
Structure-Based Virtual Screening and De Novo Design to Identify Submicromolar Inhibitors of G2019S Mutant of Leucine-Rich Repeat Kinase 2


Abstract
1. Introduction
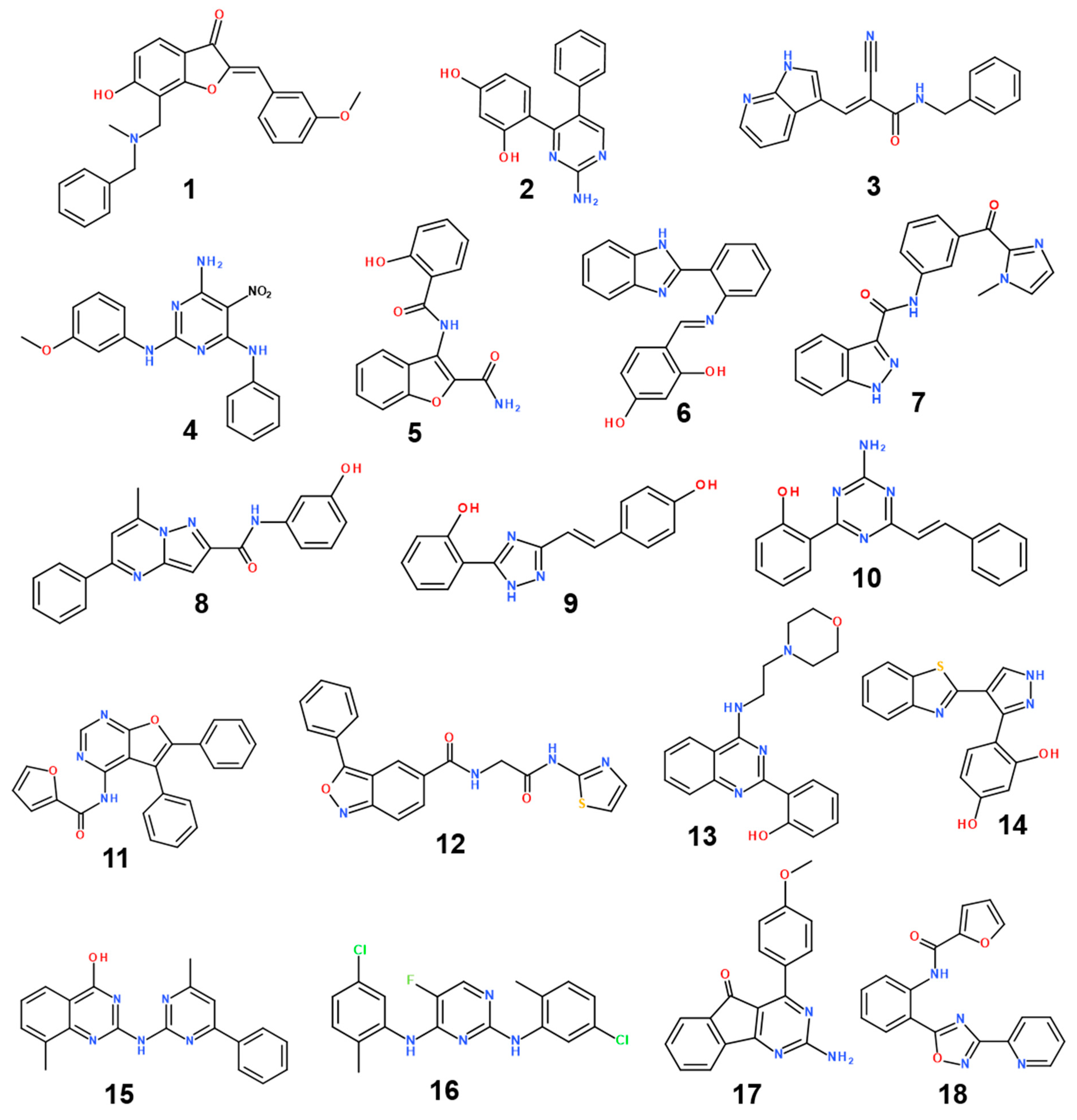
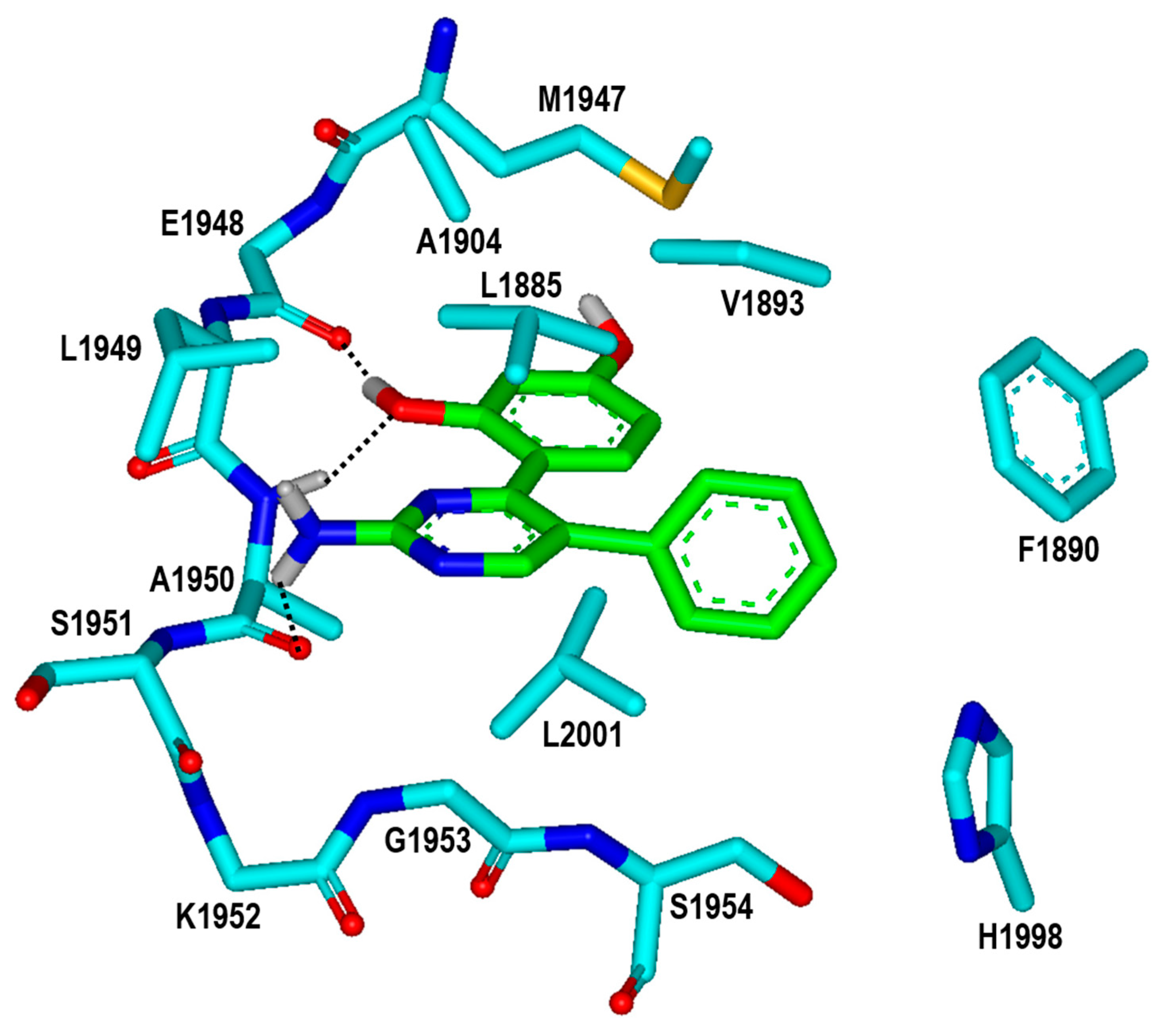
2. Results and Discussion
3. Materials and Methods
3.1. Structural Preparations of the G2019S Mutant of LRRK2
3.2. Structure-Based Virtual Screening to Identify the Inhibitors of the G2019S Mutant of LRRK2
3.3. De Novo Design to Enhance the Biochemical Potency
3.4. Molecular Dynamics Simulations
3.5. Enzyme Inhibition Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PD | Parkinson’s disease |
| LRRK2 | leucine-rich repeat kinase 2 |
| 3D | three dimensional |
| MLK1 | mixed lineage kinase 1 |
| BLAST | basic local alignment search tool |
| MW | molecular weight |
| SAR | structure–activity relationship |
| amu | atomic mass unit |
| MD | molecular dynamics |
| RMSD | root-mean-square deviation |
| ps | picosecond |
References
- Paisan-Ruız, C.; Jain, S.; Evans, E.W.; Gilks, W.P.; Simon, J.; van der Brug, M.; de Munain, A.L.; Aparicio, S.; Gil, A.M.; Khan, N.; et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 2004, 44, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Di Maio, R.; Hoffman, E.K.; Rocha, E.M.; Keeney, M.T.; Sanders, L.H.; De Miranda, B.R.; Zharikov, A.; Van Laar, A.; Stepan, A.F.; Lanz, T.A.; et al. LRRK2 activation in idiopathic Parkinson’s disease. Sci. Transl. Med. 2018, 10, eaar5429. [Google Scholar] [CrossRef] [PubMed]
- Healy, D.G.; Falchi, M.; O’Sullivan, S.S.; Bonifati, V.; Durr, A.; Bressman, S.; Brice, A.; Aasly, J.; Zabetian, C.P.; Goldwurm, S.; et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: A case-control study. Lancet Neurol. 2008, 7, 583–590. [Google Scholar] [CrossRef]
- West, A.B.; Moore, D.J.; Biskup, S.; Bugayenko, A.; Smith, W.W.; Ross, C.A.; Dawson, V.L.; Dawson, T.M. Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc. Natl. Acad. Sci. USA 2005, 102, 16842–16847. [Google Scholar] [CrossRef]
- Liu, M.; Kang, S.; Ray, S.; Jackson, J.; Zaitsev, A.D.; Gerber, S.A.; Cuny, G.D.; Glicksman, M.A. Kinetic, mechanistic, and structural modeling studies of truncated wild-type leucine-rich repeat kinase 2 and the G2019S mutant. Biochemistry 2011, 50, 9399–9408. [Google Scholar] [CrossRef]
- Cookson, M.R. The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson’s disease. Nat. Rev. Neurosci. 2010, 11, 791–797. [Google Scholar] [CrossRef]
- Volpicelli-Daley, L.A.; Abdelmotilib, H.; Liu, Z.; Stoyka, L.; Daher, J.P.; Milnerwood, A.J.; Unni, V.K.; Hirst, W.D.; Yue, Z.; Zhao, H.T.; et al. G2019S-LRRK2 expression augments alpha-synuclein sequestration into inclusions in neurons. J. Neurosci. 2016, 36, 7415–7427. [Google Scholar] [CrossRef]
- Liu, Z.; Hamamichi, S.; Lee, B.D.; Yang, D.; Ray, A.; Caldwell, G.A.; Caldwell, K.A.; Dawson, T.M.; Smith, W.W.; Dawson, V.L. Inhibitors of LRRK2 kinase attenuate neurodegeneration and Parkinson-like phenotypes in Caenorhabditis elegans and Drosophila Parkinson’s disease models. Hum. Mol. Genet. 2011, 20, 3933–3942. [Google Scholar] [CrossRef]
- De Wit, T.; Baekelandt, V.; Lobbestael, E. Inhibition of LRRK2 or casein kinase 1 results in LRRK2 protein destabilization. Mol. Neurobiol. 2019, 56, 5273–5286. [Google Scholar] [CrossRef]
- Watanabe, R.; Buschauer, R.; Böhning, J.; Audagnotto, M.; Lasker, K.; Lu, T.W.; Boassa, D.; Taylor, S.; Villa, E. The in situ structure of Parkinson’s disease-linked LRRK2. Cell 2020, 182, 1508–1518. [Google Scholar] [CrossRef]
- Myasnikov, A.; Zhu, H.; Hixson, P.; Xie, B.; Yu, K.; Pitre, A.; Peng, J.; Sun, J. Structural analysis of the full-length human LRRK2. Cell 2021, 184, 3519–3527. [Google Scholar] [CrossRef] [PubMed]
- Deniston, C.K.; Salogiannis, J.; Mathea, S.; Snead, D.M.; Lahiri, I.; Matyszewski, M.; Donosa, O.; Watanabe, R.; Böhning, J.; Shiau, A.K.; et al. Structure of LRRK2 in Parkinson’s disease and model for microtubule interaction. Nature 2020, 588, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Keylor, M.H.; Gulati, A.; Kattar, S.D.; Johnson, R.E.; Chau, R.W.; Margrey, K.A.; Ardolino, M.J.; Zarate, C.; Poremba, K.E.; Simov, V.; et al. Structure-guided discovery of aminoquinazolines as brain-penetrant and selective LRRK2 inhibitors. J. Med. Chem. 2022, 65, 838–856. [Google Scholar] [CrossRef] [PubMed]
- Leśniak, R.K.; Nichols, R.J.; Schonemann, M.; Zhao, J.; Gajera, C.R.; Lam, G.; Nguyen, K.C.; Langston, J.W.; Smith, M.; Montine, T.J. Discovery of 1H-pyrazole biaryl sulfonamides as novel G2019S-LRRK2 kinase inhibitors. ACS Med. Chem. Lett. 2022, 13, 981–988. [Google Scholar] [CrossRef]
- Williamson, D.S.; Smith, G.P.; Mikkelsen, G.K.; Jensen, T.; Acheson-Dossang, P.; Badolo, L.; Bedford, S.T.; Chell, V.; Chen, I.J.; Dokurno, P.; et al. Design and synthesis of pyrrolo[2,3-d]pyrimidine-derived leucine-rich repeat kinase 2 (LRRK2) inhibitors using a checkpoint kinase 1 (CHK1)-derived crystallographic surrogate. J. Med. Chem. 2021, 64, 10312–10332. [Google Scholar] [CrossRef]
- Helton, L.G.; Soliman, A.; von Zweydorf, F.; Kentros, M.; Manschwetus, J.T.; Hall, S.; Gilsbach, B.; Ho, F.Y.; Athanasopoulos, P.S.; Singh, R.K.; et al. Allosteric inhibition of Parkinson’s-linked LRRK2 by constrained peptides. ACS Chem. Biol. 2021, 16, 2326–2338. [Google Scholar] [CrossRef]
- Konstantinidou, M.; Oun, A.; Pathak, P.; Zhang, B.; Wang, Z.; ter Brake, F.; Dolga, A.M.; Kortholt, A.; Dömling, A. The tale of proteolysis targeting chimeras (PROTACs) for leucine-rich repeat kinase 2 (LRRK2). ChemMedChem 2021, 16, 959–965. [Google Scholar] [CrossRef]
- Zaldivar-Diez, J.; Li, L.; Garcia, A.M.; Zhao, W.N.; Medina-Menendez, C.; Haggarty, S.J.; Gil, C.; Morales, A.V.; Martinez, A. Benzothiazole-Based LRRK2 Inhibitors as Wnt Enhancers and Promoters of Oligodendrocytic Fate. J. Med. Chem. 2020, 63, 2638–2655. [Google Scholar] [CrossRef]
- Osborne, J.; Birchall, K.; Tsagris, D.J.; Lewis, S.J.; Smiljanic-Hurley, E.; Taylor, D.L.; Levy, A.; Alessi, D.R.; McIver, E.G. Discovery of potent and selective 5-azaindazole inhibitors of leucine-rich repeat kinase 2 (LRRK2)—Part 1. Bioorg. Med. Chem. Lett. 2019, 29, 668–673. [Google Scholar] [CrossRef]
- Shore, D.G.M.; Sweeney, Z.K.; Beresford, A.; Chan, B.K.; Chen, H.; Drummond, J.; Gill, A.; Kleinheinz, T.; Liu, X.; Medhurst, A.D.; et al. Discovery of potent azaindazole leucine-rich repeat kinase 2 (LRRK2) inhibitors possessing a key intramolecular hydrogen bond—Part 2. Bioorg. Med. Chem. Lett. 2019, 29, 674–680. [Google Scholar] [CrossRef]
- Garofalo, A.W.; Bright, J.; De Lombaert, S.; Toda, A.M.A.; Zobel, K.; Andreotti, D.; Beato, C.; Bernardi, S.; Budassi, F.; Caberlotto, L.; et al. Selective inhibitors of G2019S-LRRK2 kinase activity. J. Med. Chem. 2020, 63, 14821–14839. [Google Scholar] [CrossRef]
- Ding, X.; Stasi, L.P.; Dai, X.; Long, K.; Peng, C.; Zhao, B.; Wang, H.; Sun, C.; Hu, H.; Wan, Z.; et al. 5-Substituted-N-pyridazinylbenzamides as potent and selective LRRK2 inhibitors: Improved brain unbound fraction enables efficacy. Bioorg. Med. Chem. Lett. 2019, 29, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Stasi, L.P.; Ho, M.H.; Zhao, B.; Wang, H.; Long, K.; Xu, Q.; Sang, Y.; Sun, C.; Hu, H.; et al. Discovery of 4-ethoxy-7H-pyrrolo[2,3-d]pyrimidin-2-amines as potent, selective and orally bioavailable LRRK2 inhibitors. Bioorg. Med. Chem. Lett. 2018, 28, 1615–1620. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.D.; DeMong, D.E.; Greshock, T.J.; Basu, K.; Dai, X.; Harris, J.; Hruza, A.; Li, S.W.; Lin, S.I.; Liu, H.; et al. Discovery of a 3-(4-pyrimidinyl) indazole (MLi-2), an orally available and selective leucine-rich repeat kinase 2 (LRRK2) inhibitor that reduces brain kinase activity. J. Med. Chem. 2017, 60, 2983–2992. [Google Scholar] [CrossRef]
- Smith, G.P.; Badolo, L.; Chell, V.; Chen, I.J.; Christensen, K.V.; David, L.; Daechsel, J.A.; Hentzer, M.; Herzig, M.C.; Mikkelsen, G.K.; et al. The design and SAR of a novel series of 2-aminopyridine based LRRK2 inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 4500–4505. [Google Scholar] [CrossRef] [PubMed]
- Salado, I.G.; Zaldivar-Diez, J.; Sebastián-Pérez, V.; Li, L.; Geiger, L.; González, S.; Campillo, N.E.; Gil, C.; Morales, A.V.; Perez, D.I.; et al. Leucine rich repeat kinase 2 (LRRK2) inhibitors based on indolinone scaffold: Potential pro-neurogenic agents. Eur. J. Med. Chem. 2017, 138, 328–342. [Google Scholar] [CrossRef]
- Franzini, M.; Ye, X.M.; Adler, M.; Aubele, D.L.; Garofalo, A.W.; Gauby, S.; Goldbach, E.; Probst, G.D.; Quinn, K.P.; Santiago, P.; et al. Triazolopyridazine LRRK2 kinase inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 1967–1973. [Google Scholar] [CrossRef]
- Choi, H.G.; Zhang, J.; Deng, X.; Hatcher, J.M.; Patricelli, M.P.; Zhao, Z.; Alessi, D.R.; Gray, N.S. Brain Penetrant LRRK2 Inhibitor. ACS Med. Chem. Lett. 2012, 3, 658–662. [Google Scholar] [CrossRef]
- Tan, S.; Gong, X.; Liu, H.; Yao, X. Virtual Screening and Biological Activity Evaluation of New Potent Inhibitors Targeting LRRK2 Kinase Domain. ACS Chem. Neurosci. 2021, 12, 3214–3224. [Google Scholar] [CrossRef]
- Lang, C.A.; Ray, S.S.; Liu, M.; Singh, A.K.; Cuny, G.D. Discovery of LRRK2 inhibitors using sequential in silico joint pharmacophore space (JPS) and ensemble docking. Bioorg. Med. Chem. Lett. 2015, 25, 2713–2719. [Google Scholar] [CrossRef]
- Gancia, E.; De Groot, M.; Burton, B.; Clark, D.E. Discovery of LRRK2 inhibitors by using an ensemble of virtual screening methods. Bioorg. Med. Chem. Lett. 2017, 27, 2520–2527. [Google Scholar] [CrossRef] [PubMed]
- Krahn, A.I.; Wells, C.; Drewry, D.H.; Beitel, L.K.; Durcan, T.M.; Axtman, A.D. Defining the neural kinome: Strategies and opportunities for small molecule drug discovery to target neurodegenerative diseases. ACS Chem. Neurosci. 2020, 11, 1871–1886. [Google Scholar] [CrossRef]
- Shi, Y.; Mader, M. Brain penetrant kinase inhibitors: Learning from kinase neuroscience discovery. Bioorg. Med. Chem. Lett. 2018, 28, 1981–1991. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Yang, X.; Duan, Y.; Han, J.; Liao, C. Small-molecule kinase inhibitors for the treatment of nononcologic diseases. J. Med. Chem. 2021, 64, 1283–1345. [Google Scholar] [CrossRef] [PubMed]
- Warren, G.L.; Andrews, C.W.; Capelli, A.M.; Clarke, B.; LaLonde, J.; Lambert, M.H.; Lindvall, M.; Nevins, N.; Semus, S.F.; Senger, S.; et al. A critical assessment of docking programs and scoring functions. J. Med. Chem. 2006, 49, 5912–5931. [Google Scholar] [CrossRef]
- Shoichet, B.K.; Leach, A.R.; Kuntz, I.D. Ligand solvation in molecular docking. Proteins 1999, 34, 4–16. [Google Scholar] [CrossRef]
- Hudkins, R.L.; Diebold, J.L.; Tao, M.; Josef, K.A.; Park, C.H.; Angeles, T.S.; Aimone, L.D.; Husten, J.; Ator, M.A.; Meyer, S.L.; et al. Mixed-lineage kinase 1 and mixed-lineage kinase 3 subtype-selective dihydronaphthyl[3,4-a]pyrrolo[3,4-c]carbazole-5-ones: Optimization, mixed-lineage kinase 1 crystallography, and oral in vivo activity in 1-methyl-4-phenyltetrahydropyridine models. J. Med. Chem. 2008, 51, 5680–5689. [Google Scholar] [CrossRef]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36, 5–9. [Google Scholar] [CrossRef]
- Baker, D.; Sali, A. Protein structure prediction and structural genomics. Science 2001, 294, 93–96. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef]
- Yan, Y.; Wang, W.; Sun, Z.; Zhang, J.Z.H.; Ji, C. Protein–ligand empirical interaction components for virtual screening. J. Chem. Inf. Model. 2017, 57, 1793–1806. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Su, M.; Han, L.; Liu, J.; Yang, Q.; Li, Y.; Wang, R. Forging the basis for developing protein–ligand interaction scoring functions. Acc. Chem. Res. 2017, 50, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Fabian, M.A.; Biggs III, W.H.; Treiber, D.K.; Atteridge, C.E.; Azimioara, M.D.; Bendetti, M.G.; Carter, T.A.; Ciceri, P.; Edeen, P.T.; Floyd, M.; et al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 2005, 23, 329–336. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed]
- Muley, L.; Baum, B.; Smolinski, M.; Freindorf, M.; Heine, A.; Klebe, G.; Hangauer, D.G. Enhancement of hydrophobic interactions and hydrogen bond strength by cooperativity: Synthesis, modeling, and molecular dynamics simulations of a congeneric series of thrombin Inhibitors. J. Med. Chem. 2010, 53, 2126–2135. [Google Scholar] [CrossRef]
- Peng, Y.H.; Ueng, S.H.; Tseng, C.T.; Hung, M.S.; Song, J.S.; Wu, J.S.; Liao, F.Y.; Fan, Y.S.; Wu, M.H.; Hsiao, W.C.; et al. Important hydrogen bond networks in indoleamine 2,3-dioxygenase 1 (IDO1) inhibitor design revealed by crystal structures of imidazoleisoindole derivatives with IDO1. J. Med. Chem. 2016, 59, 282–293. [Google Scholar] [CrossRef]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Gasteiger, J.; Marsili, M. Iterative partial equalization of orbital electronegativity a rapid access to atomic charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Lindström, A.; Edvinsson, L.; Johansson, A.; Andersson, C.D.; Andersson, I.E.; Raubacher, F.; Linusson, A. Postprocessing of docked protein−ligand complexes using implicit solvation models. J. Chem. Inf. Model. 2011, 51, 267–282. [Google Scholar] [CrossRef]
- Mehler, E.L.; Solmajer, T. Electrostatic effects in proteins: Comparison of dielectric and charge models. Protein Eng. 1991, 4, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Stouten, P.F.W.; Frömmel, C.; Nakamura, H.; Sander, C. An effective solvation term based on atomic occupancies for use in protein simulations. Mol. Simul. 1993, 10, 97–120. [Google Scholar] [CrossRef]
- Park, H. Extended solvent-contact model approach to SAMPL4 blind prediction challenge for hydration free energies. J. Comput. Aided Mol. Des. 2014, 28, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.C.; Park, H. Accuracy enhancement in the estimation of molecular hydration free energies by implementing the intramolecular hydrogen bond effects. J. Cheminform. 2015, 7, 57. [Google Scholar] [CrossRef]
- Wang, R.; Gao, Y.; Lai, L. LigBuilder: A multi-purpose program for structure-based drug design. J. Mol. Model. 2000, 6, 498–516. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The AMBER biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Berendsen, H.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | MW (amu) | ΔGbind (kcal/mol) | IC50 (μM) | |
|---|---|---|---|---|
| G2019S Mutant | Wild Type | |||
| 1 | 401.5 | −26.16 | 0.194 ± 0.017 | 0.187 ± 0.005 |
| 2 | 279.3 | −26.02 | 0.203 ± 0.007 | 0.260 ± 0.005 |
| 3 | 316.4 | −26.03 | 0.206 ± 0.029 | 0.301 ± 0.028 |
| 4 | 352.4 | −26.44 | 0.244 ± 0.016 | 0.261 ± 0.017 |
| 5 | 296.3 | −26.08 | 0.276 ± 0.032 | 0.235 ± 0.018 |
| 6 | 329.4 | −25.91 | 0.289 ± 0.011 | 0.254 ± 0.007 |
| 7 | 345.4 | −26.02 | 0.413 ± 0.008 | ND |
| 8 | 344.4 | −26.98 | 0.494 ± 0.028 | ND |
| 9 | 279.3 | −25.91 | 0.505 ± 0.064 | ND |
| 10 | 290.3 | −26.31 | 0.563 ± 0.019 | ND |
| 11 | 381.4 | −26.86 | 0.652 ± 0.033 | ND |
| 12 | 378.4 | −27.53 | 0.681 ± 0.024 | ND |
| 13 | 350.4 | −26.92 | 0.776 ± 0.027 | ND |
| 14 | 309.4 | −26.17 | 1.918 ± 0.051 | ND |
| 15 | 343.4 | −27.01 | 1.971 ± 0.313 | ND |
| 16 | 377.3 | −26.14 | 4.408 ± 0.053 | ND |
| 17 | 303.3 | −27.24 | 4.464 ± 0.024 | ND |
| 18 | 345.4 | −26.42 | 8.942 ± 0.067 | ND |
 | |||||
|---|---|---|---|---|---|
| R1 a | R2 | R3 | R4 | IC50 (μM) | |
| 2a | CH3 | H | H | H | 0.400 ± 0.011 |
| 2b | CH3CH2 | H | H | H | 0.123 ± 0.007 |
| 2c |  | H | H | H | 0.108 ± 0.005 |
| 2d |  | H | H | H | 0.762 ± 0.025 |
| 2e |  | H | H | H | 2.627 ± 0.304 |
| 2f | CH3 | CH3O | H | H | 0.479 ± 0.009 |
| 2g | CH3CH2 | Cl | H | H | 1.115 ± 0.130 |
| 2h | CH3CH2 | Br | H | H | 1.550 ± 0.213 |
| 2i | | CH3O | H | H | 1.353 ± 0.062 |
| 2j | CH3 | CH3O | CH3O | H | 0.883 ± 0.079 |
| 2k | CH3CH2 | CH3O | CH3O | H | 0.438 ± 0.023 |
| 2l | | CH3O | CH3O | H | 1.823 ± 0.187 |
| 2m | CH3 | H | H | CH3O | 0.122 ± 0.008 |
| 2n | CH3CH2 | H | H | CH3O | 0.089 ± 0.006 |
| 2o | | H | H | CH3O | 0.316 ± 0.037 |
| 2p | | H | H | CH3O | 0.613 ± 0.056 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, H.; Kim, T.; Kim, K.; Jang, A.; Hong, S. Structure-Based Virtual Screening and De Novo Design to Identify Submicromolar Inhibitors of G2019S Mutant of Leucine-Rich Repeat Kinase 2. Int. J. Mol. Sci. 2022, 23, 12825. https://doi.org/10.3390/ijms232112825
Park H, Kim T, Kim K, Jang A, Hong S. Structure-Based Virtual Screening and De Novo Design to Identify Submicromolar Inhibitors of G2019S Mutant of Leucine-Rich Repeat Kinase 2. International Journal of Molecular Sciences. 2022; 23(21):12825. https://doi.org/10.3390/ijms232112825
Chicago/Turabian StylePark, Hwangseo, Taeho Kim, Kewon Kim, Ahyoung Jang, and Sungwoo Hong. 2022. "Structure-Based Virtual Screening and De Novo Design to Identify Submicromolar Inhibitors of G2019S Mutant of Leucine-Rich Repeat Kinase 2" International Journal of Molecular Sciences 23, no. 21: 12825. https://doi.org/10.3390/ijms232112825
APA StylePark, H., Kim, T., Kim, K., Jang, A., & Hong, S. (2022). Structure-Based Virtual Screening and De Novo Design to Identify Submicromolar Inhibitors of G2019S Mutant of Leucine-Rich Repeat Kinase 2. International Journal of Molecular Sciences, 23(21), 12825. https://doi.org/10.3390/ijms232112825