

Lung Inflammasome Activation in SARS-CoV-2 Post-Mortem Biopsies

 , , , , , , , , , ,
, , , , , , , , , ,  ,
,
Abstract
1. Introduction
2. Results
2.1. Study Sample
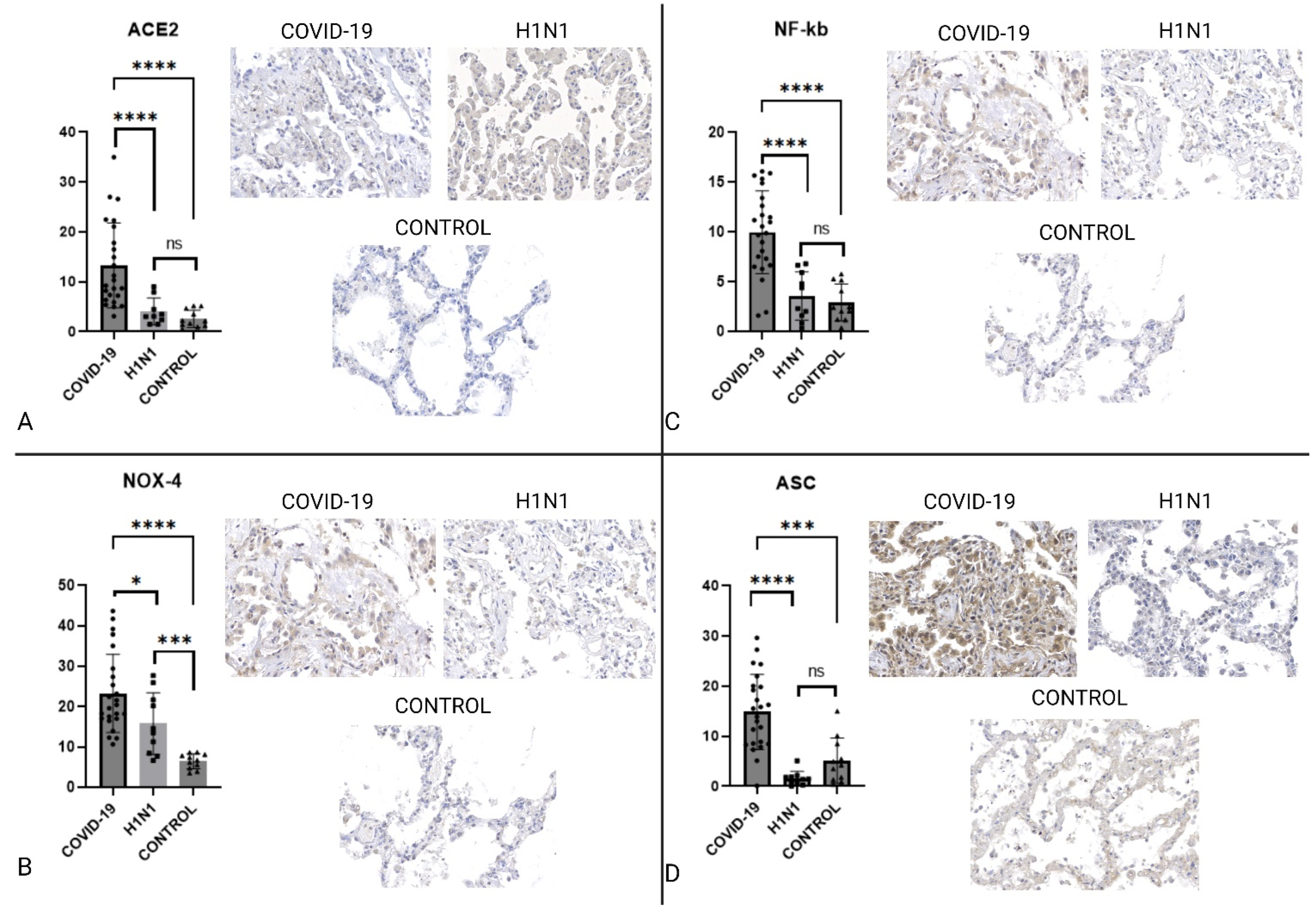
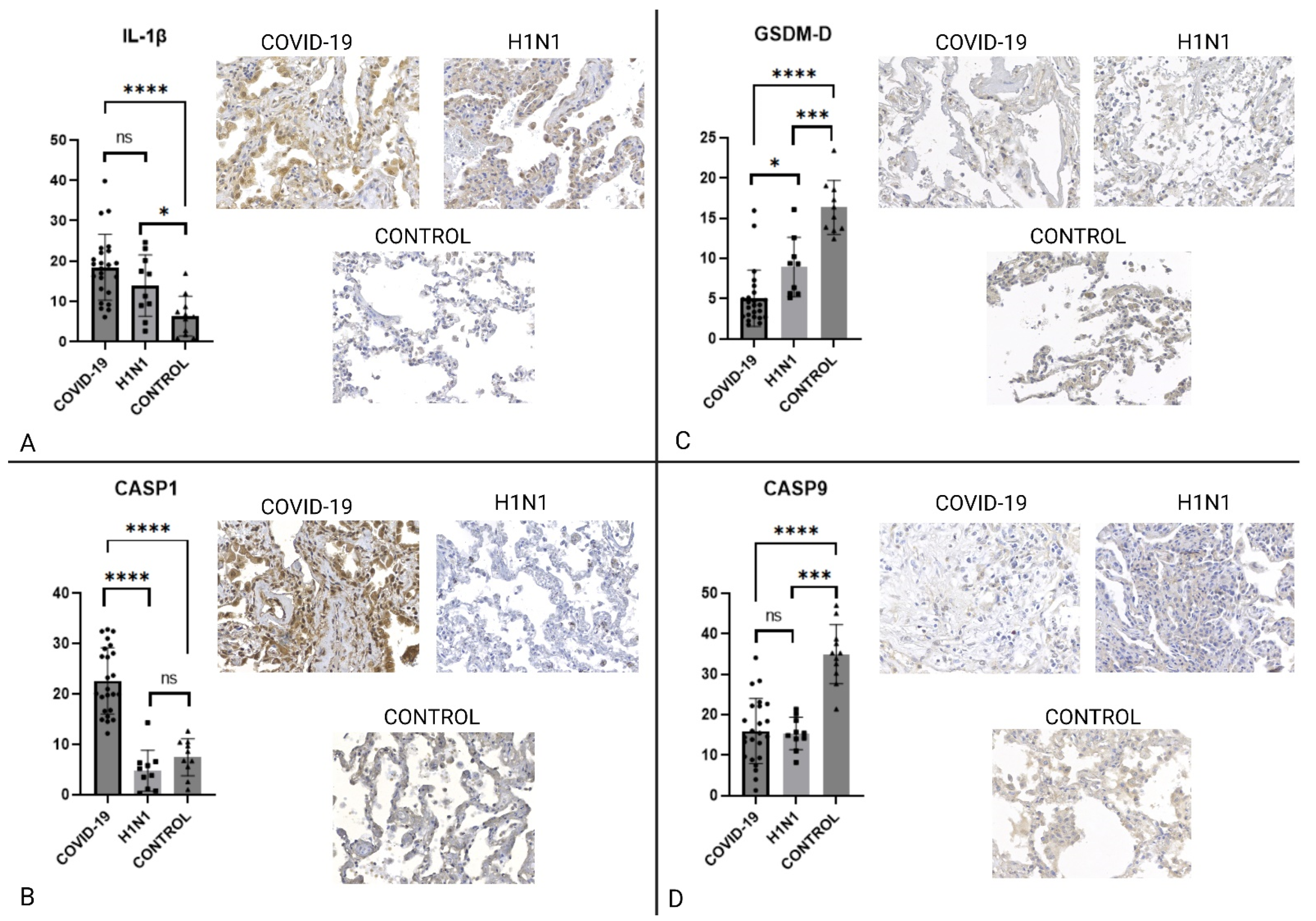
2.2. Immunohistochemistry
3. Discussion
3.1. Population
3.2. H1N1 Death Process
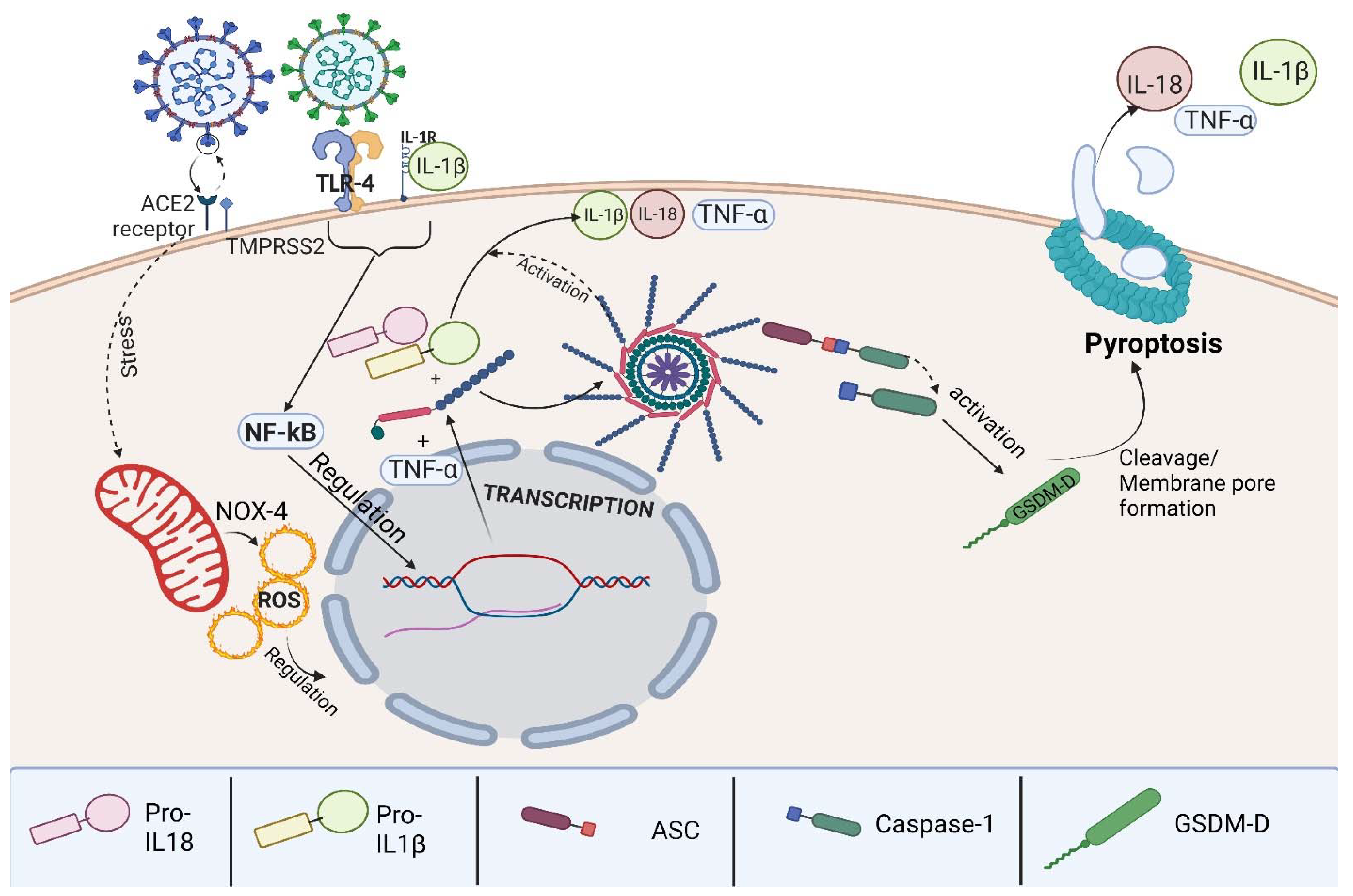
3.3. The Inflammasome Complex
3.4. Pyroptosis
3.5. Study Limitations
4. Materials and Methods
4.1. Ethics Committee Approval
Samples
4.2. Immunohistochemical Analysis
4.3. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gedefaw, L.; Ullah, S.; Leung, P.H.M.; Cai, Y.; Yip, S.P.; Huang, C.L. Inflammasome Activation-Induced Hypercoagulopathy: Impact on Cardiovascular Dysfunction Triggered in COVID-19 Patients. Cells 2021, 10, 916. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, M.Z.; Kucia, M. SARS-CoV-2 Infection and Overactivation of Nlrp3 Inflammasome as a Trigger of Cytokine “Storm” and Risk Factor for Damage of Hematopoietic Stem Cells. Leukemia 2020, 34, 1726–1729. [Google Scholar] [CrossRef]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J.; HLH Across Speciality Collaboration, UK. COVID-19: Consider Cytokine Storm Syndromes and Immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Liu, P.P.; Blet, A.; Smyth, D.; Li, H. The Science Underlying COVID-19: Implications for the Cardiovascular System. Circulation 2020, 142, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial Cell Infection and Endotheliitis in COVID-19. The Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Mü, M.A.; Drosten, C.; Pö, S.; Krü, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor Article SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 1–10. [Google Scholar] [CrossRef]
- Aboudounya, M.M.; Heads, R.J. COVID-19 and Toll-Like Receptor 4 (TLR4): SARS-CoV-2 May Bind and Activate TLR4 to Increase ACE2 Expression, Facilitating Entry and Causing Hyperinflammation. Mediat. Inflamm. 2021, 2021, 8874339. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.M.; O’Neill, L.A.J. Metabolic Regulation of NLRP3. Immunol. Rev. 2018, 281, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Verdecchia, P.; Cavallini, C.; Spanevello, A.; Angeli, F. The Pivotal Link between ACE2 Deficiency and SARS-CoV-2 Infection. Eur. J. Intern. Med. 2020, 76, 14. [Google Scholar] [CrossRef] [PubMed]
- Vora, S.M.; Lieberman, J.; Wu, H. Inflammasome Activation at the Crux of Severe COVID-19. Nat. Rev. Immunol. 2021, 21, 694–703. [Google Scholar] [CrossRef] [PubMed]
- Henao-Mejia, J.; Elinav, E.; Strowig, T.; Flavell, R.A. Inflammasomes: Far beyond Inflammation. Nat. Immunol. 2012, 13, 321–324. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wu, M. Pattern Recognition Receptors in Health and Diseases. Signal Transduct. Target. Ther. 2021, 6, 291. [Google Scholar] [CrossRef]
- Chen, M.; Wang, H.; Chen, W.; Meng, G. Regulation of Adaptive Immunity by the NLRP3 Inflammasome. Int. Immunopharmacol. 2011, 11, 549–554. [Google Scholar] [CrossRef]
- Abais, J.M.; Xia, M.; Zhang, Y.; Boini, K.M.; Li, P.-L. Redox Regulation of NLRP3 Inflammasomes: ROS as Trigger or Effector? Antioxid. Redox Signal. 2015, 22, 1111–1129. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Kanneganti, T.D. The Cell Biology of Inflammasomes: Mechanisms of Inflammasome Activation and Regulation. J. Cell Biol. 2016, 213, 617–629. [Google Scholar] [CrossRef]
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome Activation and Regulation: Toward a Better Understanding of Complex Mechanisms. Cell Discov. 2020, 6, 36. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-Activated Gasdermin D Causes Pyroptosis by Forming Membrane Pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef]
- Hartmann, C.; Miggiolaro, A.F.R.D.S.; Motta, J.D.S.; Baena Carstens, L.; Busatta Vaz De Paula, C.; Fagundes Grobe, S.; Hermann de Souza Nunes, L.; Lenci Marques, G.; Libby, P.; Zytynski Moura, L.; et al. The Pathogenesis of COVID-19 Myocardial Injury: An Immunohistochemical Study of Postmortem Biopsies. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Garg, N.J. Inflammasomes in Cardiovascular Diseases. Am. J. Cardiovasc. Dis. 2011, 1, 244–254. [Google Scholar]
- Yin, X.F.; Zhang, Q.; Chen, Z.Y.; Wang, H.F.; Li, X.; Wang, H.X.; Li, H.X.; Kang, C.M.; Chu, S.; Li, K.F.; et al. NLRP3 in Human Glioma Is Correlated with Increased WHO Grade, and Regulates Cellular Proliferation, Apoptosis and Metastasis via Epithelial-Mesenchymal Transition and the PTEN/AKT Signaling Pathway. Int. J. Oncol. 2018, 53, 973–986. [Google Scholar] [CrossRef] [PubMed]
- Zhen, Y.; Zhang, H. NLRP3 Inflammasome and Inflammatory Bowel Disease. Front. Immunol. 2019, 10, 276. [Google Scholar] [CrossRef]
- Shrivastava, G.; Valenzuela Leon, P.C.; Calvo, E. Inflammasome Fuels Dengue Severity. Front. Cell. Infect. Microbiol. 2020, 10, 489. [Google Scholar] [CrossRef]
- Halfmann, P.; Hill-Batorski, L.; Kawaoka, Y. The Induction of IL-1β Secretion Through the NLRP3 Inflammasome During Ebola Virus Infection. J. Infect. Dis. 2018, 218, S504–S507. [Google Scholar] [CrossRef]
- Chen, W.; Foo, S.S.; Zaid, A.; Teng, T.S.; Herrero, L.J.; Wolf, S.; Tharmarajah, K.; Vu, L.D.; van Vreden, C.; Taylor, A.; et al. Specific Inhibition of NLRP3 in Chikungunya Disease Reveals a Role for Inflammasomes in Alphavirus-Induced Inflammation. Nat. Microbiol. 2017, 2, 1435–1445. [Google Scholar] [CrossRef]
- Azevedo, M.L.V.; Zanchettin, A.C.; Vaz de Paula, C.B.; Motta Júnior, J.D.S.; Malaquias, M.A.S.; Raboni, S.M.; Neto, P.C.; Zeni, R.C.; Prokopenko, A.; Borges, N.H.; et al. Lung Neutrophilic Recruitment and IL-8/IL-17A Tissue Expression in COVID-19. Front. Immunol. 2021, 12, 947. [Google Scholar] [CrossRef]
- Motta Junior, J.D.S.; Miggiolaro, A.F.R.D.S.; Nagashima, S.; de Paula, C.B.V.; Baena, C.P.; Scharfstein, J.; de Noronha, L. Mast Cells in Alveolar Septa of COVID-19 Patients: A Pathogenic Pathway That May Link Interstitial Edema to Immunothrombosis. Front. Immunol. 2020, 11, 574862. [Google Scholar] [CrossRef]
- Vaz de Paula, C.B.; de Azevedo, M.L.V.; Nagashima, S.; Martins, A.P.C.; Malaquias, M.A.S.; Miggiolaro, A.F.R.D.S.; da Silva Motta Júnior, J.; Avelino, G.; do Carmo, L.A.P.; Carstens, L.B.; et al. IL-4/IL-13 Remodeling Pathway of COVID-19 Lung Injury. Sci. Rep. 2020, 10, 18689. [Google Scholar] [CrossRef] [PubMed]
- van de Veerdonk, F.L.; Netea, M.G.; Dinarello, C.A.; Joosten, L.A.B. Inflammasome Activation and IL-1β and IL-18 Processing during Infection. Trends Immunol. 2011, 32, 110–116. [Google Scholar] [CrossRef]
- Nagar, A.; Rahman, T.; Harton, J.A. The ASC Speck and NLRP3 Inflammasome Function Are Spatially and Temporally Distinct. Front. Immunol. 2021, 12, 4229. [Google Scholar] [CrossRef]
- Fernandes-Alnemri, T.; Wu, J.; Yu, J.W.; Datta, P.; Miller, B.; Jankowski, W.; Rosenberg, S.; Zhang, J.; Alnemri, E.S. The Pyroptosome: A Supramolecular Assembly of ASC Dimers Mediating Inflammatory Cell Death via Caspase-1 Activation. Cell Death Differ. 2007, 14, 1590–1604. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by Inflammatory Caspases Determines Pyroptotic Cell Death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Kuida, K. Caspase-9. Int. J. Biochem. Cell Biol. 2000, 32, 121–124. [Google Scholar] [CrossRef]
- Ribeiro dos Santos Miggiolaro, A.F.; da Silva Motta Junior, J.; Busatta Vaz de Paula, C.; Nagashima, S.; Alessandra Scaranello Malaquias, M.; Baena Carstens, L.; N Moreno-Amaral, A.; Pellegrino Baena, C.; de Noronha, L. Covid-19 Cytokine Storm in Pulmonary Tissue: Anatomopathological and Immunohistochemical Findings. Respir. Med. Case Rep. 2020, 31, 101292. [Google Scholar] [CrossRef]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host Cell Death and Inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef]
- Cunha, L.L.; Perazzio, S.F.; Azzi, J.; Cravedi, P.; Riella, L.V. Remodeling of the Immune Response With Aging: Immunosenescence and Its Potential Impact on COVID-19 Immune Response. Front. Immunol. 2020, 11, 1748. [Google Scholar] [CrossRef]
- Fulop, T.; Larbi, A.; Dupuis, G.; Page, A.; Frost, E.H.; Cohen, A.A.; Witkowski, J.M.; Franceschi, C. Immunosenescence and Inflamm-Aging as Two Sides of the Same Coin: Friends or Foes? Front. Immunol. 2018, 8, 1960. [Google Scholar] [CrossRef]
- Sharma, B.R.; Kanneganti, T.D. NLRP3 Inflammasome in Cancer and Metabolic Diseases. Nat. Immunol. 2021, 22, 550–559. [Google Scholar] [CrossRef]
- López-Reyes, A.; Martinez-Armenta, C.; Espinosa-Velázquez, R.; Vázquez-Cárdenas, P.; Cruz-Ramos, M.; Palacios-Gonzalez, B.; Gomez-Quiroz, L.E.; Martínez-Nava, G.A. NLRP3 Inflammasome: The Stormy Link Between Obesity and COVID-19. Front. Immunol. 2020, 2875. [Google Scholar] [CrossRef]
- Xing, Z.; Gao, W.; Qu, B.; Li, X.; Jin, Y.; Yang, K.; Cardona, C. Regulation of Proinflammatory Cytokine Interleukin-6 (IL-6) Induction by NF-KappaB Signaling in Pandemic H1N1 Influenza A Virus-Infected Human Bronchial Epithelial Cells (45.29). J. Immunol. 2010, 184. [Google Scholar]
- Akbari, H.; Tabrizi, R.; Lankarani, K.B.; Aria, H.; Vakili, S.; Asadian, F.; Noroozi, S.; Keshavarz, P.; Faramarz, S. The Role of Cytokine Profile and Lymphocyte Subsets in the Severity of Coronavirus Disease 2019 (COVID-19): A Systematic Review and Meta-Analysis. Life Sci. 2020, 258, 118167. [Google Scholar] [CrossRef]
- Pan, P.; Zhang, Q.; Liu, W.; Wang, W.; Lao, Z.; Zhang, W.; Shen, M.; Wan, P.; Xiao, F.; Liu, F.; et al. Dengue Virus M Protein Promotes NLRP3 Inflammasome Activation To Induce Vascular Leakage in Mice. J. Virol. 2019, 93, e00996-19. [Google Scholar] [CrossRef]
- Ratajczak, M.Z.; Bujko, K.; Ciechanowicz, A.; Sielatycka, K.; Cymer, M.; Marlicz, W.; Kucia, M. SARS-CoV-2 Entry Receptor ACE2 Is Expressed on Very Small CD45-Precursors of Hematopoietic and Endothelial Cells and in Response to Virus Spike Protein Activates the Nlrp3 Inflammasome. Stem Cell Rev. Rep. 2021, 17, 266–277. [Google Scholar] [CrossRef]
- Schimmel, L.; Chew, K.Y.; Stocks, C.J.; Yordanov, T.E.; Essebier, P.; Kulasinghe, A.; Monkman, J.; dos Santos Miggiolaro, A.F.R.; Cooper, C.; de Noronha, L.; et al. Endothelial Cells Are Not Productively Infected by SARS-CoV-2. Clin. Transl. Immunol. 2021, 10, e1350. [Google Scholar] [CrossRef]
- Nagashima, S.; Dutra, A.A.; Arantes, M.P.; Zeni, R.C.; Klein, C.K.; de Oliveira, F.C.; Piper, G.W.; Brenny, I.D.; Pereira, M.R.C.; Stocco, R.B.; et al. COVID-19 and Lung Mast Cells: The Kallikrein-Kinin Activation Pathway. Int. J. Mol. Sci. 2022, 23, 1714. [Google Scholar] [CrossRef]
- Whitsett, J.A.; Alenghat, T. Respiratory Epithelial Cells Orchestrate Pulmonary Innate Immunity. Nat. Immunol. 2014, 16, 27–35. [Google Scholar] [CrossRef]
- Reddy, S.P.; Tran, K.; Malik, A.B.; Siddiqui, M.R.; Mittal, M. Reactive Oxygen Species in Inflammation and Tissue Injury. Antioxid. Redox Signal. 2013, 20, 1126–1167. [Google Scholar] [CrossRef]
- Mussbacher, M.; Salzmann, M.; Brostjan, C.; Hoesel, B.; Schoergenhofer, C.; Datler, H.; Hohensinner, P.; Basílio, J.; Petzelbauer, P.; Assinger, A.; et al. Cell Type-Specific Roles of NF-ΚB Linking Inflammation and Thrombosis. Front. Immunol. 2019, 10, 85. [Google Scholar] [CrossRef]
- Xing, Y.; Cao, R.; Hu, H.M. TLR and NLRP3 Inflammasome-Dependent Innate Immune Responses to Tumor-Derived Autophagosomes (DRibbles). Cell Death Dis. 2016, 7, e2322. [Google Scholar] [CrossRef]
- Han, H.; Ma, Q.; Li, C.; Liu, R.; Zhao, L.; Wang, W.; Zhang, P.; Liu, X.; Gao, G.; Liu, F.; et al. Profiling Serum Cytokines in COVID-19 Patients Reveals IL-6 and IL-10 Are Disease Severity Predictors. Emerg. Microbes Infect. 2020, 9, 1123–1130. [Google Scholar] [CrossRef]
- Barbieri, S.S.; Zacchi, E.; Amadio, P.; Gianellini, S.; Mussoni, L.; Weksler, B.B.; Tremoli, E. Cytokines Present in Smokers Serum Interact with Smoke Components to Enhance Endothelial Dysfunction. Cardiovasc. Res. 2011, 90, 475–483. [Google Scholar] [CrossRef]
- Davis, B.K.; Wen, H.; Ting, J.P.Y. The Inflammasome NLRs in Immunity, Inflammation, and Associated Diseases. Annu. Rev. Immunol. 2011, 29, 707–735. [Google Scholar] [CrossRef]
- Pan, P.; Zhang, Q.; Liu, W.; Wang, W.; Yu, Z.; Lao, Z.; Zhang, W.; Shen, M.; Wan, P.; Xiao, F.; et al. Dengue Virus Infection Activates Interleukin-1β to Induce Tissue Injury and Vascular Leakage. Front. Microbiol. 2019, 10, 2637. [Google Scholar] [CrossRef]
- de Castro-Jorge, L.A.; de Carvalho, R.V.H.; Klein, T.M.; Hiroki, C.H.; Lopes, A.H.; Guimarães, R.M.; Fumagalli, M.J.; Floriano, V.G.; Agostinho, M.R.; Slhessarenko, R.D.; et al. The NLRP3 Inflammasome Is Involved with the Pathogenesis of Mayaro Virus. PLoS Pathog. 2019, 15, e1007934. [Google Scholar] [CrossRef]
- López-Bojórquez, L.N.; Arechavaleta-Velasco, F.; Vadillo-Ortega, F.; Móntes-Sánchez, D.; Ventura-Gallegos, J.L.; Zentella-Dehesa, A. NF-ΚB Translocation and Endothelial Cell Activation Is Potentiated by Macrophage-Released Signals Co-Secreted with TNF-α and IL-1β. Inflamm. Res. 2004, 53, 567–575. [Google Scholar] [CrossRef]
- Mao, L.; Kitani, A.; Strober, W.; Fuss, I.J. The Role of NLRP3 and IL-1β in the Pathogenesis of Inflammatory Bowel Disease. Front. Immunol. 2018, 9, 2566. [Google Scholar] [CrossRef]
- Fang, Y.; Tian, S.; Pan, Y.; Li, W.; Wang, Q.; Tang, Y.; Yu, T.; Wu, X.; Shi, Y.; Ma, P.; et al. Pyroptosis: A New Frontier in Cancer. Biomed. Pharmacother. 2020, 121, 109595. [Google Scholar] [CrossRef]
- Johnson, C.R.; Jarvis, W.D. Caspase-9 Regulation: An Update. Apoptosis 2004, 9, 423–427. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Characteristics | COVID-19 | H1N1 | Control | p-Value |
|---|---|---|---|---|
| Female | 9 (37.5%) | 2 (20%) | 3 (27.27%) | NS |
| Male | 15 (62.5%) | 8 (80%) | 8 (72.73%) | |
| Age (median in years) | 72.5 | 44 | 45 | COVID-19 vs. H1N1 p < 0.0001 COVID-19 vs. CONTROL p < 0.0001 |
| Time from admission to death (median in days) | 13.0 | 1.5 | 4 | COVID-19 vs. H1N1 p < 0.0005 COVID-19 vs. CONTROL p = 0.0013 |
| Duration of invasive ventilation (median in days) | 9.5 | 1.5 | N/A | p = 0.0096 |
| Death Cause | Diffuse Alveolar Damage and Disseminated Coagulopathy | Diffuse Alveolar Damage | Peritonitis, Infarction (n = 3), Neuroendocrine Carcinoma, Adenocarcinoma, Hepatic Cancer, Laryngeal Cancer, Surgical Complications, Lymphoma, Thrombosis | N/A |
| Comorbidities | Hypertension (n = 21), Chronic Cardiac disease (n = 11), | Data Not Obtained | Hypertension (n = 3), Chronic Cardiac disease (n = 5), | |
| Malignancy (n = 3) | Malignancy (n = 5) | |||
| Diabetes Mellitus type 2 (n = 11) | Diabetes Mellitus type 2 (n = 2) | |||
| Dyslipidemia (n = 17) | Dyslipidemia (n = 4) | |||
| Obesity (n = 6) | Obesity (n = 4) | |||
| Chronic Lung disease (n = 5) | Chronic Lung disease (n = 4) |
| Marker | COVID-19 × H1N1 | p-Value | COVID-19 × Control | p-Value |
|---|---|---|---|---|
| ACE2 | ↑COVID-19 | 0.0001 | ↑COVID-19 | <0.0001 |
| TLR4 | ↑H1N1 | 0.0247 | ↑CONTROL | 0.0164 |
| NLRP-3/NALP | NS | 0.4615 | NS | 0.1628 |
| IL-1β | NS | 0.1439 | ↑COVID-19 | <0.0001 |
| IL-18 | ↑COVID-19 | <0.0001 | ↑COVID-19 | 0.0004 |
| NF-κB | ↑COVID-19 | <0.0001 | ↑COVID-19 | <0.0001 |
| ASC | ↑COVID-19 | <0.0001 | ↑COVID-19 | 0.0004 |
| CASP1 | ↑COVID-19 | <0.0001 | ↑COVID-19 | <0.0001 |
| CASP9 | NS | 0.8332 | ↑CONTROL | <0.0001 |
| GSDMD | ↑H1N1 | 0.0003 | ↑CONTROL | <0.0001 |
| NOX4 | ↑COVID-19 | 0.0372 | ↑COVID-19 | <0.0001 |
| TNF-α | NS | 0.0929 | ↑COVID-19 | 0.0011 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baena Carstens, L.; Campos D’amico, R.; Fernandes de Moura, K.; Morais de Castro, E.; Centenaro, F.; Silva Barbosa, G.; Vieira Cavalcante da Silva, G.; Brenny, I.; Honório D’Agostini, J.C.; Hlatchuk, E.C.; et al. Lung Inflammasome Activation in SARS-CoV-2 Post-Mortem Biopsies. Int. J. Mol. Sci. 2022, 23, 13033. https://doi.org/10.3390/ijms232113033
Baena Carstens L, Campos D’amico R, Fernandes de Moura K, Morais de Castro E, Centenaro F, Silva Barbosa G, Vieira Cavalcante da Silva G, Brenny I, Honório D’Agostini JC, Hlatchuk EC, et al. Lung Inflammasome Activation in SARS-CoV-2 Post-Mortem Biopsies. International Journal of Molecular Sciences. 2022; 23(21):13033. https://doi.org/10.3390/ijms232113033
Chicago/Turabian StyleBaena Carstens, Lucas, Raissa Campos D’amico, Karen Fernandes de Moura, Eduardo Morais de Castro, Flávia Centenaro, Giovanna Silva Barbosa, Guilherme Vieira Cavalcante da Silva, Isadora Brenny, Júlio César Honório D’Agostini, Elisa Carolina Hlatchuk, and et al. 2022. "Lung Inflammasome Activation in SARS-CoV-2 Post-Mortem Biopsies" International Journal of Molecular Sciences 23, no. 21: 13033. https://doi.org/10.3390/ijms232113033
APA StyleBaena Carstens, L., Campos D’amico, R., Fernandes de Moura, K., Morais de Castro, E., Centenaro, F., Silva Barbosa, G., Vieira Cavalcante da Silva, G., Brenny, I., Honório D’Agostini, J. C., Hlatchuk, E. C., Pissette de Lima, S., Camargo Martins, A. P., De Castro Deus, M., Konzen Klein, C., Kubaski Benevides, A. P., Nagashima, S., Machado-Souza, C., Pinho, R. A., Pellegrino Baena, C., & de Noronha, L. (2022). Lung Inflammasome Activation in SARS-CoV-2 Post-Mortem Biopsies. International Journal of Molecular Sciences, 23(21), 13033. https://doi.org/10.3390/ijms232113033