Froment, C.; Lebaron, S.; O’Donohue, M.-F.; Kutay, U.; et al. The final step of 40S ribosomal subunit maturation is controlled by a dual key lock. Elife. 2021, 10, e61254. [Google Scholar] [CrossRef] [PubMed]
- Ganapathi, K.A.; Austin, K.M.; Lee, C.-S.; Dias, A.; Malsch, M.M.; Reed, R.; Shimamura, A. The human Shwachman-Diamond syndrome protein, SBDS, associates with ribosomal RNA. Blood 2007, 110, 1458–1465. [Google Scholar] [CrossRef] [PubMed]
- Choudhuri, A.; Maitra, U.; Evans, T. Translation initiation factor eIF3h targets specific transcripts to polysomes during embryogenesis. Proc. Natl. Acad. Sci. USA 2013, 110, 9818–9823. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Gaidamakov, S.A.; Xie, J.; Lee, J.; Martino, L.; Kozlov, G.; Crawford, A.K.; Russo, A.N.; Conte, M.R.; Gehring, K.; et al. La-related protein 4 binds poly(A), interacts with the poly(A)-binding protein MLLE domain via a variant PAM2w motif, and can promote mRNA stability. Mol. Cell Biol. 2011, 31, 542–556. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhan, X.; Kim, B.; Wu, J. A proteomic approach identifies SAFB-like transcription modulator (SLTM) as a bidirectional regulator of GLI family zinc finger transcription factors. J. Biol. Chem. 2019, 294, 5549–5561. [Google Scholar] [CrossRef]
- Zhong, Z.; Ning, J.; Boggs, E.A.; Jang, S.; Wallace, C.; Telmer, C.; Bruchez, M.P.; Ahn, J.; Engelman, A.N.; Zhang, P.; et al. Cytoplasmic CPSF6 Regulates HIV-1 Capsid Trafficking and Infection in a Cyclophilin A-Dependent Manner. MBio 2021, 12, e03142-20. [Google Scholar] [CrossRef]
- Bankar, R.; Suvarna, K.; Ghantasala, S.; Banerjee, A.; Biswas, D.; Choudhury, M.; Palanivel, V.; Salkar, A.; Verma, A.; Singh, A.; et al. Proteomic investigation reveals dominant alterations of neutrophil degranulation and mRNA translation pathways in patients with COVID-19. iScience 2021, 24, 102135. [Google Scholar] [CrossRef]
- Shen, B.; Yi, X.; Sun, Y.; Bi, X.; Du, J.; Zhang, C.; Quan, S.; Zhang, F.; Sun, R.; Qian, L.; et al. Proteomic and Metabolomic Characterization of COVID-19 Patient Sera. Cell 2020, 182, 59–72.e15. [Google Scholar] [CrossRef] [PubMed]
- Lodge, K.M.; Cowburn, A.S.; Li, W.; Condliffe, A.M. The Impact of Hypoxia on Neutrophil Degranulation and Consequences for the Host. Int. J. Mol. Sci. 2020, 21, 1183. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, S.; Sharma, K.; Silakari, O. The interplay between inflammatory pathways and COVID-19: A critical review on pathogenesis and therapeutic options. Microb Pathog. 2021, 150, 104673. [Google Scholar] [CrossRef]
- Sun, X.; Wang, T.; Cai, D.; Hu, Z.; Chen, J.; Liao, H.; Zhi, L.; Wei, H.; Zhang, Z.; Qiu, Y.; et al. Cytokine storm intervention in the early stages of COVID-19 pneumonia. Cytokine Growth Factor Rev. 2020, 53, 38–42. [Google Scholar] [CrossRef]
- Wang, J.; Jiang, M.; Chen, X.; Montaner, L.J. Cytokine storm and leukocyte changes in mild versus severe SARS-CoV-2 infection: Review of 3939 COVID-19 patients in China and emerging pathogenesis and therapy concepts. J. Leukoc. Biol. 2020, 108, 17–41. [Google Scholar] [CrossRef]
- Idrees, D.; Kumar, V. SARS-CoV-2 spike protein interactions with amyloidogenic proteins: Potential clues to neurodegeneration. Biochem. Biophys. Res. Commun. 2021, 554, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Litvinchuk, A.; Wan, Y.-W.; Swartzlander, D.B.; Chen, F.; Cole, A.; Propson, N.E.; Wang, Q.; Zhang, B.; Liu, Z.; Zheng, H. Complement C3aR Inactivation Attenuates Tau Pathology and Reverses an Immune Network Deregulated in Tauopathy Models and Alzheimer’s Disease. Neuron 2018, 100, 1337–1353.e5. [Google Scholar] [CrossRef]
- Pilotto, A.; Masciocchi, S.; Volonghi, I.; De Giuli, V.; Caprioli, F.; Mariotto, S.; Ferrari, S.; Bozzetti, S.; Imarisio, A.; Risi, B.; et al. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Encephalitis Is a Cytokine Release Syndrome: Evidences From Cerebrospinal Fluid Analyses. Clin. Infect. Dis. 2021, 73, e3019–e3026. [Google Scholar] [CrossRef]
- Paniz-Mondolfi, A.; Bryce, C.; Grimes, Z.; Gordon, R.E.; Reidy, J.; Lednicky, J.; Sordillo, E.M.; Fowkes, M. Central nervous system involvement by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). J. Med. Virol. 2020, 92, 699–702. [Google Scholar] [CrossRef]
- Katze, M.G.; Krug, R.M. Translational control in influenza virus-infected cells. Enzyme 1990, 44, 265–277. [Google Scholar] [CrossRef]
- Li, S. Regulation of Ribosomal Proteins on Viral Infection. Cells 2019, 8, 508. [Google Scholar] [CrossRef] [PubMed]
- Rider, M.A.; Cheerathodi, M.R.; Hurwitz, S.N.; Nkosi, D.; Howell, L.A.; Tremblay, D.C.; Liu, X.; Zhu, F.; Meckes, D.G., Jr. The interactome of EBV LMP1 evaluated by proximity-based BioID approach. Virology 2018, 516, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Gräßel, L.; Fast, L.A.; Scheffer, K.D.; Boukhallouk, F.; Spoden, G.A.; Tenzer, S.; Boller, K.; Bago, R.; Rajesh, S.; Overduin, M.; et al. The CD63-Syntenin-1 Complex Controls Post-Endocytic Trafficking of Oncogenic Human Papillomaviruses. Sci. Rep. 2016, 6, 32337. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, T.; Murray, J.; Walker, C.J.; Chang, H.; Tamir, S.; Hou, B.; Shacham, S.; Kauffman, M.G.; Tripp, R.A.; Landesman, Y. Selinexor, a novel selective inhibitor of nuclear export, reduces SARS-CoV-2 infection and protects the respiratory system in vivo. Antivir. Res. 2021, 192, 105115. [Google Scholar] [CrossRef] [PubMed]
- Freundt, E.C.; Yu, L.; Park, E.; Lenardo, M.J.; Xu, X.-N. Molecular determinants for subcellular localization of the severe acute respiratory syndrome coronavirus open reading frame 3b protein. J. Virol. 2009, 83, 6631–6640. [Google Scholar] [CrossRef]
- Konno, Y.; Kimura, I.; Uriu, K.; Fukushi, M.; Irie, T.; Koyanagi, Y.; Sauter, D.; Gifford, R.J.; USFQ-COVID19 Consortium; Nakagawa, S.; et al. SARS-CoV-2 ORF3b Is a Potent Interferon Antagonist Whose Activity Is Increased by a Naturally Occurring Elongation Variant. Cell Rep. 2020, 32, 108185. [Google Scholar] [CrossRef]
- Li, J.-Y.; Liao, C.-H.; Wang, Q.; Tan, Y.-J.; Luo, R.; Qiu, Y.; Ge, X.-Y. The ORF6, ORF8 and nucleocapsid proteins of SARS-CoV-2 inhibit type I interferon signaling pathway. Virus Res. 2020, 286, 198074. [Google Scholar] [CrossRef]
- Jiang, H.-W.; Zhang, H.-N.; Meng, Q.-F.; Xie, J.; Li, Y.; Chen, H.; Zheng, Y.-X.; Wang, X.-N.; Qi, H.; Zhang, J.; et al. SARS-CoV-2 Orf9b suppresses type I interferon responses by targeting TOM70. Cell Mol. Immunol. 2020, 17, 998–1000. [Google Scholar] [CrossRef]
- Mathew, C.; Ghildyal, R. CRM1 Inhibitors for Antiviral Therapy. Front. Microbiol. 2017, 8, 1171. [Google Scholar] [CrossRef]
- Uddin, M.H.; Zonder, J.A.; Azmi, A.S. Exportin 1 inhibition as antiviral therapy. Drug Discov. Today 2020, 25, 1775–1781. [Google Scholar] [CrossRef]
- Luo, H. Interplay between the virus and the ubiquitin-proteasome system: Molecular mechanism of viral pathogenesis. Curr. Opin. Virol. 2016, 17, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Huibregtse, J.M.; Vierstra, R.D.; Howley, P.M. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 1993, 75, 495–505. [Google Scholar] [CrossRef]
- Querido, E.; Blanchette, P.; Yan, Q.; Kamura, T.; Morrison, M.; Boivin, D.; Kaelin, W.G.; Conaway, R.C.; Conaway, J.W.; Branton, P.E. Degradation of p53 by adenovirus E4orf6 and E1B55K proteins occurs via a novel mechanism involving a Cullin-containing complex. Genes Dev. 2001, 15, 3104–3117. [Google Scholar] [CrossRef]
- Frontera, J.A.; Boutajangout, A.; Masurkar, A.V.; Betensky, R.A.; Ge, Y.; Vedvyas, A.; Debure, L.; Moreira, A.; Lewis, A.; Huang, J.; et al. Comparison of serum neurodegenerative biomarkers among hospitalized COVID-19 patients versus non-COVID subjects with normal cognition, mild cognitive impairment, or Alzheimer’s dementia. Alzheimers Dement. 2022, 18, 899–910. [Google Scholar] [CrossRef]
- Hong, S.-W.; Kim, S.-M.; Jin, D.-H.; Kim, Y.S.; Hur, D.Y. RPS27a enhances EBV-encoded LMP1-mediated proliferation and invasion by stabilizing of LMP1. Biochem. Biophys. Res. Commun. 2017, 491, 303–309. [Google Scholar] [CrossRef]
- Fatima, G.; Mathan, G.; Kumar, V. The HBx protein of hepatitis B virus regulates the expression, intracellular distribution and functions of ribosomal protein S27a. J. Gen. Virol. 2012, 93, 706–715. [Google Scholar] [CrossRef] [PubMed]
- Jouffe, C.; Cretenet, G.; Symul, L.; Martin, E.; Atger, F.; Naef, F.; Gachon, F. The circadian clock coordinates ribosome biogenesis. PLoS Biol. 2013, 11, e1001455. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.-X.; DeVine, T.; Challagundla, K.B.; Dai, M.-S. Interplay between ribosomal protein S27a and MDM2 protein in p53 activation in response to ribosomal stress. J. Biol. Chem. 2011, 286, 22730–22741. [Google Scholar] [CrossRef]
- Zhang, J.; Buranjiang, G.; Mutalifu, Z.; Jin, H.; Yao, L. KIF14 affects cell cycle arrest and cell viability in cervical cancer by regulating the p27Kip1 pathway. World J. Surg. Oncol. 2022, 20, 125. [Google Scholar] [CrossRef]
- Feuer, R.; Mena, I.; Pagarigan, R.; Slifka, M.K.; Whitton, J.L. Cell cycle status affects coxsackievirus replication, persistence, and reactivation in vitro. J. Virol. 2002, 76, 4430–4440. [Google Scholar] [CrossRef]
- Chen, H.; Wurm, T.; Britton, P.; Brooks, G.; Hiscox, J.A. Interaction of the coronavirus nucleoprotein with nucleolar antigens and the host cell. J. Virol. 2002, 76, 5233–5250. [Google Scholar] [CrossRef] [PubMed]
- Oughtred, R.; Rust, J.; Chang, C.; Breitkreutz, B.-J.; Stark, C.; Willems, A.; Boucher, L.; Leung, G.; Kolas, N.; Zhang, F.; et al. The BioGRID database: A comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci. 2021, 30, 187–200. [Google Scholar] [CrossRef]
- Oh, S.J.; Shin, O.S. SARS-CoV-2 Nucleocapsid Protein Targets RIG-I-Like Receptor Pathways to Inhibit the Induction of Interferon Response. Cells 2021, 10, 530. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Sui, L.; Wu, P.; Wang, W.; Wang, Z.; Yu, Y.; Hou, Z.; Tan, G.; Liu, Q.; Wang, G. A dual-role of SARS-CoV-2 nucleocapsid protein in regulating innate immune response. Signal Transduct Target Ther. 2021, 6, 331. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Ye, Q.; Lu, S.; Corbett, K.D. Structural basis for SARS-CoV-2 Nucleocapsid protein recognition by single-domain antibodies. Front. Immunol. 2021, 12, 719037. [Google Scholar] [CrossRef]
- Ryan, V.H.; Fawzi, N.L. Physiological, Pathological, and Targetable Membraneless Organelles in Neurons. Trends Neurosci. 2019, 42, 693–708. [Google Scholar] [CrossRef]
- White, J.P.; Lloyd, R.E. Regulation of stress granules in virus systems. Trends Microbiol. 2012, 20, 175–183. [Google Scholar] [CrossRef]
- Linero, F.N.; Thomas, M.G.; Boccaccio, G.L.; Scolaro, L.A. Junin virus infection impairs stress-granule formation in Vero cells treated with arsenite via inhibition of eIF2α phosphorylation. J. Gen. Virol. 2011, 92, 2889–2899. [Google Scholar] [CrossRef]
- Bocchetti, M.; Scrima, M.; Melisi, F.; Luce, A.; Sperlongano, R.; Caraglia, M.; Zappavigna, S.; Cossu, A.M. LncRNAs and Immunity: Coding the Immune System with Noncoding Oligonucleotides. Int. J. Mol. Sci. 2021, 22, 1741. [Google Scholar] [CrossRef]
- Devadoss, D.; Acharya, A.; Manevski, M.; Houserova, D.; Cioffi, M.D.; Pandey, K.; Nair, M.; Chapagain, P.; Mirsaeidi, M.; Borchert, G.M.; et al. Immunomodulatory LncRNA on antisense strand of ICAM-1 augments SARS-CoV-2 infection-associated airway mucoinflammatory phenotype. iScience 2022, 25, 104685. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Han, D.; Pan, L.; Sun, J. The positive feedback between lncRNA TNK2-AS1 and STAT3 enhances angiogenesis in non-small cell lung cancer. Biochem. Biophys. Res. Commun. 2018, 507, 185–192. [Google Scholar] [CrossRef] [PubMed]
- You, Y.; Zhao, X.; Wu, Y.; Mao, J.; Ge, L.; Guo, J.; Zhao, C.; Chen, D.; Song, Z. Integrated Transcriptome Profiling Revealed That Elevated Long Non-Coding RNA-AC007278.2 Expression Repressed CCR7 Transcription in Systemic Lupus Erythematosus. Front. Immunol. 2021, 12, 615859. [Google Scholar] [CrossRef] [PubMed]
- Hung, J.; Scanlon, J.P.; Mahmoud, A.D.; Rodor, J.; Ballantyne, M.; Fontaine, M.A.C.; Temmerman, L.; Kaczynski, J.; Connor, K.L.; Bhushan, R.; et al. Novel Plaque Enriched Long Noncoding RNA in Atherosclerotic Macrophage Regulation (PELATON). Arterioscler Thromb. Vasc. Biol. 2020, 40, 697–713. [Google Scholar] [CrossRef]
- Deng, X.; Bi, Q.; Chen, S.; Chen, X.; Li, S.; Zhong, Z.; Guo, W.; Li, X.; Deng, Y.; Yang, Y. Identification of a Five-Autophagy-Related-lncRNA Signature as a Novel Prognostic Biomarker for Hepatocellular Carcinoma. Front. Mol. Biosci. 2020, 7, 611626. [Google Scholar] [CrossRef]
- Zheng, Y.; Yu, K.; Huang, C.; Liu, L.; Zhao, H.; Huo, M.; Zhang, J. Integrated bioinformatics analysis reveals role of the LINC01093/miR-96-5p/ZFAND5/NF-κB signaling axis in hepatocellular carcinoma. Exp. Ther. Med. 2019, 18, 3853–3860. [Google Scholar] [CrossRef]
- Chen, J.; Li, P.; Chen, Z.; Wang, S.; Tang, S.; Chen, X.; Chen, Z.; Zhou, J. Elevated LINC01550 induces the apoptosis and cell cycle arrest of melanoma. Med. Oncol. 2021, 38, 32. [Google Scholar] [CrossRef]
- Huang, K.; Wang, C.; Vagts, C.; Raguveer, V.; Finn, P.W.; Perkins, D.L. Long non-coding RNAs (lncRNAs) NEAT1 and MALAT1 are differentially expressed in severe COVID-19 patients: An integrated single-cell analysis. PLoS ONE 2022, 17, e0261242. [Google Scholar] [CrossRef]
- Frye, M.; Harada, B.T.; Behm, M.; He, C. RNA modifications modulate gene expression during development. Science 2018, 361, 1346–1349. [Google Scholar] [CrossRef]
- Slobodin, B.; Sehrawat, U.; Lev, A.; Hayat, D.; Zuckerman, B.; Fraticelli, D.; Ogran, A.; Ben-Shmuel, A.; Bar-David, E.; Levy, H.; et al. Cap-independent translation and a precisely located RNA sequence enable SARS-CoV-2 to control host translation and escape anti-viral response. Nucleic Acids Res. 2022, 50, 8080–8092. [Google Scholar] [CrossRef]
- Paramasivam, A. RNA 2′-O-methylation modification and its implication in COVID-19 immunity. Cell Death Discov. 2020, 6, 118. [Google Scholar] [CrossRef] [PubMed]
- Krafcikova, P.; Silhan, J.; Nencka, R.; Boura, E. Structural analysis of the SARS-CoV-2 methyltransferase complex involved in RNA cap creation bound to sinefungin. Nat. Commun. 2020, 11, 3717. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Falcon, S.; Gentleman, R. Using GOstats to test gene lists for GO term association. Bioinformatics 2007, 23, 257–258. [Google Scholar] [CrossRef]
- Luo, W.; Brouwer, C. Pathview: An R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics 2013, 29, 1830–1831. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Teng, X.; Chen, X.; Xue, H.; Tang, Y.; Zhang, P.; Kang, Q.; Hao, Y.; Chen, R.; Zhao, Y.; He, S. NPInter v4.0: An integrated database of ncRNA interactions. Nucleic Acids Res. 2020, 48, D160–D165. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Laboratory and Clinical Features | Severe (n = 12) | Mild (n = 5) | p-Value |
|---|---|---|---|
| SARS-Cov-2 E viral load (median) * | 6620 | 13.8 | 0.0059 |
| Hospitalization (in days; median) | 14 | NA | - |
| Hypertension | (10) 83.3 % | (1) 20.0 % | - |
| Diabetes | (9) 75.0 | 0 | - |
| Obesity | 0 | 0 | - |
| Dyslipidemia | 0 | 0 | - |
| Pneumonia | (12) 100% | 0 | - |
| Oxygen supplementation | (12) 100% | 0 | - |
| SOFA index (median) | 6 | NA | - |
| Neurofilament plasma level (median) | 91.80 pg/mL | 14.52 pg/mL | 0.0275 |
| Glial fibrillary acid protein plasma level (median) | 346.0 pg/mL | 143.9 pg/mL | 0.0517 |
| Ubiquitin carboxy-terminal hydroxylase L1 plasma level (median) | 100.1 pg/mL | 38.83 pg/mL | 0.0133 |
| TAU total protein plasma level (median) | 6.668 pg/mL | 2.599 pg/mL | 0.0133 |
| Lethality | (6) 50% | 0 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cavalcante, L.T.d.F.; da Fonseca, G.C.; Amado Leon, L.A.; Salvio, A.L.; Brustolini, O.J.; Gerber, A.L.; Guimarães, A.P.d.C.; Marques, C.A.B.; Fernandes, R.A.; Ramos Filho, C.H.F.; et al. Buffy Coat Transcriptomic Analysis Reveals Alterations in Host Cell Protein Synthesis and Cell Cycle in Severe COVID-19 Patients. Int. J. Mol. Sci. 2022, 23, 13588. https://doi.org/10.3390/ijms232113588
Cavalcante LTdF, da Fonseca GC, Amado Leon LA, Salvio AL, Brustolini OJ, Gerber AL, Guimarães APdC, Marques CAB, Fernandes RA, Ramos Filho CHF, et al. Buffy Coat Transcriptomic Analysis Reveals Alterations in Host Cell Protein Synthesis and Cell Cycle in Severe COVID-19 Patients. International Journal of Molecular Sciences. 2022; 23(21):13588. https://doi.org/10.3390/ijms232113588
Chicago/Turabian StyleCavalcante, Liliane Tavares de Faria, Guilherme Cordenonsi da Fonseca, Luciane Almeida Amado Leon, Andreza Lemos Salvio, Otávio José Brustolini, Alexandra Lehmkuhl Gerber, Ana Paula de Campos Guimarães, Carla Augusta Barreto Marques, Renan Amphilophio Fernandes, Carlos Henrique Ferreira Ramos Filho, and et al. 2022. "Buffy Coat Transcriptomic Analysis Reveals Alterations in Host Cell Protein Synthesis and Cell Cycle in Severe COVID-19 Patients" International Journal of Molecular Sciences 23, no. 21: 13588. https://doi.org/10.3390/ijms232113588
APA StyleCavalcante, L. T. d. F., da Fonseca, G. C., Amado Leon, L. A., Salvio, A. L., Brustolini, O. J., Gerber, A. L., Guimarães, A. P. d. C., Marques, C. A. B., Fernandes, R. A., Ramos Filho, C. H. F., Kader, R. L., Pimentel Amaro, M., da Costa Gonçalves, J. P., Vieira Alves-Leon, S., & Vasconcelos, A. T. R. (2022). Buffy Coat Transcriptomic Analysis Reveals Alterations in Host Cell Protein Synthesis and Cell Cycle in Severe COVID-19 Patients. International Journal of Molecular Sciences, 23(21), 13588. https://doi.org/10.3390/ijms232113588