
Potential Therapeutic Effects of Thiazolidinedione on Malignant Glioma

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Arrest of Glioma Growth by Ciglitazone Either In Vitro and Vivo Assessment
2.2. Antiangiogenesis Effects of Ciglitazone on Malignant Glioma
2.3. Abolishment of Glioma Growth by Ciglitazone through STAT3 Pathway in Both In Vitro and In Vivo Experiments
2.4. Ciglitazone Induced Strong Interactions of p-STAT3 and SHP-2 in U87 Cells
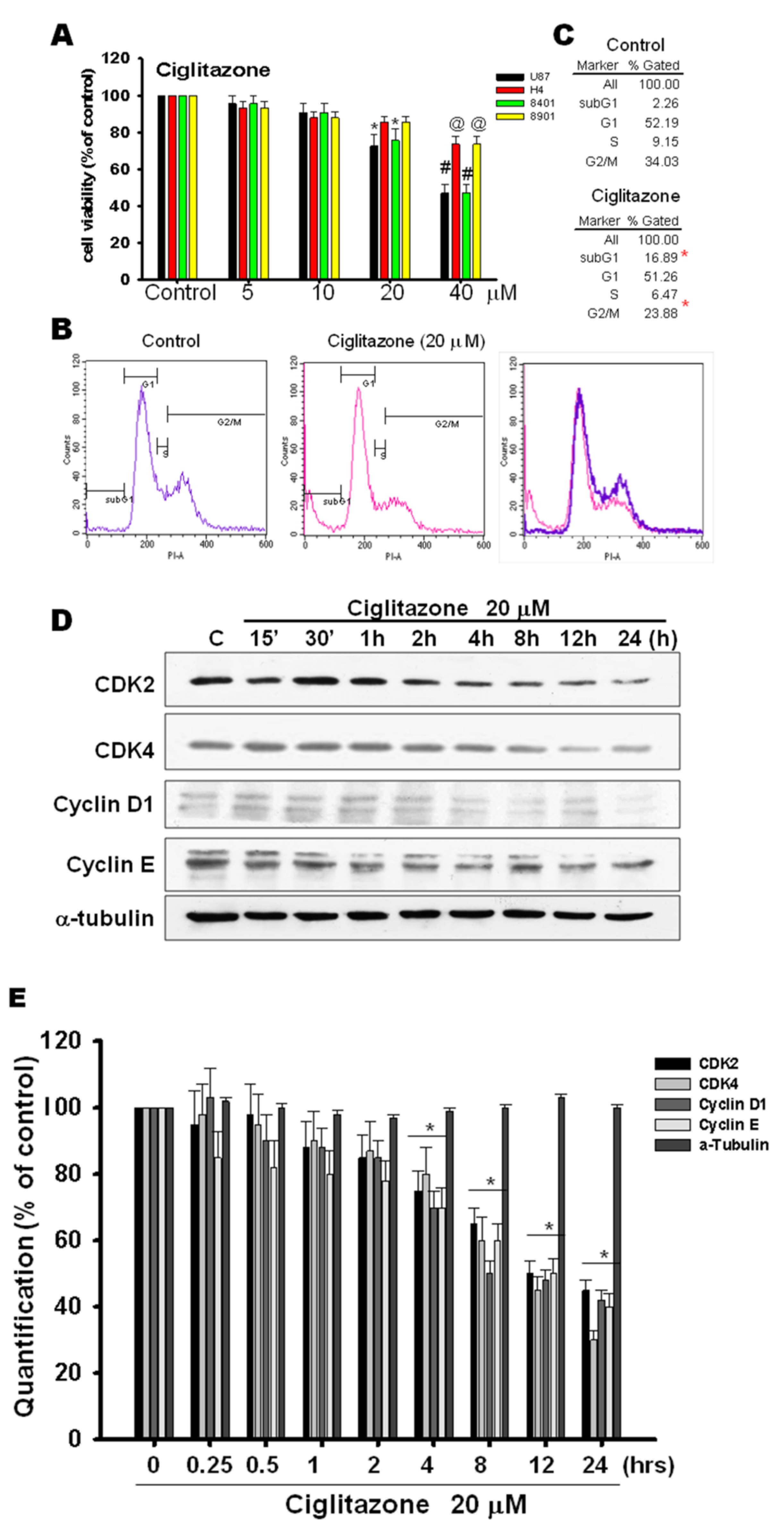
2.5. Glioma Tumor Growths Arrested by Ciglitazone Involved in Cell Cycle Associated with Proteasome and Lysosome Activity
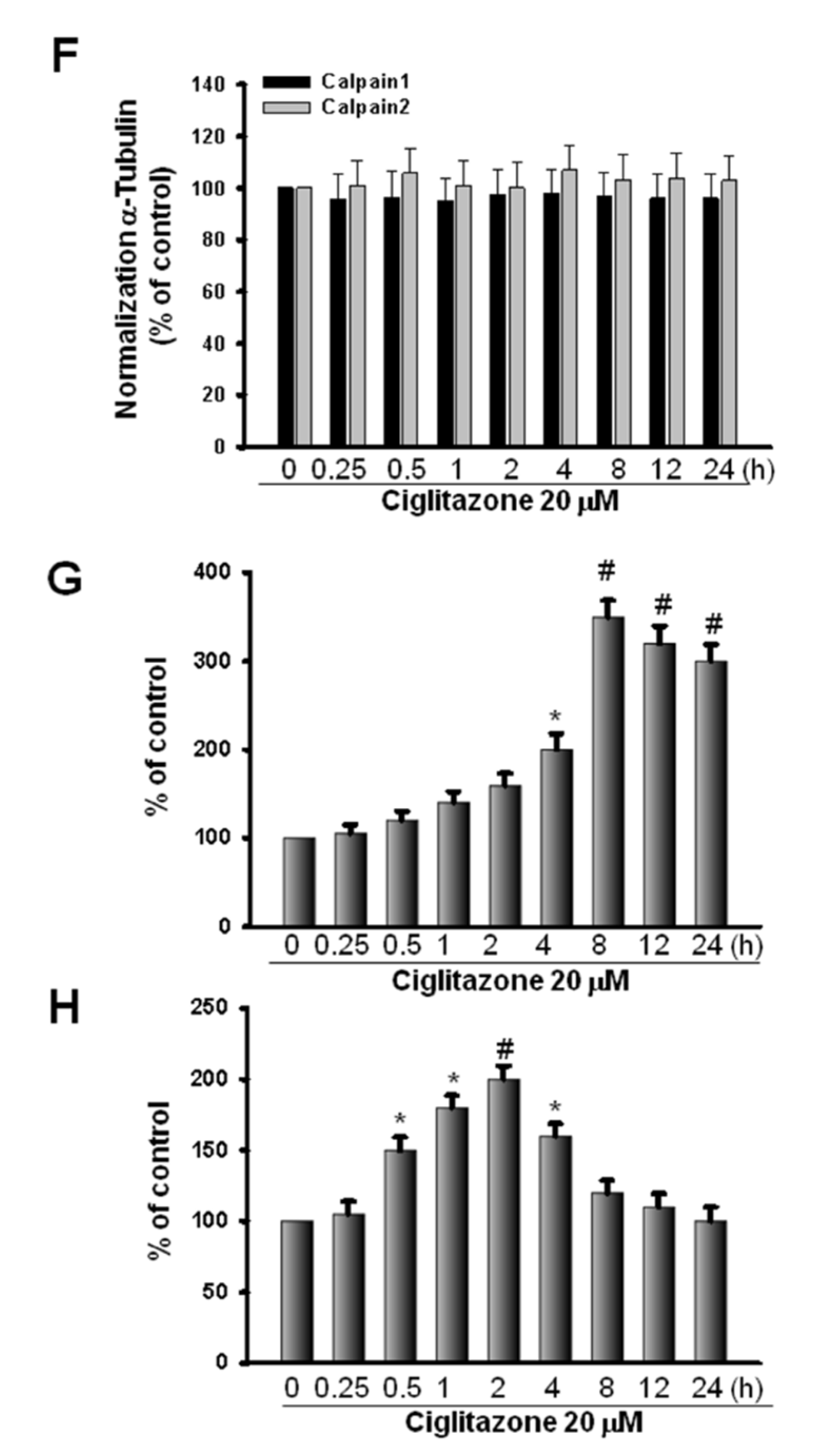
2.6. Induction of Proteasome and Lysosome Formation by Ciglitazone
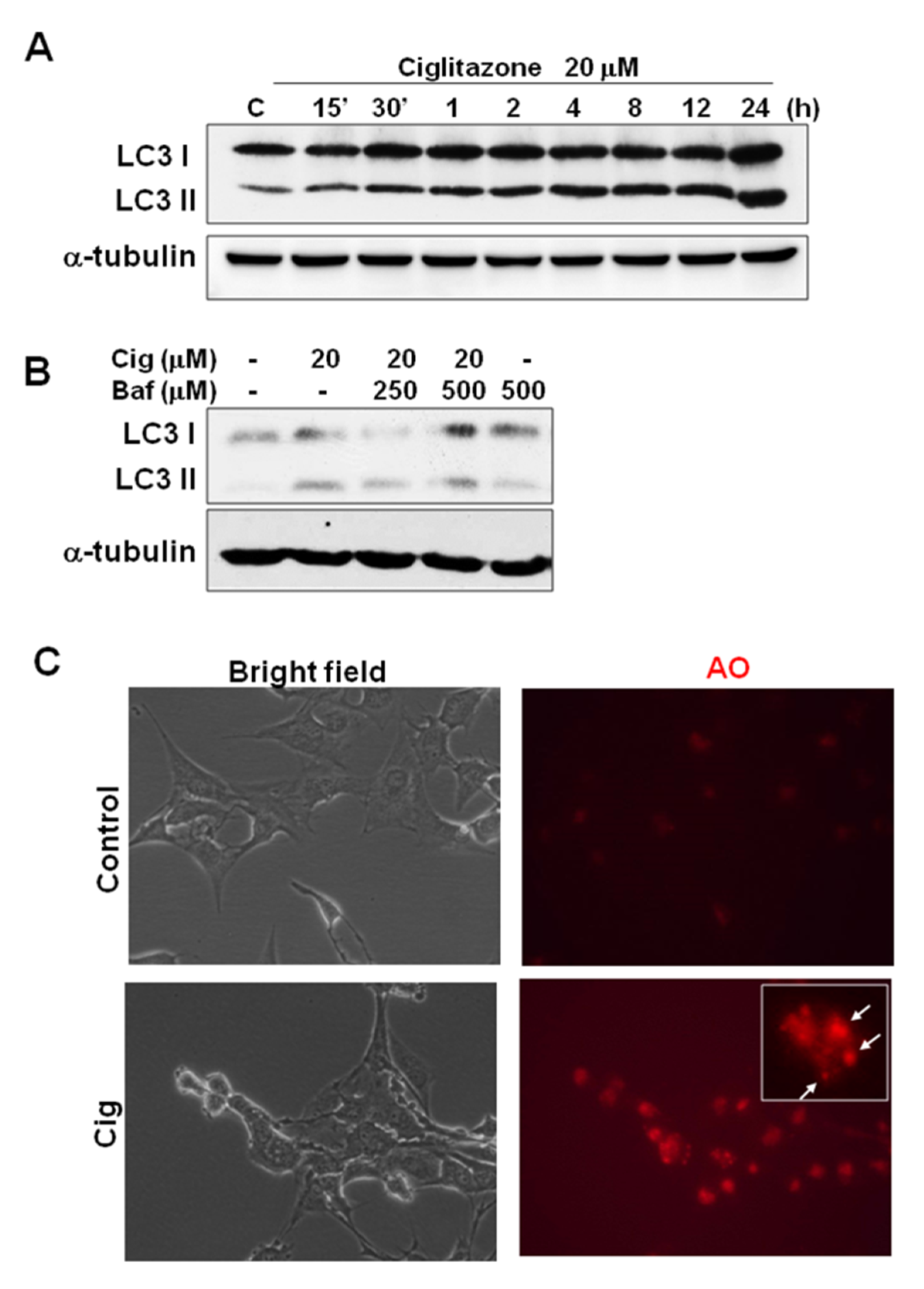
2.7. Ciglitazone-Induced Autophagy in U87 Cells through Overexpressing LC3
3. Discussion
4. Materials and Methods
4.1. Subcutaneous U87 MG Xenograft
4.2. Cell Culture
4.3. Matrigel Plug Assay
4.4. Ex Vivo Vessel Sprouting Aortic Ring Assay
4.5. Immunoprecipitations Analyses
4.6. Western Blot Analyses
4.7. Protein Tyrosine Phosphatase Activity Assay
4.8. Immunohistochemistry
4.9. Histological Examination
4.10. Zymography
4.11. Soft Agar Assay
4.12. Wound Healing Assay
4.13. Acridine Orange Staining
4.14. Flow Cytochemistry
4.15. MTT Assay
4.16. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ostrom, Q.T.; Cote, D.J.; Ascha, M.; Kruchko, C.; Barnholtz-Sloan, J.S. Adult Glioma Incidence and Survival by Race or Ethnicity in the United States From 2000 to 2014. JAMA Oncol. 2018, 4, 1254–1262. [Google Scholar] [CrossRef] [PubMed]
- Zanders, E.D.; Svensson, F.; Bailey, D.S. Therapy for glioblastoma: Is it working? Drug Discov. Today 2019, 24, 1193–1201. [Google Scholar] [CrossRef]
- Fine, H.A. New strategies in glioblastoma: Exploiting the new biology. Clin Cancer Res Off. J. Am. Assoc. Cancer Res. 2015, 21, 1984–1988. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Morosetti, R.; Servidei, T.; Mirabella, M.; Rutella, S.; Mangiola, A.; Maira, G.; Mastrangelo, R.; Koeffler, H.P. The PPARgamma ligands PGJ2 and rosiglitazone show a differential ability to inhibit proliferation and to induce apoptosis and differentiation of human glioblastoma cell lines. Int. J. Oncol. 2004, 25, 493–502. [Google Scholar] [PubMed]
- Pérez-Ortiz, J.M.; Tranque, P.; Vaquero, C.F.; Domingo, B.; Molina, F.; Calvo, S.; Jordán, J.; Ceña, V.; Llopis, J. Glitazones differentially regulate primary astrocyte and glioma cell survival. Involvement of reactive oxygen species and peroxisome proliferator-activated receptor-gamma. J. Biol. Chem. 2004, 279, 8976–8985. [Google Scholar] [CrossRef] [PubMed]
- Grommes, C.; Conway, D.S.; Alshekhlee, A.; Barnholtz-Sloan, J.S. Inverse association of PPARγ agonists use and high grade glioma development. J. Neurooncol. 2010, 100, 233–239. [Google Scholar] [CrossRef]
- Ellis, H.P.; Kurian, K.M. Biological Rationale for the Use of PPARγ Agonists in Glioblastoma. Front. Oncol. 2014, 4, 52. [Google Scholar] [CrossRef]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Bromberg, J.; Darnell, J.E., Jr. The role of STATs in transcriptional control and their impact on cellular function. Oncogene 2000, 19, 2468–2473. [Google Scholar] [CrossRef]
- Luwor, R.B.; Stylli, S.S.; Kaye, A.H. The role of Stat3 in glioblastoma multiforme. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas. 2013, 20, 907–911. [Google Scholar] [CrossRef]
- Lin, G.S.; Yang, L.J.; Wang, X.F.; Chen, Y.P.; Tang, W.L.; Chen, L.; Lin, Z.X. STAT3 Tyr705 phosphorylation affects clinical outcome in patients with newly diagnosed supratentorial glioblastoma. Med. Oncol. 2014, 31, 924. [Google Scholar] [CrossRef] [PubMed]
- Chearwae, W.; Bright, J.J. PPARgamma agonists inhibit growth and expansion of CD133+ brain tumour stem cells. Br. J. Cancer 2008, 99, 2044–2053. [Google Scholar] [CrossRef] [PubMed]
- Niu, G.; Wright, K.L.; Huang, M.; Song, L.; Haura, E.; Turkson, J.; Zhang, S.; Wang, T.; Sinibaldi, D.; Coppola, D.; et al. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene 2002, 21, 2000–2008. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.H.; Kuo, M.L.; Chen, C.A.; Chou, C.H.; Lai, K.B.; Lee, C.N.; Hsieh, C.Y. Interleukin-6 promotes cervical tumor growth by VEGF-dependent angiogenesis via a STAT3 pathway. Oncogene 2003, 22, 1517–1527. [Google Scholar] [CrossRef]
- Yahata, Y.; Shirakata, Y.; Tokumaru, S.; Yamasaki, K.; Sayama, K.; Hanakawa, Y.; Detmar, M.; Hashimoto, K. Nuclear translocation of phosphorylated STAT3 is essential for vascular endothelial growth factor-induced human dermal microvascular endothelial cell migration and tube formation. J. Biol. Chem. 2003, 278, 40026–40031. [Google Scholar] [CrossRef]
- Brantley, E.C.; Benveniste, E.N. Signal transducer and activator of transcription-3: A molecular hub for signaling pathways in gliomas. Mol. Cancer Res. MCR 2008, 6, 675–684. [Google Scholar] [CrossRef]
- Schwartz, G.K.; Shah, M.A. Targeting the cell cycle: A new approach to cancer therapy. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 9408–9421. [Google Scholar] [CrossRef]
- Henson, J.W.; Schnitker, B.L.; Correa, K.M.; von Deimling, A.; Fassbender, F.; Xu, H.J.; Benedict, W.F.; Yandell, D.W.; Louis, D.N. The retinoblastoma gene is involved in malignant progression of astrocytomas. Ann. Neurol. 1994, 36, 714–721. [Google Scholar] [CrossRef]
- Sidransky, D.; Mikkelsen, T.; Schwechheimer, K.; Rosenblum, M.L.; Cavanee, W.; Vogelstein, B. Clonal expansion of p53 mutant cells is associated with brain tumour progression. Nature 1992, 355, 846–847. [Google Scholar] [CrossRef]
- Lam, P.Y.; Di Tomaso, E.; Ng, H.K.; Pang, J.C.; Roussel, M.F.; Hjelm, N.M. Expression of p19INK4d, CDK4, CDK6 in glioblastoma multiforme. Br. J. Neurosurg. 2000, 14, 28–32. [Google Scholar]
- Grommes, C.; Landreth, G.E.; Heneka, M.T. Antineoplastic effects of peroxisome proliferator-activated receptor gamma agonists. Lancet Oncol. 2004, 5, 419–429. [Google Scholar] [CrossRef]
- Chattopadhyay, N.; Singh, D.P.; Heese, O.; Godbole, M.M.; Sinohara, T.; Black, P.M.; Brown, E.M. Expression of peroxisome proliferator-activated receptors (PPARS) in human astrocytic cells: PPARgamma agonists as inducers of apoptosis. J. Neurosci. Res. 2000, 61, 67–74. [Google Scholar] [CrossRef]
- Toyoda, M.; Takagi, H.; Horiguchi, N.; Kakizaki, S.; Sato, K.; Takayama, H.; Mori, M. A ligand for peroxisome proliferator activated receptor gamma inhibits cell growth and induces apoptosis in human liver cancer cells. Gut 2002, 50, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Rusinova, R.; Herold, K.F.; Sanford, R.L.; Greathouse, D.V.; Hemmings, H.C., Jr.; Andersen, O.S. Thiazolidinedione insulin sensitizers alter lipid bilayer properties and voltage-dependent sodium channel function: Implications for drug discovery. J. Gen. Physiol. 2011, 138, 249–270. [Google Scholar] [CrossRef] [PubMed]
- Xin, X.; Yang, S.; Kowalski, J.; Gerritsen, M.E. Peroxisome proliferator-activated receptor gamma ligands are potent inhibitors of angiogenesis in vitro and in vivo. J. Biol. Chem. 1999, 274, 9116–9121. [Google Scholar] [CrossRef]
- Kim, J.E.; Patel, M.; Ruzevick, J.; Jackson, C.M.; Lim, M. STAT3 Activation in Glioblastoma: Biochemical and Therapeutic Implications. Cancers 2014, 6, 376–395. [Google Scholar] [CrossRef]
- He, G.; Thuillier, P.; Fischer, S.M. Troglitazone inhibits cyclin D1 expression and cell cycling independently of PPARgamma in normal mouse skin keratinocytes. J. Investig. Dermatol. 2004, 123, 1110–1119. [Google Scholar] [CrossRef]
- Bromberg, J.F.; Horvath, C.M.; Besser, D.; Lathem, W.W.; Darnell, J.E., Jr. Stat3 activation is required for cellular transformation by v-src. Mol. Cell. Biol. 1998, 18, 2553–2558. [Google Scholar] [CrossRef]
- Bromberg, J.F.; Wrzeszczynska, M.H.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; Darnell, J.E., Jr. Stat3 as an oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef]
- Calò, V.; Migliavacca, M.; Bazan, V.; Macaluso, M.; Buscemi, M.; Gebbia, N.; Russo, A. STAT proteins: From normal control of cellular events to tumorigenesis. J. Cell. Physiol. 2003, 197, 157–168. [Google Scholar] [CrossRef]
- Birner, P.; Toumangelova-Uzeir, K.; Natchev, S.; Guentchev, M. STAT3 tyrosine phosphorylation influences survival in glioblastoma. J. Neurooncol. 2010, 100, 339–343. [Google Scholar] [PubMed]
- Rahaman, S.O.; Harbor, P.C.; Chernova, O.; Barnett, G.H.; Vogelbaum, M.A.; Haque, S.J. Inhibition of constitutively active Stat3 suppresses proliferation and induces apoptosis in glioblastoma multiforme cells. Oncogene 2002, 21, 8404–8413. [Google Scholar] [PubMed]
- Dasgupta, A.; Raychaudhuri, B.; Haqqi, T.; Prayson, R.; Van Meir, E.G.; Vogelbaum, M.; Haque, S.J. Stat3 activation is required for the growth of U87 cell-derived tumours in mice. Eur. J. Cancer (Oxf. Engl. 1990) 2009, 45, 677–684. [Google Scholar] [CrossRef]
- Akasaki, Y.; Liu, G.; Matundan, H.H.; Ng, H.; Yuan, X.; Zeng, Z.; Black, K.L.; Yu, J.S. A peroxisome proliferator-activated receptor-gamma agonist, troglitazone, facilitates caspase-8 and -9 activities by increasing the enzymatic activity of protein-tyrosine phosphatase-1B on human glioma cells. J. Biol. Chem. 2006, 281, 6165–6174. [Google Scholar] [PubMed]
- Adams, J. The proteasome: A suitable antineoplastic target. Nat. Rev. Cancer 2004, 4, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Chondrogianni, N.; Fragoulis, E.G.; Gonos, E.S. Protein degradation during aging: The lysosome-, the calpain- and the proteasome-dependent cellular proteolytic systems. Biogerontology 2002, 3, 121–123. [Google Scholar] [CrossRef] [PubMed]
- Lai, E.; Teodoro, T.; Volchuk, A. Endoplasmic reticulum stress: Signaling the unfolded protein response. Physiology 2007, 22, 193–201. [Google Scholar]
- Qin, C.; Burghardt, R.; Smith, R.; Wormke, M.; Stewart, J.; Safe, S. Peroxisome proliferator-activated receptor gamma agonists induce proteasome-dependent degradation of cyclin D1 and estrogen receptor alpha in MCF-7 breast cancer cells. Cancer Res. 2003, 63, 958–964. [Google Scholar]
- Zander, T.; Kraus, J.A.; Grommes, C.; Schlegel, U.; Feinstein, D.; Klockgether, T.; Landreth, G.; Koenigsknecht, J.; Heneka, M.T. Induction of apoptosis in human and rat glioma by agonists of the nuclear receptor PPARgamma. J. Neurochem. 2002, 81, 1052–1060. [Google Scholar]
- Strakova, N.; Ehrmann, J.; Dzubak, P.; Bouchal, J.; Kolar, Z. The synthetic ligand of peroxisome proliferator-activated receptor-gamma ciglitazone affects human glioblastoma cell lines. J. Pharmacol. Exp. Ther. 2004, 309, 1239–1247. [Google Scholar] [CrossRef]
- Singletary, K.; Milner, J. Diet, autophagy, and cancer: A review. Cancer Epidemiol. Biomark. Prev. A Publ. Am. Assoc. Cancer Res. Cosponsored Am. Soc. Prev. Oncol. 2008, 17, 1596–1610. [Google Scholar] [CrossRef] [PubMed]
- Panigrahy, D.; Singer, S.; Shen, L.Q.; Butterfield, C.E.; Freedman, D.A.; Chen, E.J.; Moses, M.A.; Kilroy, S.; Duensing, S.; Fletcher, C.; et al. PPARgamma ligands inhibit primary tumor growth and metastasis by inhibiting angiogenesis. J. Clin. Investig. 2002, 110, 923–932. [Google Scholar] [CrossRef] [PubMed]
- Sheu, W.H.; Ou, H.C.; Chou, F.P.; Lin, T.M.; Yang, C.H. Rosiglitazone inhibits endothelial proliferation and angiogenesis. Life Sci. 2006, 78, 1520–1528. [Google Scholar] [CrossRef] [PubMed]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sheu, M.-L.; Pan, L.-Y.; Hu, H.-Y.; Su, H.-L.; Sheehan, J.; Tsou, H.-K.; Pan, H.-C. Potential Therapeutic Effects of Thiazolidinedione on Malignant Glioma. Int. J. Mol. Sci. 2022, 23, 13510. https://doi.org/10.3390/ijms232113510
Sheu M-L, Pan L-Y, Hu H-Y, Su H-L, Sheehan J, Tsou H-K, Pan H-C. Potential Therapeutic Effects of Thiazolidinedione on Malignant Glioma. International Journal of Molecular Sciences. 2022; 23(21):13510. https://doi.org/10.3390/ijms232113510
Chicago/Turabian StyleSheu, Meei-Ling, Liang-Yi Pan, Huai-Yun Hu, Hong-Lin Su, Jason Sheehan, Hsi-Kai Tsou, and Hung-Chuan Pan. 2022. "Potential Therapeutic Effects of Thiazolidinedione on Malignant Glioma" International Journal of Molecular Sciences 23, no. 21: 13510. https://doi.org/10.3390/ijms232113510
APA StyleSheu, M.-L., Pan, L.-Y., Hu, H.-Y., Su, H.-L., Sheehan, J., Tsou, H.-K., & Pan, H.-C. (2022). Potential Therapeutic Effects of Thiazolidinedione on Malignant Glioma. International Journal of Molecular Sciences, 23(21), 13510. https://doi.org/10.3390/ijms232113510