

Neuropeptide Y Promotes Human M2 Macrophage Polarization and Enhances p62/SQSTM1-Dependent Autophagy and NRF2 Activation

 , , ,
, , , 
 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
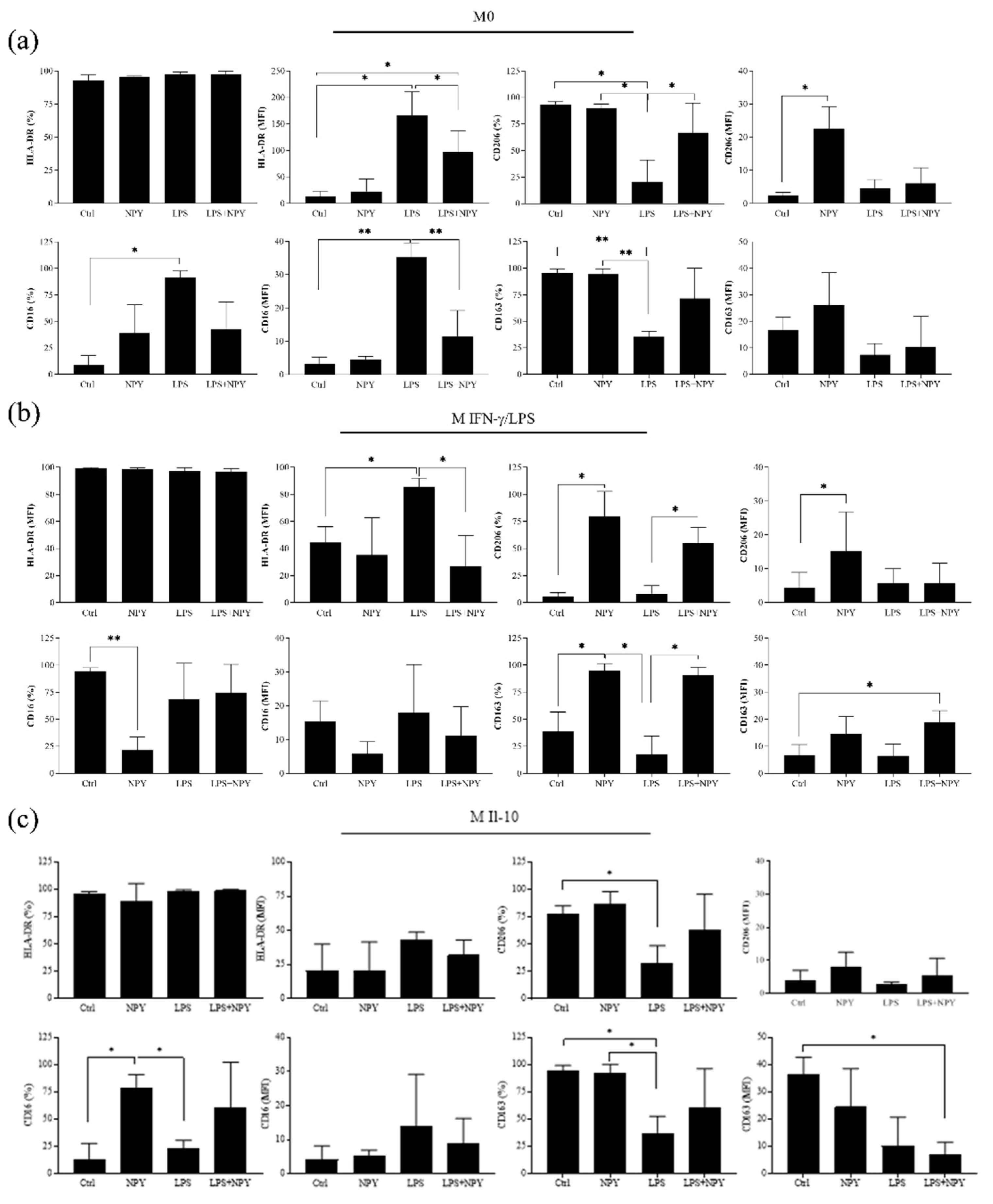
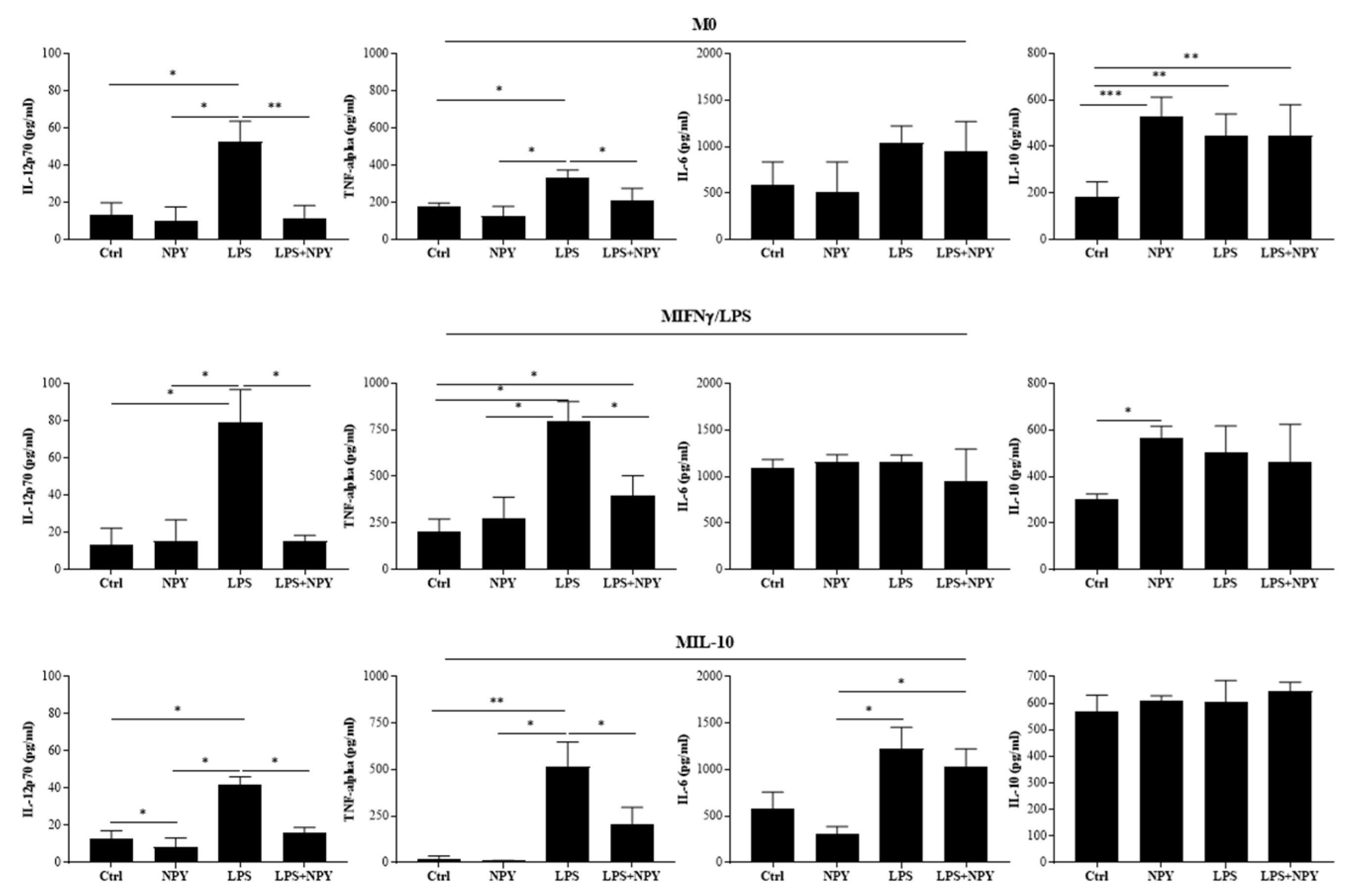
2.1. NPY Skews M(IFN-γ/LPS) Macrophages towards Anti-Inflammatory Phenotype
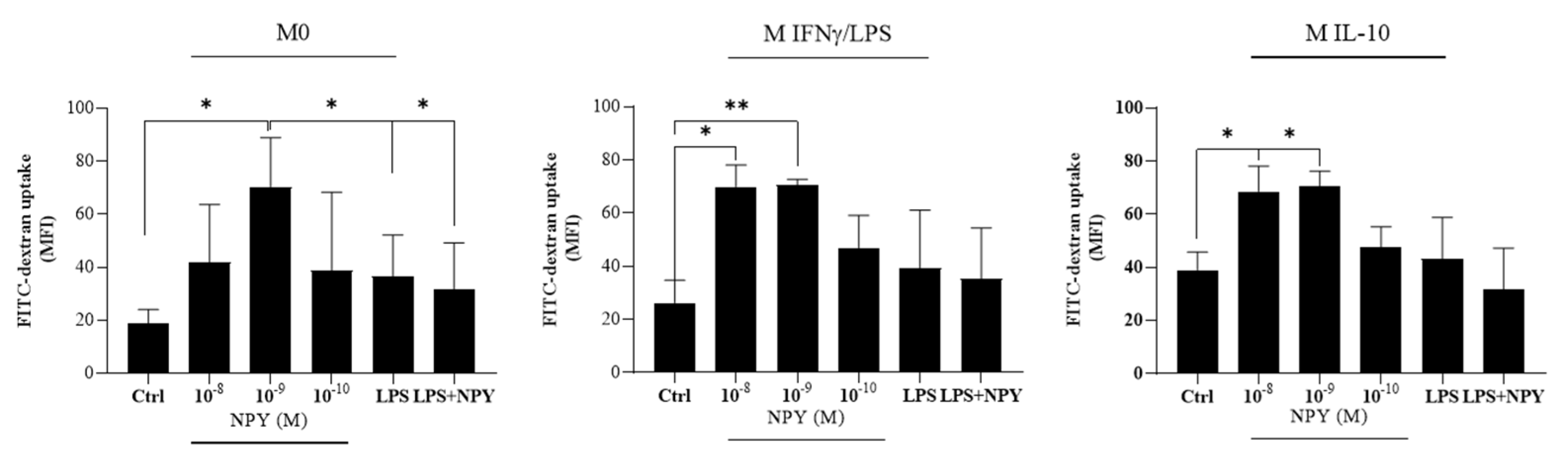
2.2. Neuropeptide Y Promotes Endocytosis in Macrophages
2.3. Neuropeptide Y Prevents 7-Keto-Cholesterol-Induced Foam Cell Formation in Macrophages
2.4. Neuropeptide Y Enhances Autophagosome Formation and p62 Accumulation in Macrophages
2.5. Neuropeptide Y Promotes Human NRF2/HO-1 Activation in M0 Macrophages
3. Discussion
4. Materials and Methods
4.1. In Vitro Cell Stimulation/Polarization
4.2. Flow Cytometric Analysis of Macrophage Surface Markers and Endocytosis
4.3. Secretome Profile of Cytokines in Macrophage Culture Supernatants
4.4. Flow Cytometric Analysis of Intracellular Lipid Levels
4.5. Evaluation of Autophagy Markers and NRF2/HO-1
4.5.1. Indirect Immunofluorescence Labeling
4.5.2. LysoTracker Green Labeling
4.5.3. Western Blot Analysis of Macrophage Lysates
4.5.4. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guerrini, V.; Gennaro, M.L. Foam Cells: One Size Doesn’t Fit All. Trends Immunol. 2019, 40, 1163–1179. [Google Scholar] [CrossRef]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Progress and challenges in translating the biology of atherosclerosis. Nature 2011, 473, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Gattinoni, L.; Chiumello, D.; Caironi, P.; Busana, M.; Romitti, F.; Brazzi, L.; Camporota, L. COVID-19 pneumonia: Different respiratory treatments for different phenotypes? Intensive Care Med. 2020, 46, 1099–1102. [Google Scholar] [CrossRef] [PubMed]
- Gui, T.; Shimokado, A.; Sun, Y.; Akasaka, T.; Muragaki, Y. Diverse roles of macrophages in atherosclerosis: From inflammatory biology to biomarker discovery. Mediat. Inflamm. 2012, 2012, 693083. [Google Scholar] [CrossRef]
- Stout, R.D.; Jiang, C.; Matta, B.; Tietzel, I.; Watkins, S.K.; Suttles, J. Macrophages sequentially change their functional phenotype in response to changes in microenvironmental influences. J. Immunol. 2005, 175, 342–349. [Google Scholar] [CrossRef]
- Gordon, S.; Plüddemann, A.; Martinez Estrada, F. Macrophage heterogeneity in tissues: Phenotypic diversity and functions. Immunol. Rev. 2014, 262, 36–55. [Google Scholar] [CrossRef] [PubMed]
- Locati, M.; Mantovani, A.; Sica, A. Macrophage activation and polarization as an adaptive component of innate immunity. Adv. Immunol. 2013, 120, 163–184. [Google Scholar]
- Gordon, S.; Martinez, F.O. Alternative activation of macrophages: Mechanism and functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef]
- Odegaard, J.I.; Chawla, A. Alternative macrophage activation and metabolism. Annu. Rev. Pathol. 2011, 6, 275–297. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Businaro, R.; Tagliani, A.; Buttari, B.; Profumo, E.; Ippoliti, F.; Di Cristofano, C.; Capoano, R.; Salvati, B.; Riganò, R. Cellular and molecular players in the atherosclerotic plaque progression. Ann. N. Y. Acad. Sci. 2012, 1262, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Calcagni, E.; Elenkov, I. Stress system activity, innate and T helper cytokines, and susceptibility to immune-related diseases. Ann. N. Y. Acad. Sci. 2006, 1069, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Koller, A.; Brunner, S.M.; Bianchini, R.; Ramspacher, A.; Emberger, M.; Locker, F.; Schlager, S.; Kofler, B. Galanin is a potent modulator of cytokine and chemokine expression in human macrophages. Sci. Rep. 2019, 9, 7237. [Google Scholar] [CrossRef]
- Wang, Q.; Whim, M.D. Stress-induced changes in adrenal neuropeptide Y expression are regulated by a negative feedback loop. J. Neurochem. 2013, 125, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Wheway, J.; Herzog, H.; Mackay, F. NPY and Receptors in Immune and Inflammatory Diseases. Curr. Top. Med. Chem. 2007, 7, 1743–1752. [Google Scholar] [CrossRef]
- Dimitrijević, M.; Stanojević, S.; Mićić, S.; Vujić, V.; Kovačević-Jovanović, V.; Mitić, K.; von Hörsten, S.; Kosec, D. Neuropeptide Y (NPY) modulates oxidative burst and nitric oxide production in carrageenan-elicited granulocytes from rat air pouch. Peptides 2006, 27, 3208–3215. [Google Scholar] [CrossRef]
- Bedoui, S.; Kromer, A.; Gebhardt, T.; Jacobs, R.; Raber, K.; Dimitrijevic, M.; Heine, J.; von Hörsten, S. Neuropeptide Y receptor-specifically modulates human neutrophil function. J. Neuroimmunol. 2008, 195, 88–95. [Google Scholar] [CrossRef]
- Dimitrijević, M.; Stanojević, S.; Mitić, K.; Kuštrimović, N.; Vujić, V.; Miletić, T.; Kovačević-Jovanović, V. Modulation of granulocyte functions by peptide YY in the rat: Age-related differences in Y receptors expression and plasma dipeptidyl peptidase 4 activity. Regul. Pept. 2010, 159, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Dimitrijević, M.; Stanojević, S.; Vujić, V.; Beck-Sickinger, A.; Von Hörsten, S. Neuropeptide Y and its receptor subtypes specifically modulate rat peritoneal macrophage functions in vitro: Counter regulation through Y1 and Y2/5 receptors. Regul. Pept. 2005, 124, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Dimitrijevi, M.; Stanojevi, S.; Vuji, V.; Kovačevi-Jovanovi, V.; Beck-Sickinger, A.; Demuth, H.U.; Hörsten, S. Von Effect of neuropeptide Y on inflammatory paw edema in the rat: Involvement of peripheral NPY Y1 and Y5 receptors and interaction with dipeptidyl-peptidase IV (CD26). J. Neuroimmunol. 2002, 129, 35–42. [Google Scholar] [CrossRef]
- Zhou, J.R.; Xu, Z.; Jiang, C.L. Neuropeptide Y promotes TGF-beta1 production in RAW264.7 cells by activating PI3K pathway via Y1 receptor. Neurosci. Bull. 2008, 24, 155–159. [Google Scholar] [CrossRef]
- Dimitrijević, M.; Stanojević, S. The intriguing mission of neuropeptide Y in the immune system. Amino Acids 2013, 45, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Buttari, B.; Profumo, E.; Domenici, G.; Tagliani, A.; Ippoliti, F.; Bonini, S.; Businaro, R.; Elenkov, I.; Riganò, R. Neuropeptide Y induces potent migration of human immature dendritic cells and promotes a Th2 polarization. FASEB J. 2014, 28, 3038–3049. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Sun, W.; Zhang, C.; Song, Z.; Lin, S. The role of neuropeptide Y in the pathophysiology of atherosclerotic cardiovascular disease. Int. J. Cardiol. 2016, 220, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Kuo, L.E.; Zukowska, Z. Stress, NPY and vascular remodeling: Implications for stress-related diseases. Peptides 2007, 28, 435–440. [Google Scholar] [CrossRef]
- Jääskeläinen, A.E.; Seppälä, S.; Kakko, T.; Jaakkola, U.; Kallio, J. Systemic treatment with neuropeptide Y receptor Y1-antagonist enhances atherosclerosis and stimulates IL-12 expression in ApoE deficient mice. Neuropeptides 2013, 47, 67–73. [Google Scholar] [CrossRef]
- Rosano, G.M.C.; Tousoulis, D.; McFadden, E.; Clarke, J.; Davies, G.J.; Kaski, J.C. Effects of neuropeptide Y on coronary artery vasomotion in patients with microvascular angina. Int. J. Cardiol. 2017, 238, 123–127. [Google Scholar] [CrossRef]
- Qin, Y.Y.; Huang, X.R.; Zhang, J.; Wu, W.; Chen, J.; Wan, S.; Yu, X.Y.; Lan, H.Y. Neuropeptide Y attenuates cardiac remodeling and deterioration of function following myocardial infarction. Mol. Ther. 2022, 30, 881–897. [Google Scholar] [CrossRef]
- Aveleira, C.A.; Botelho, M.; Carmo-Silva, S.; Pascoal, J.F.; Ferreira-Marques, M.; Nóbrega, C.; Cortes, L.; Valero, J.; Sousa-Ferreira, L.; Álvaro, A.R.; et al. Neuropeptide Y stimulates autophagy in hypothalamic neurons. Proc. Natl. Acad. Sci. USA 2015, 112, E1642–E1651. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.; Cuervo, A.M. Integration of clearance mechanisms: The proteasome and autophagy. Cold Spring Harb. Perspect. Biol. 2010, 2, a006734. [Google Scholar] [CrossRef] [PubMed]
- Takahama, M.; Akira, S.; Saitoh, T. Autophagy limits activation of the inflammasomes. Immunol. Rev. 2018, 281, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.; Wang, H.; Suttles, J.; Graves, D.T.; Martin, M. Mammalian target of rapamycin complex 2 (mTORC2) negatively regulates toll-like receptor 4-mediated inflammatory response via FoxO1. J. Biol. Chem. 2011, 286, 44295–44305. [Google Scholar] [CrossRef] [PubMed]
- Festuccia, W.T.; Pouliot, P.; Bakan, I.; Sabatini, D.M.; Laplante, M. Myeloid-specific rictor deletion induces M1 macrophage polarization and potentiates in vivo pro-inflammatory response to lipopolysaccharide. PLoS ONE 2014, 9, e95432. [Google Scholar] [CrossRef]
- Liu, K.; Zhao, E.; Ilyas, G.; Lalazar, G.; Lin, Y.; Haseeb, M.; Tanaka, K.E.; Czaja, M.J. Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy 2015, 11, 271–284. [Google Scholar] [CrossRef]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef]
- Buttari, B.; Segoni, L.; Profumo, E.; D’Arcangelo, D.; Rossi, S.; Facchiano, F.; Businaro, R.; Iuliano, L.; Rigano, R. 7-Oxo-cholesterol potentiates pro-inflammatory signaling in human M1 and M2 macrophages. Biochem. Pharmacol. 2013, 86, 130–137. [Google Scholar] [CrossRef]
- Yuan, X.M.; Sultana, N.; Siraj, N.; Ward, L.J.; Ghafouri, B.; Li, W. Autophagy induction protects against 7-oxysterol- induced cell death via lysosomal pathway and oxidative stress. J. Cell Death 2016, 9, 1–7. [Google Scholar] [CrossRef]
- Saha, S.; Profumo, E.; Togna, A.R.; Riganò, R.; Saso, L.; Buttari, B. Lupeol Counteracts the Proinflammatory Signalling Triggered in Macrophages by 7-Keto-Cholesterol: New Perspectives in the Therapy of Atherosclerosis. Oxid. Med. Cell. Longev. 2020, 2020, 1232816. [Google Scholar] [CrossRef]
- Wang, X.; Li, L.; Li, M.; Dang, X.; Wan, L.; Wang, N.; Bi, X.; Gu, C.; Qiu, S.; Niu, X.; et al. Knockdown of mTOR by lentivirus-mediated RNA interference suppresses atherosclerosis and stabilizes plaques via a decrease of macrophages by autophagy in apolipoprotein E-deficient mice. Int. J. Mol. Med. 2013, 32, 1215–1221. [Google Scholar] [CrossRef]
- Bjørkøy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Øvervatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef]
- Saha, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation. Molecules 2020, 25, 5474. [Google Scholar] [CrossRef] [PubMed]
- Cuculi, F.; Herring, N.; De Caterina, A.R.; Banning, A.P.; Prendergast, B.D.; Forfar, J.C.; Choudhury, R.P.; Channon, K.M.; Kharbanda, R.K. Relationship of plasma neuropeptide Y with angiographic, electrocardiographic and coronary physiology indices of reperfusion during ST elevation myocardial infarction. Heart 2013, 99, 1198–1203. [Google Scholar] [CrossRef] [PubMed]
- Ullman, B.; Hulting, J.; Lundberg, J.M. Prognostic value of plasma neuropeptide-Y in coronary care unit patients with and without acute myocardial infarction. Eur. Heart J. 1994, 15, 454–461. [Google Scholar] [CrossRef]
- Ajijola, O.A.; Chatterjee, N.A.; Gonzales, M.J.; Gornbein, J.; Liu, K.; Li, D.; Paterson, D.J.; Shivkumar, K.; Singh, J.P.; Herring, N. Coronary Sinus Neuropeptide Y Levels and Adverse Outcomes in Patients With Stable Chronic Heart Failure. JAMA Cardiol. 2020, 5, 318–325. [Google Scholar] [CrossRef]
- Ferreira, R.; Santos, T.; Viegas, M.; Cortes, L.; Bernardino, L.; Vieira, O.V.; Malva, J.O. Neuropeptide Y inhibits interleukin-1β-induced phagocytosis by microglial cells. J. Neuroinflamm. 2011, 8, 169. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.S.; Chen, P.N.; Yu, C.H.; Liao, J.M.; Kuo, D.Y. Inhibiting neuropeptide Y Y1 receptor modulates melanocortin receptor- and NF-κB-mediated feeding behavior in phenylpropanolamine-treated rats. Horm. Behav. 2013, 64, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.C.; Liu, Y.B.; Liu, W.F.; Zhou, Y.Y.; He, H.F.; Lin, S. Neuropeptide Y Is an Immunomodulatory Factor: Direct and Indirect. Front. Immunol. 2020, 11, 580378. [Google Scholar] [CrossRef]
- Dimitrijević, M.; Stanojević, S.; Mitić, K.; Kuštrimović, N.; Vujić, V.; Miletić, T.; Kovačević-Jovanović, V. The anti-inflammatory effect of neuropeptide Y (NPY) in rats is dependent on dipeptidyl peptidase 4 (DP4) activity and age. Peptides 2008, 29, 2179–2187. [Google Scholar] [CrossRef]
- Liu, M.; Liu, H.; Liang, F.; Song, X.Q.; Hu, P.A. Neuropeptide Y promotes adipogenic differentiation in primary cultured human adipose-derived stem cells. Endocr. J. 2018, 65, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Pons, J.; Kitlinska, J.; Jacques, D.; Perreault, C.; Nader, M.; Everhart, L.; Zhang, Y.; Zukowska, Z. Interactions of multiple signaling pathways in neuropeptide Y-mediated bimodal vascular smooth muscle cell growth. Can. J. Physiol. Pharmacol. 2008, 86, 438–448. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Jin, D.; Wu, J.Q.; Xu, Z.Y.; Fu, S.; Mei, G.; Zou, Z.L.; Ma, S. hui Neuropeptide Y stimulates osteoblastic differentiation and VEGF expression of bone marrow mesenchymal stem cells related to canonical Wnt signaling activating in vitro. Neuropeptides 2016, 56, 105–113. [Google Scholar] [CrossRef]
- Ferreira, R.; Santos, T.; Cortes, L.; Cochaud, S.; Agasse, F.; Silva, A.P.; Xapelli, S.; Malva, J.O. Neuropeptide Y inhibits interleukin-1 beta-induced microglia motility. J. Neurochem. 2012, 120, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Leof, E.B. Growth factor receptor signalling: Location, location, location. Trends Cell Biol. 2000, 10, 343–348. [Google Scholar] [CrossRef]
- Hayden, J.M.; Brachova, L.; Higgins, K.; Obermiller, L.; Sevanian, A.; Khandrika, S.; Reaven, P.D. Induction of monocyte differentiation and foam cell formation in vitro by 7-ketocholesterol. J. Lipid Res. 2002, 43, 26–35. [Google Scholar] [CrossRef]
- Ouimet, M.; Marcel, Y.L. Regulation of lipid droplet cholesterol efflux from macrophage foam cells. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 575–581. [Google Scholar] [CrossRef]
- Moore, K.J.; Koplev, S.; Fisher, E.A.; Tabas, I.; Björkegren, J.L.M.; Doran, A.C.; Kovacic, J.C. Macrophage Trafficking, Inflammatory Resolution, and Genomics in Atherosclerosis: JACC Macrophage in CVD Series (Part 2). J. Am. Coll. Cardiol. 2018, 72, 2181–2197. [Google Scholar] [CrossRef]
- Choi, B.; Shin, M.K.; Kim, E.Y.; Park, J.E.; Lee, H.; Kim, S.W.; Song, J.K.; Chang, E.J. Elevated Neuropeptide Y in Endothelial Dysfunction Promotes Macrophage Infiltration and Smooth Muscle Foam Cell Formation. Front. Immunol. 2019, 10, 1701. [Google Scholar] [CrossRef]
- Lu, G.; Zhang, R.; Geng, S.; Peng, L.; Jayaraman, P.; Chen, C.; Xu, F.; Yang, J.; Li, Q.; Zheng, H.; et al. Myeloid cell-derived inducible nitric oxide synthase suppresses M1 macrophage polarization. Nat. Commun. 2015, 6, 6676. [Google Scholar] [CrossRef]
- Martinet, W.; De Meyer, G.R.Y. Autophagy in atherosclerosis: A cell survival and death phenomenon with therapeutic potential. Circ. Res. 2009, 104, 304–317. [Google Scholar] [CrossRef] [PubMed]
- De Meyer, G.R.Y.; Grootaert, M.O.J.; Michiels, C.F.; Kurdi, A.; Schrijvers, D.M.; Martinet, W. Autophagy in vascular disease. Circ. Res. 2015, 116, 468–479. [Google Scholar] [CrossRef] [PubMed]
- Grootaert, M.O.J.; da Costa Martins, P.A.; Bitsch, N.; Pintelon, I.; de Meyer, G.R.Y.; Martinet, W.; Schrijvers, D.M. Defective autophagy in vascular smooth muscle cells accelerates senescence and promotes neointima formation and atherogenesis. Autophagy 2015, 11, 2014–2032. [Google Scholar] [CrossRef]
- Bardag-Gorce, F.; Francis, T.; Nan, L.; Li, J.; Yan, H.L.; French, B.A.; French, S.W. Modifications in P62 occur due to proteasome inhibition in alcoholic liver disease. Life Sci. 2005, 77, 2594–2602. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef] [PubMed]
- Baird, L.; Dinkova-Kostova, A.T. The cytoprotective role of the Keap1-Nrf2 pathway. Arch. Toxicol. 2011, 85, 241–272. [Google Scholar] [CrossRef] [PubMed]
- Rojo, A.I.; Innamorato, N.G.; Martín-Moreno, A.M.; De Ceballos, M.L.; Yamamoto, M.; Cuadrado, A. Nrf2 regulates microglial dynamics and neuroinflammation in experimental Parkinson’s disease. Glia 2010, 58, 588–598. [Google Scholar]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef] [PubMed]
- Funakoshi-Hirose, I.; Aki, T.; Unuma, K.; Funakoshi, T.; Noritake, K.; Uemura, K. Distinct effects of methamphetamine on autophagy-lysosome and ubiquitin-proteasome systems in HL-1 cultured mouse atrial cardiomyocytes. Toxicology 2013, 312, 74–82. [Google Scholar] [PubMed]
- Ooi, B.K.; Goh, H.; Yap, W.H. Molecular Sciences Oxidative Stress in Cardiovascular Diseases: Involvement of Nrf2 Antioxidant Redox Signaling in Macrophage Foam Cells Formation. Int. J. Mol. Sci. 2017, 18, 2336. [Google Scholar] [CrossRef]
- Sussan, T.E.; Jun, J.; Thimmulappa, R.; Bedja, D.; Antero, M.; Gabrielson, K.L.; Polotsky, V.Y.; Biswal, S. Disruption of Nrf2, a key inducer of antioxidant defenses, attenuates ApoE-mediated atherosclerosis in mice. PLoS ONE 2008, 3, e3791. [Google Scholar] [CrossRef] [PubMed]
- Barajas, B.; Che, N.; Yin, F.; Rowshanrad, A.; Orozco, L.D.; Gong, K.W.; Wang, X.; Castellani, L.W.; Reue, K.; Lusis, A.J.; et al. NF-E2-related factor 2 promotes atherosclerosis by effects on plasma lipoproteins and cholesterol transport that overshadow antioxidant protection. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 58–66. [Google Scholar] [CrossRef]
- Freigang, S.; Ampenberger, F.; Spohn, G.; Heer, S.; Shamshiev, A.T.; Kisielow, J.; Hersberger, M.; Yamamoto, M.; Bachmann, M.F.; Kopf, M. Nrf2 is essential for cholesterol crystal-induced inflammasome activation and exacerbation of atherosclerosis. Eur. J. Immunol. 2011, 41, 2040–2051. [Google Scholar] [CrossRef] [PubMed]
- Harada, N.; Ito, K.; Hosoya, T.; Mimura, J.; Maruyama, A.; Noguchi, N.; Yagami, K.I.; Morito, N.; Takahashi, S.; Maher, J.M.; et al. Nrf2 in bone marrow-derived cells positively contributes to the advanced stage of atherosclerotic plaque formation. Free Radic. Biol. Med. 2012, 53, 2256–2262. [Google Scholar] [CrossRef] [PubMed]
- Ruotsalainen, A.K.; Lappalainen, J.P.; Heiskanen, E.; Merentie, M.; Sihvola, V.; Näpänkangas, J.; Lottonen-Raikaslehto, L.; Kansanen, E.; Adinolfi, S.; Kaarniranta, K.; et al. Nuclear factor E2-related factor 2 deficiency impairs atherosclerotic lesion development but promotes features of plaque instability in hypercholesterolaemic mice. Cardiovasc. Res. 2019, 115, 243–254. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Profumo, E.; Maggi, E.; Arese, M.; Di Cristofano, C.; Salvati, B.; Saso, L.; Businaro, R.; Buttari, B. Neuropeptide Y Promotes Human M2 Macrophage Polarization and Enhances p62/SQSTM1-Dependent Autophagy and NRF2 Activation. Int. J. Mol. Sci. 2022, 23, 13009. https://doi.org/10.3390/ijms232113009
Profumo E, Maggi E, Arese M, Di Cristofano C, Salvati B, Saso L, Businaro R, Buttari B. Neuropeptide Y Promotes Human M2 Macrophage Polarization and Enhances p62/SQSTM1-Dependent Autophagy and NRF2 Activation. International Journal of Molecular Sciences. 2022; 23(21):13009. https://doi.org/10.3390/ijms232113009
Chicago/Turabian StyleProfumo, Elisabetta, Elisa Maggi, Marzia Arese, Claudio Di Cristofano, Bruno Salvati, Luciano Saso, Rita Businaro, and Brigitta Buttari. 2022. "Neuropeptide Y Promotes Human M2 Macrophage Polarization and Enhances p62/SQSTM1-Dependent Autophagy and NRF2 Activation" International Journal of Molecular Sciences 23, no. 21: 13009. https://doi.org/10.3390/ijms232113009
APA StyleProfumo, E., Maggi, E., Arese, M., Di Cristofano, C., Salvati, B., Saso, L., Businaro, R., & Buttari, B. (2022). Neuropeptide Y Promotes Human M2 Macrophage Polarization and Enhances p62/SQSTM1-Dependent Autophagy and NRF2 Activation. International Journal of Molecular Sciences, 23(21), 13009. https://doi.org/10.3390/ijms232113009