

Synthesis, Anticancer and Antitubercular Properties of New Chalcones and Their Nitrogen-Containing Five-Membered Heterocyclic Hybrids Bearing Sulfonamide Moiety
 , ,
, ,  , , , , , , ,
, , , , , , ,
Abstract
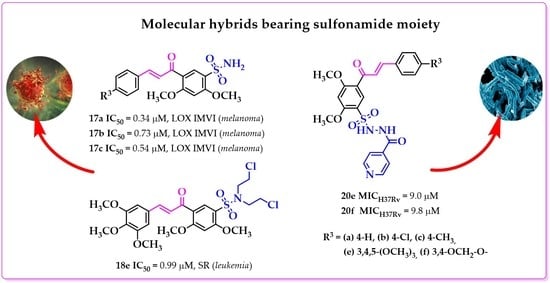
1. Introduction
2. Results and Discussion
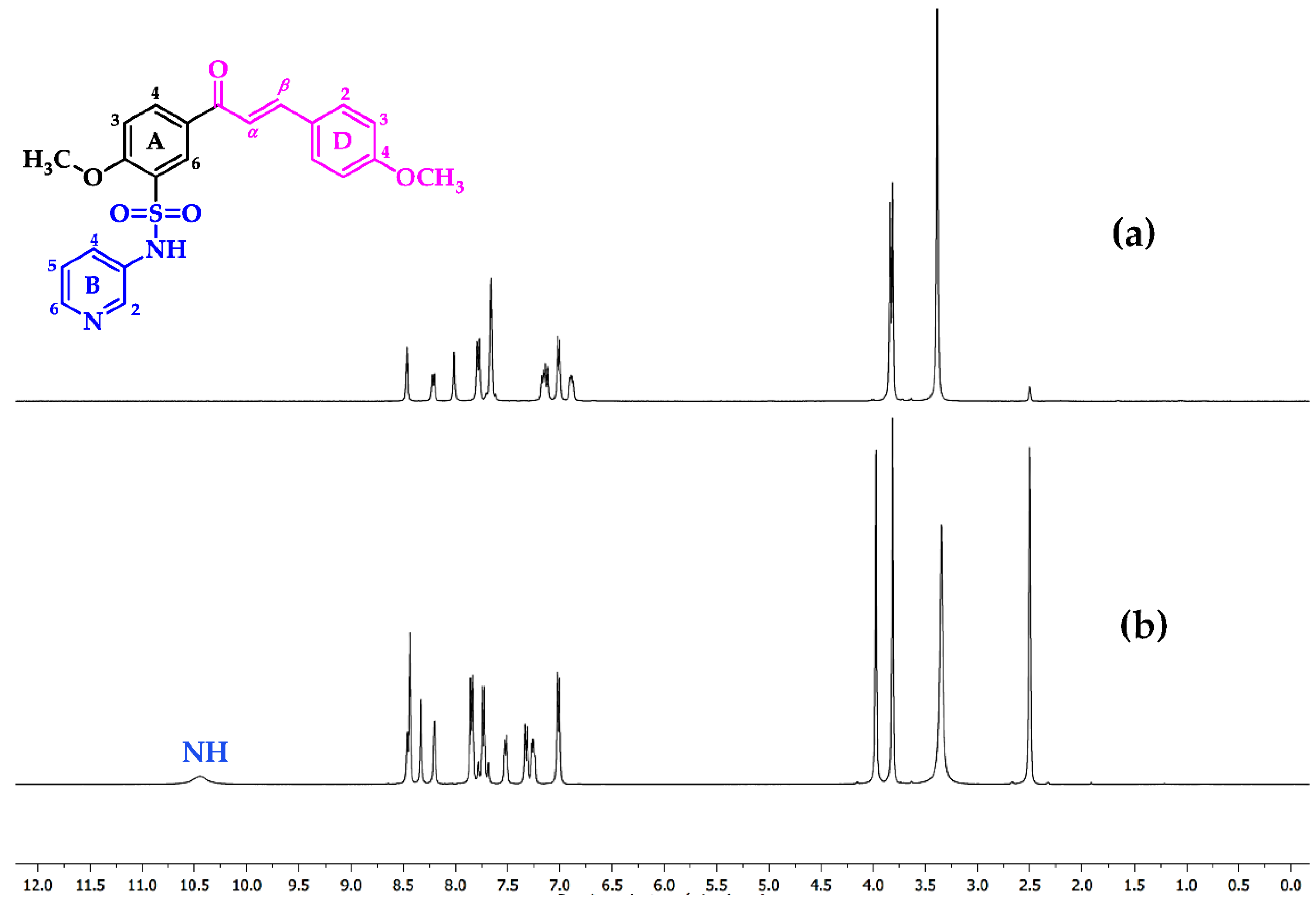
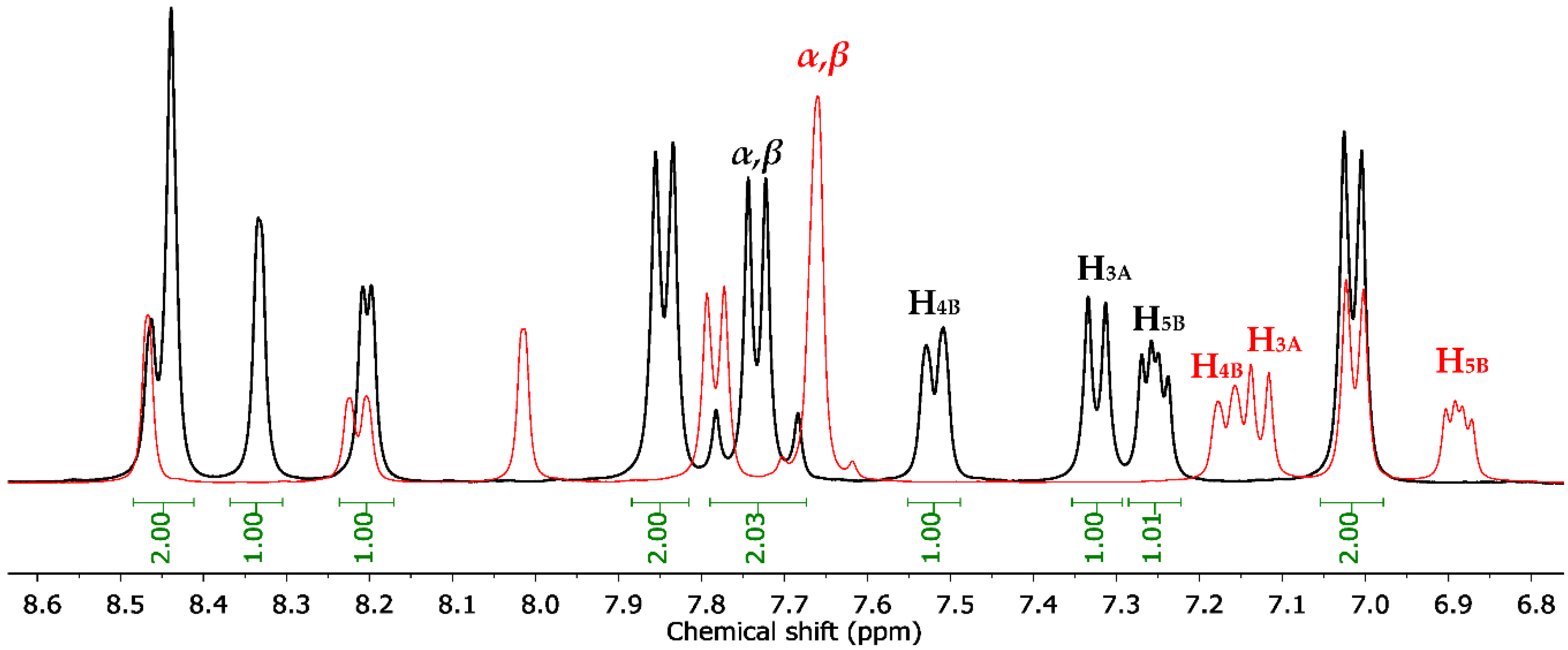
2.1. Chemistry
2.2. Anticancer Activity
2.3. Antituberculosis Activity, Cytotoxicity and Synergism
3. Materials and Methods
3.1. General
3.2. Chemistry
3.2.1. General Procedure for the Synthesis of Acetophenone 5b
1-(2,4-Dimethoxyphenyl)ethan-1-one (5b)
3.2.2. General Procedure for the Synthesis of Benzenesulfonyl Chloride 6b
5-Acetyl-2,4-dimethoxybenzenesulfonyl Chloride (6b)
3.2.3. General Procedure for the Synthesis of Sulfonamides 8a-b
5-Acetyl-2-methoxybenzenesulfonamide (8a)
5-Acetyl-2,4-dimethoxybenzenesulfonamide (8b)
3.2.4. General Procedure for the Synthesis of Sulfonamide 10
5-Acetyl-2-methoxy-N-(pyridin-3-yl)benzenesulfonamide (10)
3.2.5. General Procedure for the Synthesis of Sulfonamide 12
5-Acetyl-N,N-bis(2-chloroethyl)-2,4-dimethoxybenzenesulfonamide (12)
3.2.6. General Procedure for the Synthesis of Sulfonamides 14a-b
5-Acetyl-N’-isonicotinoyl-2-methoxybenzenesulfonohydrazide (14a)
5-Acetyl-N’-isonicotinoyl-2,4-dimethoxybenzenesulfonohydrazide (14b)
3.2.7. General Procedure for the Synthesis of Chalcone–Sulfonamide Hybrids 16a-f and 17a-f
5-Cinnamoyl-2-methoxy-N-(pyridin-3-yl)benzenesulfonamide (16a)
(E)-5-(3-(4-Chlorophenyl)acryloyl)-2-methoxy-N-(pyridin-3-yl)benzenesulfonamide (16b)
(E)-2-Methoxy-N-(pyridin-3-yl)-5-(3-(p-tolyl)acryloyl)benzenesulfonamide (16c)
(E)-2-Methoxy-5-(3-(4-methoxyphenyl)acryloyl)-N-(pyridin-3-yl)benzenesulfonamide (16d)
(E)-2-Methoxy-N-(pyridin-3-yl)-5-(3-(3,4,5-trimethoxyphenyl)acryloyl)benzenesulfonamide (16e)
(E)-5-(3-(Benzo[d][1,3]dioxol-5-yl)acryloyl)-2-methoxy-N-(pyridin-3-yl)benzenesulfonamide (16f)
5-Cinnamoyl-2,4-dimethoxybenzenesulfonamide (17a)
(E)-5-(3-(4-Chlorophenyl)acryloyl)-2,4-dimethoxybenzenesulfonamide (17b)
(E)-2,4-Dimethoxy-5-(3-(p-tolyl)acryloyl)benzenesulfonamide (17c)
(E)-2,4-Dimethoxy-5-(3-(4-methoxyphenyl)acryloyl)benzenesulfonamide (17d)
(E)-2,4-Dimethoxy-5-(3-(3,4,5-trimethoxyphenyl)acryloyl)benzenesulfonamide (17e)
(E)-5-(3-(Benzo[d][1,3]dioxol-5-yl)acryloyl)-2,4-dimethoxybenzenesulfonamide (17f)
3.2.8. General Procedure for the Synthesis of Chalcone–Sulfonamide Hybrids 18a-f
N,N-bis(2-chloroethyl)-5-cinnamoyl-2,4-dimethoxybenzenesulfonamide (18a)
(E)-N,N-bis(2-Chloroethyl)-5-(3-(4-chlorophenyl)acryloyl)-2,4-dimethoxybenzenesulfonamide (18b)
(E)-N,N-bis(2-Chloroethyl)-2,4-dimethoxy-5-(3-(p-tolyl)acryloyl)benzenesulfonamide (18c)
(E)-N,N-bis(2-Chloroethyl)-2,4-dimethoxy-5-(3-(4-methoxyphenyl)acryloyl)benzenesulfonamide (18d)
(E)-N,N-bis(2-Chloroethyl)-2,4-dimethoxy-5-(3-(3,4,5-trimethoxyphenyl)acryloyl)benzenesulfonamide (18e)
(E)-5-(3-(Benzo[d][1,3]dioxol-5-yl)acryloyl)-N,N-bis(2-chloroethyl)-2,4-dimethoxybenzenesulfonamide (18f)
3.2.9. General Procedure for the Synthesis of Chalcone–Sulfonamide Hybrids 19a-f
5-Cinnamoyl-N’-isonicotinoyl-2-methoxybenzenesulfonohydrazide (19a)
(E)-5-(3-(4-Chlorophenyl)acryloyl)-N’-isonicotinoyl-2-methoxybenzenesulfonohydrazide (19b)
(E)-N’-Isonicotinoyl-2-methoxy-5-(3-(p-tolyl)acryloyl)benzenesulfonohydrazide (19c)
(E)-N’-Isonicotinoyl-2-methoxy-5-(3-(4-methoxyphenyl)acryloyl)benzenesulfonohydrazide (19d)
(E)-N’-Isonicotinoyl-2-methoxy-5-(3-(3,4,5-trimethoxyphenyl)acryloyl)benzenesulfonohydrazide (19e)
(E)-5-(3-(Benzo[d][1,3]dioxol-5-yl)acryloyl)-N’-isonicotinoyl-2-methoxybenzenesulfonohydrazide (19f)
3.2.10. General Procedure for the Synthesis of Chalcone–Sulfonamide Hybrids 20a-f
5-Cinnamoyl-N’-isonicotinoyl-2,4-dimethoxybenzenesulfonohydrazide (20a)
(E)-5-(3-(4-Chlorophenyl)acryloyl)-N’-isonicotinoyl-2,4-dimethoxybenzenesulfonohydrazide (20b)
(E)-N’-Isonicotinoyl-2,4-dimethoxy-5-(3-(p-tolyl)acryloyl)benzenesulfonohydrazide (20c)
(E)-N’-Isonicotinoyl-2,4-dimethoxy-5-(3-(4-methoxyphenyl)acryloyl)benzenesulfonohydrazide (20d)
(E)-N’-Isonicotinoyl-2,4-dimethoxy-5-(3-(3,4,5-trimethoxyphenyl)acryloyl)benzenesulfonohydrazide (20e)
(E)-5-(3-(Benzo[d][1,3]dioxol-5-yl)acryloyl)-N’-isonicotinoyl-2,4-dimethoxybenzenesulfonohydrazide (20f)
3.2.11. General Procedure for the Synthesis of Pyrazoline–Sulfonamide Hybrids 22a-d
5-(5-(4-Chlorophenyl)-1-formyl-4,5-dihydro-1H-pyrazol-3-yl)-2-methoxybenzenesulfonamide (22a)
5-(5-(4-Chlorophenyl)-1-formyl-4,5-dihydro-1H-pyrazol-3-yl)-2-methoxy-N-(pyridin-3-yl)benzenesulfonamide (22b)
5-(5-(4-Chlorophenyl)-1-formyl-4,5-dihydro-1H-pyrazol-3-yl)-2,4-dimethoxybenzenesulfonamide (22c)
N,N-bis(2-Chloroethyl)-5-(5-(4-chlorophenyl)-1-formyl-4,5-dihydro-1H-pyrazol-3-yl)-2,4-dimethoxybenzenesulfonamide (22d)
3.2.12. General Procedure for the Synthesis of Pyrazoline–Sulfonamide Hybrids 23a-d
5-(1-Acetyl-5-(4-chlorophenyl)-4,5-dihydro-1H-pyrazol-3-yl)-2-methoxybenzenesulfonamide (23a)
5-(L-acetyl-5-(4-chlorophenyl)-4,5-dihydro-1H-pyrazol-3-yl)-2-methoxy-N-(pyridin-3-yl)benzenesulfonamide (23b)
5-(L-acetyl-5-(4-chlorophenyl)-4,5-dihydro-1H-pyrazol-3-yl)-2,4-dimethoxybenzenesulfonamide (23c)
5-(L-acetyl-5-(4-chlorophenyl)-4,5-dihydro-1H-pyrazol-3-yl)-N,N-bis(2-chloroethyl)-2,4-dimethoxybenzenesulfonamide (23d)
3.2.13. General Procedure for the Synthesis of Pyrazoline–Sulfonamide Hybrids 24a-b
5-(5-(4-Chlorophenyl)-1-formyl-4,5-dihydro-1H-pyrazol-3-yl)-N’-isonicotinoyl-2-methoxybenzenesulfonohydrazide (24a)
5-(5-(4-Chlorophenyl)-1-formyl-4,5-dihydro-1H-pyrazol-3-yl)-N’-isonicotinoyl-2,4-dimethoxybenzenesulfonohydrazide (24b)
3.2.14. General Procedure for the Synthesis of Pyrazoline–Sulfonamide Hybrids 25a-b
5-(1-Acetyl-5-(4-chlorophenyl)-4,5-dihydro-1H-pyrazol-3-yl)-N’-isonicotinoyl-2-methoxybenzenesulfonohydrazide (25a)
5-(L-acetyl-5-(4-chlorophenyl)-4,5-dihydro-1H-pyrazol-3-yl)-N’-isonicotinoyl-2,4-dimethoxybenzenesulfonohydrazide (25b)
3.2.15. General Procedure for the Synthesis of Carbothioamide–Sulfonamide Hybrids 27a-f
5-(4-Chlorophenyl)-3-(4-methoxy-3-sulfamoylphenyl)-4,5-dihydro-1H-pyrazole-1-carbothioamide (27a)
5-(4-Chlorophenyl)-3-(4-methoxy-3-(N-(pyridin-3-yl)sulfamoyl)phenyl)-4,5-dihydro-1H-pyrazole-1-carbothioamide (27b)
5-(4-Chlorophenyl)-3-(3-((2-isonicotinoylhydrazineyl)sulfonyl)-4-methoxyphenyl)-4,5-dihydro-1H-pyrazole-1-carbothioamide (27c)
5-(4-Chlorophenyl)-3-(2,4-dimethoxy-5-sulfamoylphenyl)-4,5-dihydro-1H-pyrazole-1-carbothioamide (27d)
3-(5-(N,N-bis(2-Chloroethyl)sulfamoyl)-2,4-dimethoxyphenyl)-5-(4-chlorophenyl)-4,5-dihydro-1H-pyrazole-1-carbothioamide (27e)
5-(4-Chlorophenyl)-3-(5-((2-isonicotinoylhydrazineyl)sulfonyl)-2,4-dimethoxyphenyl)-4,5-dihydro-1H-pyrazole-1-carbothioamide (27f)
3.3. Anticancer Activity
3.4. Antituberculosis Activity
3.5. Cytotoxicity on Vero Cell Line
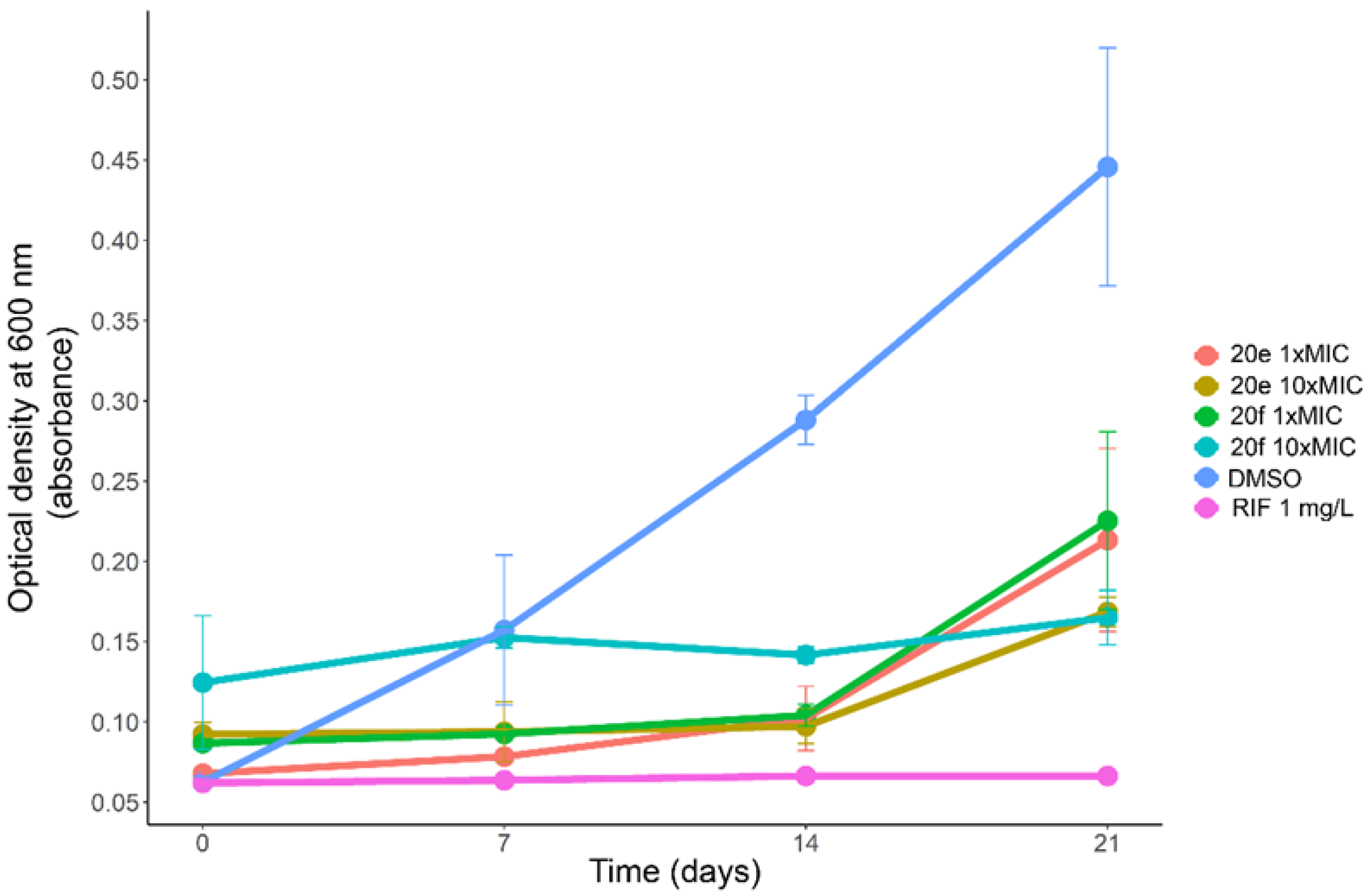
3.6. Synergism
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Santucci, C.; Carioli, G.; Bertuccio, P.; Malvezzi, M.; Pastorino, U.; Boffetta, P.; Negri, E.; Bosetti, C.; La Vecchia, C. Progress in Cancer Mortality, Incidence, and Survival: A Global Overview. Eur. J. Cancer Prev. 2020, 29, 367–381. [Google Scholar] [CrossRef] [PubMed]
- Shalini; Kumar, V. Have Molecular Hybrids Delivered Effective Anti-Cancer Treatments and What Should Future Drug Discovery Focus On? Expert Opin. Drug Discov. 2021, 16, 335–363. [Google Scholar] [CrossRef]
- Global Tuberculosis Report. Available online: https://www.who.int/teams/global-tuberculosis-programme/tb-reports/global-tuberculosis-report-2021 (accessed on 31 August 2022).
- Peloquin, C.A.; Davies, G.R. The Treatment of Tuberculosis. Clin. Pharmacol. Ther. 2021, 110, 1455–1466. [Google Scholar] [CrossRef]
- Furin, J.; Cox, H.; Pai, M. Tuberculosis. Lancet 2019, 393, 1642–1656. [Google Scholar] [CrossRef]
- Natarajan, A.; Beena, P.M.; Devnikar, A.V.; Mali, S. A Systemic Review on Tuberculosis. Indian J. Tuberc. 2020, 67, 295–311. [Google Scholar] [CrossRef] [PubMed]
- Apaydın, S.; Török, M. Sulfonamide Derivatives as Multi-Target Agents for Complex Diseases. Bioorg. Med. Chem. Lett. 2019, 29, 2042–2050. [Google Scholar] [CrossRef] [PubMed]
- Scozzafava, A.; Carta, F.; Supuran, C.T. Secondary and Tertiary Sulfonamides: A Patent Review. Expert Opin. Ther. Pat. 2013, 23, 203–213. [Google Scholar] [CrossRef]
- Zhao, C.; Rakesh, K.P.; Ravidar, L.; Fang, W.Y.; Qin, H.L. Pharmaceutical and Medicinal Significance of Sulfur (SVI)-Containing Motifs for Drug Discovery: A Critical Review. Eur. J. Med. Chem. 2019, 162, 679–734. [Google Scholar] [CrossRef]
- Catarro, M.; Serrano, J.L.; Ramos, S.S.; Silvestre, S.; Almeida, P. Nimesulide Analogues: From Anti-Inflammatory to Antitumor Agents. Bioorg. Chem. 2019, 88, 102966. [Google Scholar] [CrossRef]
- Roell, D.; Baniahmad, A. The Natural Compounds Atraric Acid and N-Butylbenzene-Sulfonamide as Antagonists of the Human Androgen Receptor and Inhibitors of Prostate Cancer Cell Growth. Mol. Cell. Endocrinol. 2011, 332, 1–8. [Google Scholar] [CrossRef]
- Carta, F.; Supuran, C.T.; Scozzafava, A. Sulfonamides and Their Isosters as Carbonic Anhydrase Inhibitors. Future Med. Chem. 2014, 6, 1149–1165. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Kazuhiko, I.; Takaya, N.; Yamaguchi, Y.; Kishii, R.; Kohno, Y.; Kurasaki, H. Sulfonamide-Based Non-Alkyne LpxC Inhibitors as Gram-Negative Antibacterial Agents. Bioorg. Med. Chem. Lett. 2017, 27, 1045–1049. [Google Scholar] [CrossRef] [PubMed]
- Tassini, S.; Sun, L.; Lanko, K.; Crespan, E.; Langron, E.; Falchi, F.; Kissova, M.; Armijos-Rivera, J.I.; Delang, L.; Mirabelli, C.; et al. Discovery of Multitarget Agents Active as Broad-Spectrum Antivirals and Correctors of Cystic Fibrosis Transmembrane Conductance Regulator for Associated Pulmonary Diseases. J. Med. Chem. 2017, 60, 1400–1416. [Google Scholar] [CrossRef] [PubMed]
- Thareja, S.; Kokil, G.R.; Aggarwal, S.; Bhardwaj, T.R.; Kumar, M. Sulphonamides as Inhibitors of Protein Tyrosine Phosphatase 1B: A Three-Dimensional Quantitative Structure-Activity Relationship Study Using Self-Organizing Molecular Field Analysis Approach. Chem. Pharm. Bull. 2010, 58, 526–532. [Google Scholar] [CrossRef]
- Supuran, C.T.; Capasso, C. The η-Class Carbonic Anhydrases as Drug Targets for Antimalarial Agents. Expert Opin. Ther. Targets 2015, 19, 551–563. [Google Scholar] [CrossRef]
- Singh, P.; Anand, A.; Kumar, V. Recent Developments in Biological Activities of Chalcones: A Mini Review. Eur. J. Med. Chem. 2014, 85, 758–777. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.R.; Jiang, K.; Yang, J.L.; Shi, Y.P. Flavonoids as Key Bioactive Components of Oxytropis Falcata Bunge, a Traditional Anti-Inflammatory and Analgesic Tibetan Medicine. Nat. Prod. Res. 2020, 34, 3335–3352. [Google Scholar] [CrossRef]
- Sadula, A.; Peddaboina, U.R.; Prameela Subhashini, N.J. Synthesis and Characterization of Novel Chalcone Linked Imidazolones as Potential Antimicrobial and Antioxidant Agents. Med. Chem. Res. 2015, 24, 851–859. [Google Scholar] [CrossRef]
- Wang, L.; Yang, X.; Zhang, Y.; Chen, R.; Cui, Y.; Wang, Q. Anti-Inflammatory Chalcone-Isoflavone Dimers and Chalcone Dimers from Caragana Jubata. J. Nat. Prod. 2019, 82, 2761–2767. [Google Scholar] [CrossRef]
- Kontogiorgis, C.; Mantzanidou, M.; Hadjipavlou-Litina, D. Chalcones and Their Potential Role in Inflammation. Mini-Rev. Med. Chem. 2008, 8, 1224–1242. [Google Scholar] [CrossRef]
- Mateeva, N.; Eyunni, S.V.K.; Redda, K.K.; Ononuju, U.; Hansberry, T.D.; Aikens, C.; Nag, A. Functional Evaluation of Synthetic Flavonoids and Chalcones for Potential Antiviral and Anticancer Properties. Bioorg. Med. Chem. Lett. 2017, 27, 2350–2356. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, D.K.; Bharti, S.K. Therapeutic Potential of Chalcones as Cardiovascular Agents. Life Sci. 2016, 148, 154–172. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, D.K.; Asati, V.; Bharti, S.K. Chalcones and Their Therapeutic Targets for the Management of Diabetes: Structural and Pharmacological Perspectives. Eur. J. Med. Chem. 2015, 92, 839–865. [Google Scholar] [CrossRef] [PubMed]
- Gomes, M.N.; Braga, R.C.; Grzelak, E.M.; Neves, B.J.; Muratov, E.; Ma, R.; Klein, L.L.; Cho, S.; Oliveira, G.R.; Franzblau, S.G.; et al. QSAR-Driven Design, Synthesis and Discovery of Potent Chalcone Derivatives with Antitubercular Activity. Eur. J. Med. Chem. 2017, 137, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Ejaz, S.A.; Saeed, A.; Siddique, M.N.; un Nisa, Z.; Khan, S.; Lecka, J.; Sévigny, J.; Iqbal, J. Synthesis, Characterization and Biological Evaluation of Novel Chalcone Sulfonamide Hybrids as Potent Intestinal Alkaline Phosphatase Inhibitors. Bioorg. Chem. 2017, 70, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Gomes, M.N.; Muratov, E.N.; Pereira, M.; Peixoto, J.C.; Rosseto, L.P.; Cravo, P.V.L.; Andrade, C.H.; Neves, B.J. Chalcone Derivatives: Promising Starting Points for Drug Design. Molecules 2017, 22, 1210. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, C.; Zhang, W.; Sheng, C.; Zhang, W.; Xing, C.; Miao, Z. Chalcone: A Privileged Structure in Medicinal Chemistry. Chem. Rev. 2017, 117, 7762–7810. [Google Scholar] [CrossRef]
- Bashir, R.; Ovais, S.; Yaseen, S.; Hamid, H.; Alam, M.S.; Samim, M.; Singh, S.; Javed, K. Synthesis of Some New 1,3,5-Trisubstituted Pyrazolines Bearing Benzene Sulfonamide as Anticancer and Anti-Inflammatory Agents. Bioorg. Med. Chem. Lett. 2011, 21, 4301–4305. [Google Scholar] [CrossRef]
- Tucker, J.W.; Chenard, L.; Young, J.M. Selective Access to Heterocyclic Sulfonamides and Sulfonyl Fluorides via a Parallel Medicinal Chemistry Enabled Method. ACS Comb. Sci. 2015, 17, 653–657. [Google Scholar] [CrossRef]
- Kucukoglu, K.; Oral, F.; Aydin, T.; Yamali, C.; Algul, O.; Sakagami, H.; Gulcin, I.; Supuran, C.T.; Gul, H.I. Synthesis, Cytotoxicity and Carbonic Anhydrase Inhibitory Activities of New Pyrazolines. J. Enzyme Inhib. Med. Chem. 2016, 31, 20–24. [Google Scholar] [CrossRef]
- Yamali, C.; Gul, H.I.; Kazaz, C.; Levent, S.; Gulcin, I. Synthesis, Structure Elucidation, and in Vitro Pharmacological Evaluation of Novel Polyfluoro Substituted Pyrazoline Type Sulfonamides as Multi-Target Agents for Inhibition of Acetylcholinesterase and Carbonic Anhydrase I and II Enzymes. Bioorg. Chem. 2020, 96, 103627. [Google Scholar] [CrossRef] [PubMed]
- Shaaban, M.R.; Mayhoub, A.S.; Farag, A.M. Recent Advances in the Therapeutic Applications of Pyrazolines. Expert Opin. Ther. Pat. 2012, 22, 253–291. [Google Scholar] [CrossRef] [PubMed]
- Acharya, P.T.; Bhavsar, Z.A.; Jethava, D.J.; Patel, D.B.; Patel, H.D. A Review on Development of Bio-Active Thiosemicarbazide Derivatives: Recent Advances. J. Mol. Struct. 2021, 1226, 129268. [Google Scholar] [CrossRef]
- Tilekar, K.; Upadhyay, N.; Meyer-Almes, F.J.; Loiodice, F.; Anisimova, N.Y.; Spirina, T.S.; Sokolova, D.V.; Smirnova, G.B.; Choe, J.Y.; Pokrovsky, V.S.; et al. Synthesis and Biological Evaluation of Pyrazoline and Pyrrolidine-2,5-Dione Hybrids as Potential Antitumor Agents. ChemMedChem 2020, 15, 1813–1825. [Google Scholar] [CrossRef]
- Montoya, A.; Quiroga, J.; Abonia, R.; Nogueras, M.; Cobo, J.; Insuasty, B. Synthesis and in Vitro Antitumor Activity of a Novel Series of 2-Pyrazoline Derivatives Bearing the 4-Aryloxy-7-Chloroquinoline Fragment. Molecules 2014, 19, 18656–18675. [Google Scholar] [CrossRef]
- Chen, K.; Zhang, Y.L.; Fan, J.; Ma, X.; Qin, Y.J.; Zhu, H.L. Novel Nicotinoyl Pyrazoline Derivates Bearing N-Methyl Indole Moiety as Antitumor Agents: Design, Synthesis and Evaluation. Eur. J. Med. Chem. 2018, 156, 722–737. [Google Scholar] [CrossRef]
- Nehra, B.; Rulhania, S.; Jaswal, S.; Kumar, B.; Singh, G.; Monga, V. Recent Advancements in the Development of Bioactive Pyrazoline Derivatives. Eur. J. Med. Chem. 2020, 205, 112666. [Google Scholar] [CrossRef]
- Iñiguez, M.A.; Punzón, C.; Cacheiro-Llaguno, C.; Díaz-Muñoz, M.D.; Duque, J.; Cuberes, R.; Alvarez, I.; Andrés, E.M.; Buxens, J.; Buschmann, H.; et al. Cyclooxygenase-Independent Inhibitory Effects on T Cell Activation of Novel 4,5-Dihydro-3 Trifluoromethyl Pyrazole Cyclooxygenase-2 Inhibitors. Int. Immunopharmacol. 2010, 10, 1295–1304. [Google Scholar] [CrossRef]
- Kharbanda, C.; Alam, M.S.; Hamid, H.; Javed, K.; Bano, S.; Dhulap, A.; Ali, Y.; Nazreen, S.; Haider, S. Synthesis and Evaluation of Pyrazolines Bearing Benzothiazole as Anti-Inflammatory Agents. Bioorg. Med. Chem. 2014, 22, 5804–5812. [Google Scholar] [CrossRef]
- Faidallah, H.M.; Al-Mohammadi, M.M.; Alamry, K.A.; Khan, K.A. Synthesis and Biological Evaluation of Fluoropyrazolesulfonylurea and Thiourea Derivatives as Possible Antidiabetic Agents. J. Enzyme Inhib. Med. Chem. 2016, 31, 157–163. [Google Scholar] [CrossRef]
- Özdemir, A.; Altintop, M.D.; Kaplancikli, Z.A.; Can, Ö.D.; Özkay, Ü.D.; Turan-Zitouni, G. Synthesis and Evaluation of New 1,5-Diaryl-3-[4-(Methylsulfonyl)Phenyl]-4,5-Dihydro-1 H-Pyrazole Derivatives as Potential Antidepressant Agents. Molecules 2015, 20, 2668–2684. [Google Scholar] [CrossRef] [PubMed]
- Insuasty, B.; Montoya, A.; Becerra, D.; Quiroga, J.; Abonia, R.; Robledo, S.; Vélez, I.D.; Upegui, Y.; Nogueras, M.; Cobo, J. Synthesis of Novel Analogs of 2-Pyrazoline Obtained from [(7-Chloroquinolin-4-Yl)Amino]Chalcones and Hydrazine as Potential Antitumor and Antimalarial Agents. Eur. J. Med. Chem. 2013, 67, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Fioravanti, R.; Desideri, N.; Carta, A.; Atzori, E.M.; Delogu, I.; Collu, G.; Loddo, R. Inhibitors of Yellow Fever Virus Replication Based on 1,3,5-Triphenyl-4,5-Dihydropyrazole Scaffold: Design, Synthesis and Antiviral Evaluation. Eur. J. Med. Chem. 2017, 141, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Hassan, G.S.; Rahman, D.E.A.; Saleh, D.O.; Jaleel, G.A.R.A. Benzofuran-Morpholinomethyl-Pyrazoline Hybrids as a New Class of Vasorelaxant Agents: Synthesis and Quantitative Structure-Activity Relationship Study. Chem. Pharm. Bull. 2014, 62, 1238–1251. [Google Scholar] [CrossRef]
- Shin, S.Y.; Ahn, S.; Yoon, H.; Jung, H.; Jung, Y.; Koh, D.; Lee, Y.H.; Lim, Y. Colorectal Anticancer Activities of Polymethoxylated 3-Naphthyl-5-Phenylpyrazoline-Carbothioamides. Bioorg. Med. Chem. Lett. 2016, 26, 4301–4309. [Google Scholar] [CrossRef]
- Kim, B.S.; Shin, S.Y.; Ahn, S.; Koh, D.; Lee, Y.H.; Lim, Y. Biological Evaluation of 2-Pyrazolinyl-1-Carbothioamide Derivatives against HCT116 Human Colorectal Cancer Cell Lines and Elucidation on QSAR and Molecular Binding Modes. Bioorg. Med. Chem. 2017, 25, 5423–5432. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, B.S.; Ahn, S.; Koh, D.; Lee, Y.H.; Shin, S.Y.; Lim, Y. Anticancer and Structure-Activity Relationship Evaluation of 3-(Naphthalen-2-Yl)-N,5-Diphenyl-Pyrazoline-1-Carbothioamide Analogs of Chalcone. Bioorg. Chem. 2016, 68, 166–176. [Google Scholar] [CrossRef]
- Wang, H.; Zheng, J.; Xu, W.; Chen, C.; Wei, D.; Ni, W.; Pan, Y. A New Series of Cytotoxic Pyrazoline Derivatives as Potential Anticancer Agents That Induce Cell Cycle Arrest and Apoptosis. Molecules 2017, 22, 1635. [Google Scholar] [CrossRef]
- Zaki, Y.H.; Al-Gendey, M.S.; Abdelhamid, A.O. A Facile Synthesis, and Antimicrobial and Anticancer Activities of Some Pyridines, Thioamides, Thiazole, Urea, Quinazoline, β-Naphthyl Carbamate, and Pyrano[2,3-d]Thiazole Derivatives. Chem. Cent. J. 2018, 12, 1–14. [Google Scholar] [CrossRef]
- Siddiqui, N.; Alam, P.; Ahsan, W. Design, Synthesis, and In-Vivo Pharmacological Screening of N,3-(Substituted Diphenyl)-5-Phenyl-1 H-Pyrazoline-1- Carbothioamide Derivatives. Arch. Pharm. 2009, 342, 173–181. [Google Scholar] [CrossRef]
- Mathew, B.; Ahsan, M.J. Carboxamide/Carbothioamide Anticonvulsant Analogues Towards GABA-Aminotransferase- An in Silico Approach. Cent. Nerv. Syst. Agents Med. Chem. 2014, 14, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, M.; Jain, P. Synthetic and Biological Studies of Pyrazolines and Related Heterocyclic Compounds. Arab. J. Chem. 2014, 7, 553–596. [Google Scholar] [CrossRef]
- Abdelhamid, A.O.; El Sayed, I.E.; Hussein, M.Z.; Mangoud, M.M. Synthesis and Antimicrobial Activity of Some New Thiadiazoles, Thioamides, 5-Arylazothiazoles and Pyrimido[4,5-d][1,2,4]Triazolo[4,3-a]Pyrimidines. Molecules 2016, 21, 1072. [Google Scholar] [CrossRef] [PubMed]
- Alex, J.M.; Kumar, R. 4,5-Dihydro-1H-Pyrazole: An Indispensable Scaffold. J. Enzyme Inhib. Med. Chem. 2014, 29, 427–442. [Google Scholar] [CrossRef] [PubMed]
- Castaño, L.F.; Cuartas, V.; Bernal, A.; Insuasty, A.; Guzman, J.; Vidal, O.; Rubio, V.; Puerto, G.; Lukáč, P.; Vimberg, V.; et al. New Chalcone-Sulfonamide Hybrids Exhibiting Anticancer and Antituberculosis Activity. Eur. J. Med. Chem. 2019, 176, 50–60. [Google Scholar] [CrossRef]
- Riaz, S.; Khan, I.U.; Bajda, M.; Ashraf, M.; Shaukat, A.; Rehman, T.U.; Mutahir, S.; Hussain, S.; Yar, M.; Khan, I.U.; et al. Pyridine Sulfonamide as a Small Key Organic Molecule for the Potential Treatment of Type-II Diabetes Mellitus and Alzheimer’s Disease: In Vitro Studies against Yeast α-Glucosidase, Acetylcholinesterase and Butyrylcholinesterase. Bioorg. Chem. 2015, 63, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Mótyán, G.; Kovács, F.; Wölfling, J.; Gyovai, A.; Zupkó, I.; Frank, É. Microwave-Assisted Stereoselective Approach to Novel Steroidal Ring D-Fused 2-Pyrazolines and an Evaluation of Their Cell-Growth Inhibitory Effects in Vitro. Steroids 2016, 112, 36–46. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef]
- Zhao, L.; Wang, Y.; Cao, D.; Chen, T.; Wang, Q.; Li, Y.; Xu, Y.; Zhang, N.; Wang, X.; Chen, D.; et al. Fragment-Based Drug Discovery of 2-Thiazolidinones as BRD4 Inhibitors: Structure-Based Optimization. J. Med. Chem. 2015, 58, 1281–1297. [Google Scholar] [CrossRef]
- Evangelopoulos, D.; Bhakta, S. Rapid methods for testing inhibitors of mycobacterial growth. in Antibiotic Resistance Protocols. Methods Mol. Biol. 2010, 642, 193–201. [Google Scholar] [CrossRef]
- Brudey, K.; Driscoll, J.R.; Rigouts, L.; Prodinger, W.M.; Gori, A.; Al-Hajoj, S.A.; Allix, C.; Aristimuño, L.; Arora, J.; Baumanis, V.; et al. Mycobacterium tuberculosis complex genetic diversity: Mining the fourth international spoligotyping database (SpolDB4) for classification, population genetics and epidemiology. BMC Microbiol. 2006, 6, 1–17. [Google Scholar] [CrossRef]
- Puerto, G.; Erazo, L.; Wintaco, M.; Castro, C.; Ribón, W.; Guerrero, M.I. Mycobacterium tuberculosis Genotypes Determined by Spoligotyping to Be Circulating in Colombia between 1999 and 2012 and Their Possible Associations with Transmission and Susceptibility to First-Line Drugs. PLoS ONE 2015, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Realpe, T.; Correa, N.; Rozo, J.C.; Ferro, B.E.; Gómez, V.; Zapata, E.; Wellman, R.; Puerta, G.; Castro, C.; Nieto, L.M.; et al. Population Structure among Mycobacterium tuberculosis Isolates from Pulmonary Tuberculosis Patients in Colombia. PLoS ONE 2014, 9, e93848. [Google Scholar] [CrossRef] [PubMed]
- Reynaud, Y.; Millet, J.; Rastogi, N. Genetic Structuration, Demography and Evolutionary History of Mycobacterium tuberculosis LAM9 Sublineage in the Americas as Two Distinct Subpopulations Revealed by Bayesian Analyses. PLoS ONE 2015, 10, 1–15. [Google Scholar] [CrossRef]
- Cubillos, A.; Sandoval, A.; Ritacco, V.; López, B.; Robledo, J.; Correa, N.; Hernandez-Neuta, I.; Zambrano, M.M.; Del Portillo, P. Genomic signatures of the Haarlem lineage of Mycobacterium tuberculosis: Implications of strain genetic variation in drug and vaccine development. J. Clin. Microbiol. 2010, 48, 3614–3623. [Google Scholar] [CrossRef]
- National Cancer Insititute. DCTD Division of Cancer Treatment & Diagnosis. Available online: https://dtp.cancer.gov/discovery_development/nci-60/methodology.htm (accessed on 27 June 2022).
- Guzman, J.D.; Mortazavi, P.N.; Munshi, T.; Evangelopoulos, D.; McHugh, T.D.; Gibbons, S.; Malkinson, J.; Bhakta, S. 2-Hydroxy-Substituted Cinnamic Acids and Acetanilides Are Selective Growth Inhibitors of Mycobacterium Tuberculosis. Med. Chem. Comm. 2014, 5, 47–50. [Google Scholar] [CrossRef]
- Chen, T.; Li, Q.; Guo, L.; Yu, L.; Li, Z.; Guo, H.; Li, H.; Zhao, M.; Chen, L.; Chen, X.; et al. Lower cytotoxicity, high stability, and long-term antibacterial activity of a poly (methacrylic acid)/isoniazid/rifampin nanogel against multidrug-resistant intestinal Mycobacterium tuberculosis. Mater. Sci. Eng. C 2016, 58, 659–665. [Google Scholar] [CrossRef]
- Kabongo, P.N.; Eloff, J.N.; Obi, C.L.; Mcgaw, L.J. Antimycobacterial Activity and Low Cytotoxicity of Leaf Extracts of Some African Anacardiaceae Tree Species. Phytother. Res. 2016, 30, 2001–2011. [Google Scholar] [CrossRef]
- Ying, R.; Huang, X.; Gao, Y.; Wang, J.; Liu, Y.; Sha, W.; Yang, H. In vitro Synergism of Six Antituberculosis Agents Against Drug-Resistant Mycobacterium tuberculosis Isolated from Retreatment Tuberculosis Patients. Infect. Drug Resist. 2021, 14, 3729–3736. [Google Scholar] [CrossRef]
- Caleffi, K.R.; Maltempe, F.G.; Siqueira, V.L.D.; Cardoso, R.F. Fast detection of drug interaction in Mycobacterium tuberculosis by a checkerboard resazurin method. Tuberculosis 2013, 93, 660–663. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Panel/Cell Line | Compound | |||||||
|---|---|---|---|---|---|---|---|---|
| 17a | 17b | 17c | 18e | |||||
| IC50 b | LC50 c | IC50 | LC50 | IC50 | LC50 | IC50 | LC50 | |
| Leukemia | ||||||||
| CCRF-CEM | 3.93 | >100 | 1.52 | >100 | 3.99 | >100 | 1.69 | >100 |
| HL-60(TB) | 2.27 | >100 | 1.91 | >100 | 2.52 | >100 | 1.60 | >100 |
| K-562 | 1.50 | >100 | 1.85 | >100 | 2.43 | >100 | 1.30 | >100 |
| MOLT-4 | 10.1 | >100 | 2.83 | >100 | 3.07 | >100 | 2.04 | >100 |
| RPMI-8226 | 2.98 | >100 | 1.86 | >100 | 3.20 | >100 | 2.52 | >100 |
| SR | 1.73 | >100 | 2.11 | >100 | 2.26 | >100 | 0.99 | >100 |
| Non-Small Cell Lung Cancer | ||||||||
| A549/ATCC | 10.5 | 49.0 | 3.17 | >100 | 2.75 | 34.8 | 3.11 | >100 |
| EKVX | 14.2 | 53.6 | 3.88 | >100 | 4.33 | 43.3 | 2.69 | >100 |
| HOP-62 | 9.51 | 51.9 | 2.5 | 25.9 | 2.90 | 29.5 | 3.77 | >100 |
| HOP-92 | 2.06 | 37.1 | 1.46 | 24.9 | 2.30 | 35.4 | >100 | >100 |
| NCI-H226 | 10.0 | 69.2 | 2.56 | >100 | 4.14 | 90.8 | 2.61 | >100 |
| NCI-H322M | 10.7 | 47.6 | 2.31 | 38.7 | 2.87 | 34.7 | 3.00 | >100 |
| NCI-H460 | 1.95 | 51.2 | 2.84 | >100 | 2.24 | 44.3 | 1.95 | >100 |
| NCI-H522 | 6.56 | 49.7 | 1.63 | 7.14 | 2.48 | 32.5 | 1.66 | 8.59 |
| Colon Cancer | ||||||||
| COLO 205 | 10.6 | 54.2 | 1.66 | 5.88 | 3.33 | 41.7 | 4.46 | 8.39 |
| HCC-2998 | 12.5 | 54.7 | 2.41 | 22.3 | 2.39 | 22.3 | 3.54 | 6.35 |
| HCT-116 | 1.49 | 67.2 | 0.75 | 5.13 | 30.6 | 58.0 | 2.82 | 6.47 |
| HCT-15 | 3.55 | 57.5 | 1.46 | >100 | 2.56 | 74.8 | 3.98 | >100 |
| HT29 | 4.02 | 42.5 | 2.18 | 9.97 | 3.50 | 64.7 | 5.92 | >100 |
| KM12 | 3.31 | 62.3 | 1.82 | >100 | 3.13 | >100 | 4.25 | 9.40 |
| SW-620 | 2.68 | 48.5 | 2.49 | 95.9 | 3.43 | 75.8 | 25.1 | >100 |
| CNS Cancer | ||||||||
| SF-268 | 5.88 | 62.7 | 3.75 | >100 | 3.20 | >100 | 7.70 | >100 |
| SF-295 | 15.7 | 58.2 | 3.88 | 57.6 | 3.89 | 43.2 | 35.4 | >100 |
| SF-539 | 2.44 | 30.1 | 2.14 | 12.6 | 2.17 | 14.4 | 3.61 | 7.51 |
| SNB-19 | 14.0 | 58.0 | 3.17 | >100 | 3.65 | 45.3 | 5.71 | 45.7 |
| SNB-75 | 2.08 | 26.9 | 1.50 | 6.95 | 1.84 | 18.2 | 8.53 | >100 |
| U251 | 2.92 | 39.2 | 2.18 | >100 | 2.66 | 32.7 | 3.68 | 7.61 |
| Melanoma | ||||||||
| LOX IMVI | 0.34 | 41.4 | 0.73 | 7.03 | 0.54 | 76.2 | 2.89 | 5.79 |
| MALME-3M | 13.9 | 61.9 | 2.79 | 87.4 | 8.55 | 59.5 | 7.18 | >100 |
| M14 | 7.51 | 60.5 | 1.47 | 6.70 | 3.25 | 56.0 | 5.45 | >100 |
| MDA-MB-435 | 2.12 | 35.9 | 2.08 | 29.3 | 2.27 | 25.4 | 7.51 | >100 |
| SK-MEL-2 | 12.6 | 55.9 | 11.5 | 57.2 | 11.5 | 55.7 | 4.65 | 27.6 |
| SK-MEL-28 | 12.5 | 51.0 | 3.03 | 33.4 | 4.09 | 40.8 | 3.54 | 6.88 |
| SK-MEL-5 | 17.6 | 59.7 | 4.93 | 43.0 | 7.81 | 49.4 | 3.64 | 7.19 |
| UACC-257 | 9.96 | 47.8 | 5.46 | 71.6 | 3.86 | 41.9 | 3.89 | 9.14 |
| UACC-62 | 12.2 | 59.4 | 2.67 | 40.8 | 3.63 | 46.2 | 4.94 | >100 |
| Ovarian Cancer | ||||||||
| IGROV1 | 9.74 | 77.0 | 2.95 | >100 | 3.56 | 72.4 | 9.05 | >100 |
| OVCAR-3 | 2.71 | 38.3 | 3.51 | 66.8 | 3.05 | >100 | 6.62 | >100 |
| OVCAR-4 | 11.8 | 55.9 | 3.54 | >100 | 3.66 | 53.2 | >100 | >100 |
| OVCAR-5 | 13.2 | 53.7 | 3.18 | 39.0 | 2.57 | 27.2 | 6.80 | >100 |
| OVCAR-8 | 11.3 | 51.3 | 3.01 | >100 | 2.17 | 15.8 | >100 | >100 |
| NCI/ADR-RES | 15.5 | >100 | 3.97 | >100 | 5.25 | >100 | >100 | >100 |
| SK-OV-3 | 14.4 | 52.9 | 2.80 | 35.4 | 3.18 | 32.5 | 13.0 | >100 |
| Renal Cancer | ||||||||
| 786-0 | 4.82 | 63.2 | 1.90 | 9.83 | 3.85 | >100 | 5.66 | >100 |
| A498 | 16.9 | 55.8 | 18.6 | 62.2 | 16.2 | 58.1 | 7.23 | 65.5 |
| ACHN | 6.99 | 44.8 | 3.16 | 33.4 | 3.86 | 37.3 | 6.16 | >100 |
| CAKI-1 | 1.86 | 39.5 | 1.89 | 32.9 | 2.42 | 35.2 | >100 | >100 |
| RXF 393 | 1.90 | 30.0 | 1.62 | 6.89 | 2.24 | 28.8 | 2.83 | 6.44 |
| SN12C | 3.49 | 43.8 | 2.48 | 60.2 | 2.95 | 38.8 | 13.0 | >100 |
| TK-10 | 12.9 | 51.5 | 2.91 | 33.4 | 3.41 | 39.6 | 75.0 | >100 |
| UO-31 | 4.76 | 43.5 | 2.36 | 33.1 | 3.46 | 38.1 | 2.92 | 5.51 |
| Prostate Cancer | ||||||||
| PC-3 | 5.95 | 50.4 | 1.60 | 20.8 | 3.42 | 55.1 | >100 | >100 |
| DU-145 | 1.88 | 38.1 | 3.44 | 42.2 | 4.23 | 65.1 | 14.8 | >100 |
| Breast Cancer | ||||||||
| MCF7 | 0.97 | 62.5 | 4.11 | 40.3 | 2.48 | 54.1 | 4.36 | 35.5 |
| MDA-MB-231/ATCC | 11.9 | 61.6 | 18.9 | >100 | 3.41 | 60.0 | >100 | >100 |
| HS 578T | 5.89 | >100 | >100 | >100 | 3.50 | >100 | >100 | >100 |
| BT-549 | 2.65 | 40.6 | 2.73 | 5.97 | 2.18 | 31.6 | 2.92 | 6.24 |
| T-47D | 2.09 | 51.6 | 12.4 | 64.6 | 2.32 | 44.8 | >100 | >100 |
| MDA-MB-468 | 1.20 | 43.9 | 3.08 | 6.54 | 1.96 | 54.3 | 3.66 | 8.84 |
| Compound | MIC Against M. bovis BCG (µM) | MIC Against M. tuberculosis H37Rv (µM) | Cytotoxicity IC50 on Vero Cell Line (µM) | Selectivity Index (SI) (SI = IC50/MIC) |
|---|---|---|---|---|
| 17a | >29 | 29 | 1.06 ± 1.21 | 0.04 |
| 17f | >25 | 25 | 1.70 ± 1.74 | 0.07 |
| 19a | >23 | 23 | 6.32 ± 0.82 | 0.28 |
| 19d | >21 | 21 | 2.19 ± 0.68 | 0.10 |
| 20a | 11 | 11 | 6.05 ± 1.39 | 0.28 |
| 20d | >20 | 20 | 1.00 ± 1.20 | 0.05 |
| 20e | 9.0 | 9.0 | 0.26 ± 0.69 | 0.03 |
| 20f | 20 | 9.8 | 2.55 ± 0.93 | 0.26 |
| Isoniazid | 0.36 | 0.36 | nd |
| Compound | a | b | c | d | e | f |
|---|---|---|---|---|---|---|
| 17a | 29 | >29 | >29 | 29 | >29 | 29 |
| 17f | >2 | >2 | >2 | >2 | >25 | >25 |
| 19a | >23 | >23 | 23 | 23 | >23 | <11 |
| 19d | 21 | >21 | 21 | 21 | >21 | 21 |
| 20a | >21 | >21 | 21 | <11 | >21 | <11 * |
| 20d | >20 | >20 | >20 | >20 | >20 | 20 |
| 20e | >18 | >18 | <9.0 * | <9.0 * | >18 | <9.0 * |
| 20f | >20 | >20 | <10 | 20 | >20 | <10 * |
| Levofloxacin | 2.8 | 2.8 | 2.8 | 2.8 | 2.8 | 2.8 |
| Combination | MIC mg/L | FICI Index | |
|---|---|---|---|
| Alone | Combination | ||
| RIF | 0.19 | 0.048 | 1.25 |
| 20a | 5 | 5 | |
| INH | 0.012 | 0.006 | 0.75 |
| 20a | 5 | 1.25 | |
| LVX | 0.5 | 0.5 | 1.25 |
| 20a | 5 | 1.25 | |
| AMK | 1.5 | 1.5 | 2.00 |
| 20a | 5 | 5 | |
| RIF | 0.39 | 0.048 | 0.37 * |
| 20f | 5 | 1.25 | |
| INH | 0.012 | 0.006 | 1.00 |
| 20f | 5 | 2.5 | |
| LVX | 0.5 | 0.25 | 1.00 |
| 20f | 5 | 2.5 | |
| AMK | 1.5 | 1.5 | 2.00 |
| 20f | 5 | 5 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castaño, L.F.; Quiroga, J.; Abonia, R.; Insuasty, D.; Vidal, O.M.; Seña, R.; Rubio, V.; Puerto, G.; Nogueras, M.; Cobo, J.; et al. Synthesis, Anticancer and Antitubercular Properties of New Chalcones and Their Nitrogen-Containing Five-Membered Heterocyclic Hybrids Bearing Sulfonamide Moiety. Int. J. Mol. Sci. 2022, 23, 12589. https://doi.org/10.3390/ijms232012589
Castaño LF, Quiroga J, Abonia R, Insuasty D, Vidal OM, Seña R, Rubio V, Puerto G, Nogueras M, Cobo J, et al. Synthesis, Anticancer and Antitubercular Properties of New Chalcones and Their Nitrogen-Containing Five-Membered Heterocyclic Hybrids Bearing Sulfonamide Moiety. International Journal of Molecular Sciences. 2022; 23(20):12589. https://doi.org/10.3390/ijms232012589
Chicago/Turabian StyleCastaño, Lina Fernanda, Jairo Quiroga, Rodrigo Abonia, Daniel Insuasty, Oscar M. Vidal, Rosalia Seña, Vivian Rubio, Gloria Puerto, Manuel Nogueras, Justo Cobo, and et al. 2022. "Synthesis, Anticancer and Antitubercular Properties of New Chalcones and Their Nitrogen-Containing Five-Membered Heterocyclic Hybrids Bearing Sulfonamide Moiety" International Journal of Molecular Sciences 23, no. 20: 12589. https://doi.org/10.3390/ijms232012589
APA StyleCastaño, L. F., Quiroga, J., Abonia, R., Insuasty, D., Vidal, O. M., Seña, R., Rubio, V., Puerto, G., Nogueras, M., Cobo, J., Guzman, J., Insuasty, A., & Insuasty, B. (2022). Synthesis, Anticancer and Antitubercular Properties of New Chalcones and Their Nitrogen-Containing Five-Membered Heterocyclic Hybrids Bearing Sulfonamide Moiety. International Journal of Molecular Sciences, 23(20), 12589. https://doi.org/10.3390/ijms232012589