
Candesartan Does Not Activate PPARγ and Its Target Genes in Early Gestation Trophoblasts

 , , and
, , and 
Abstract
1. Introduction
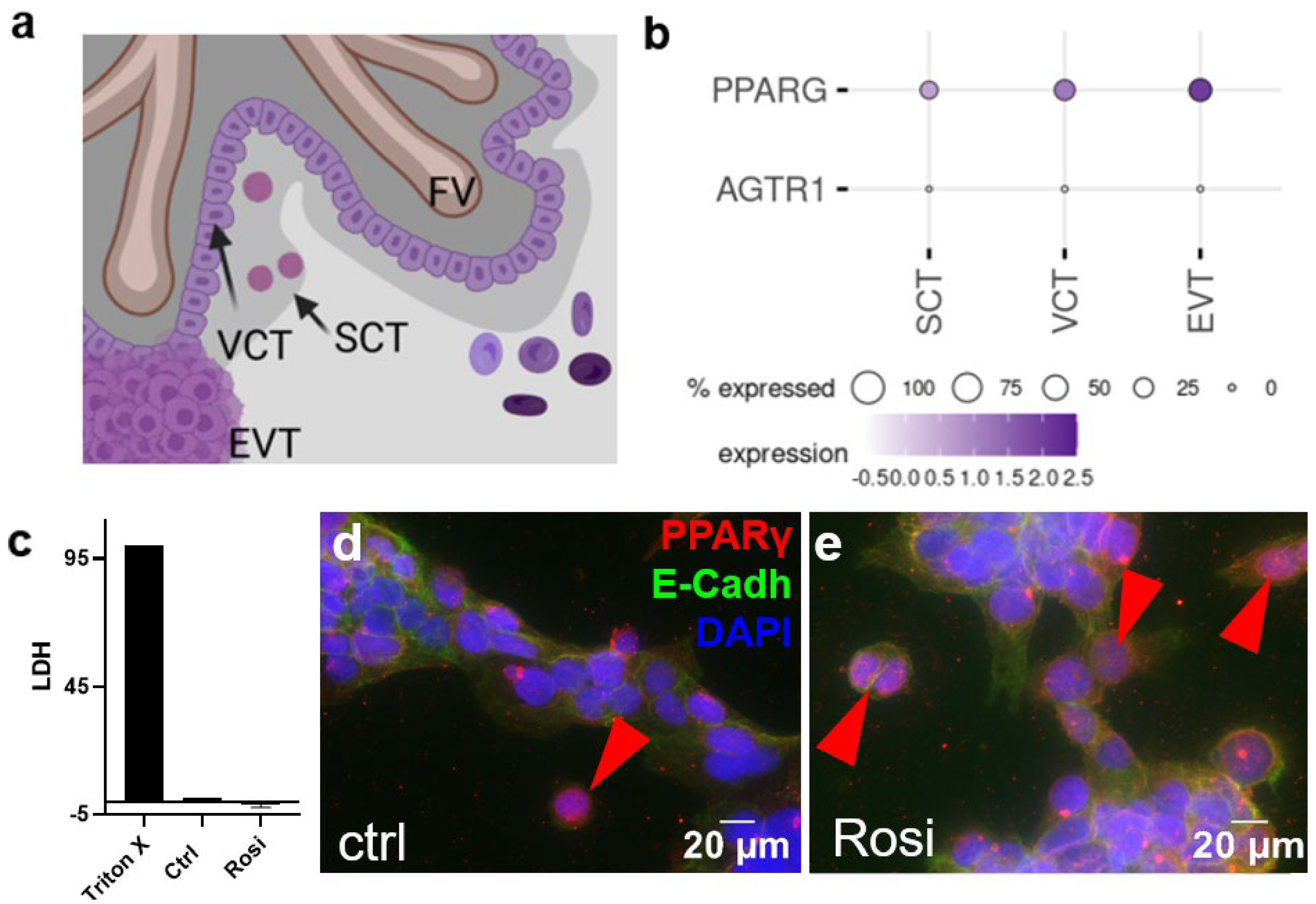
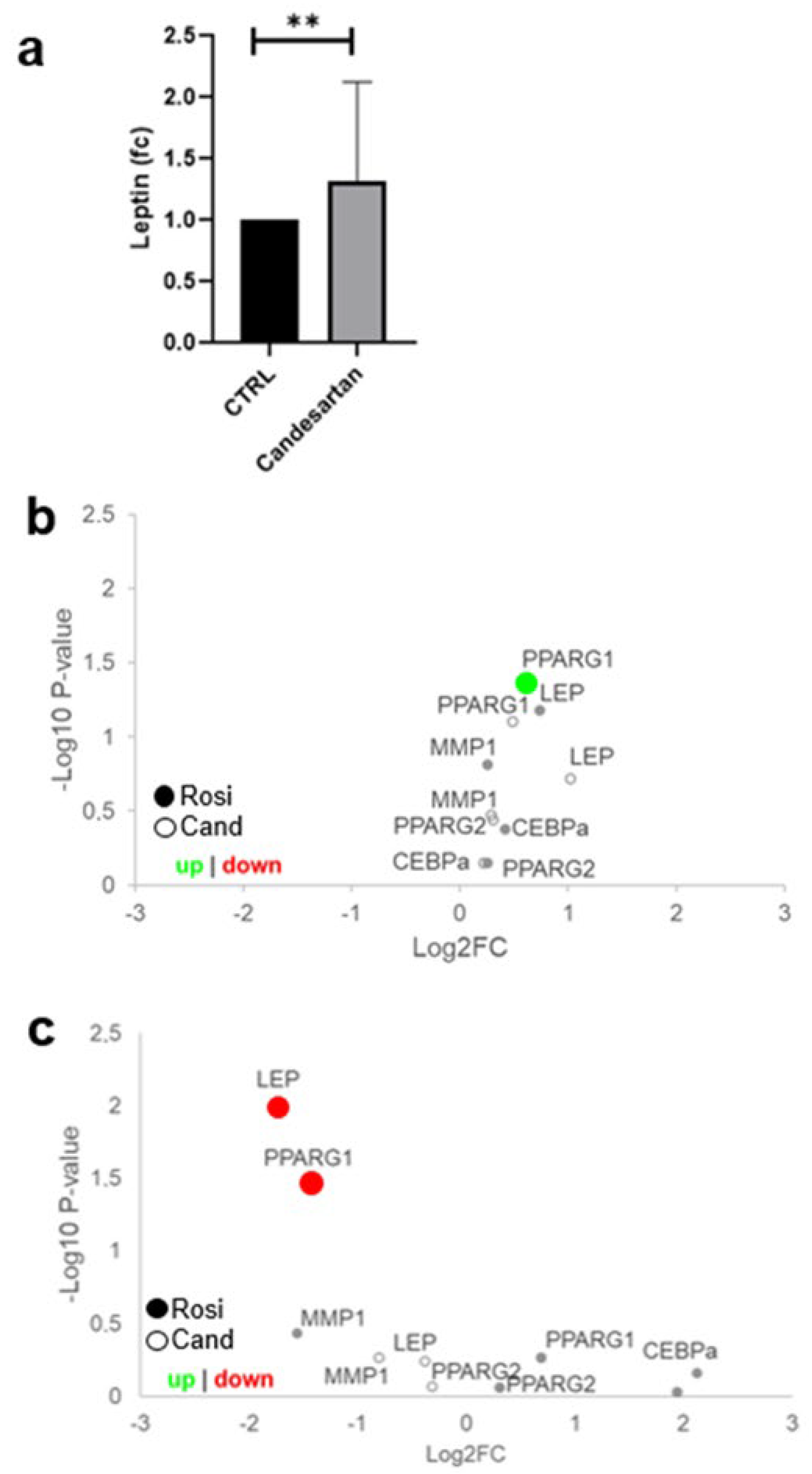
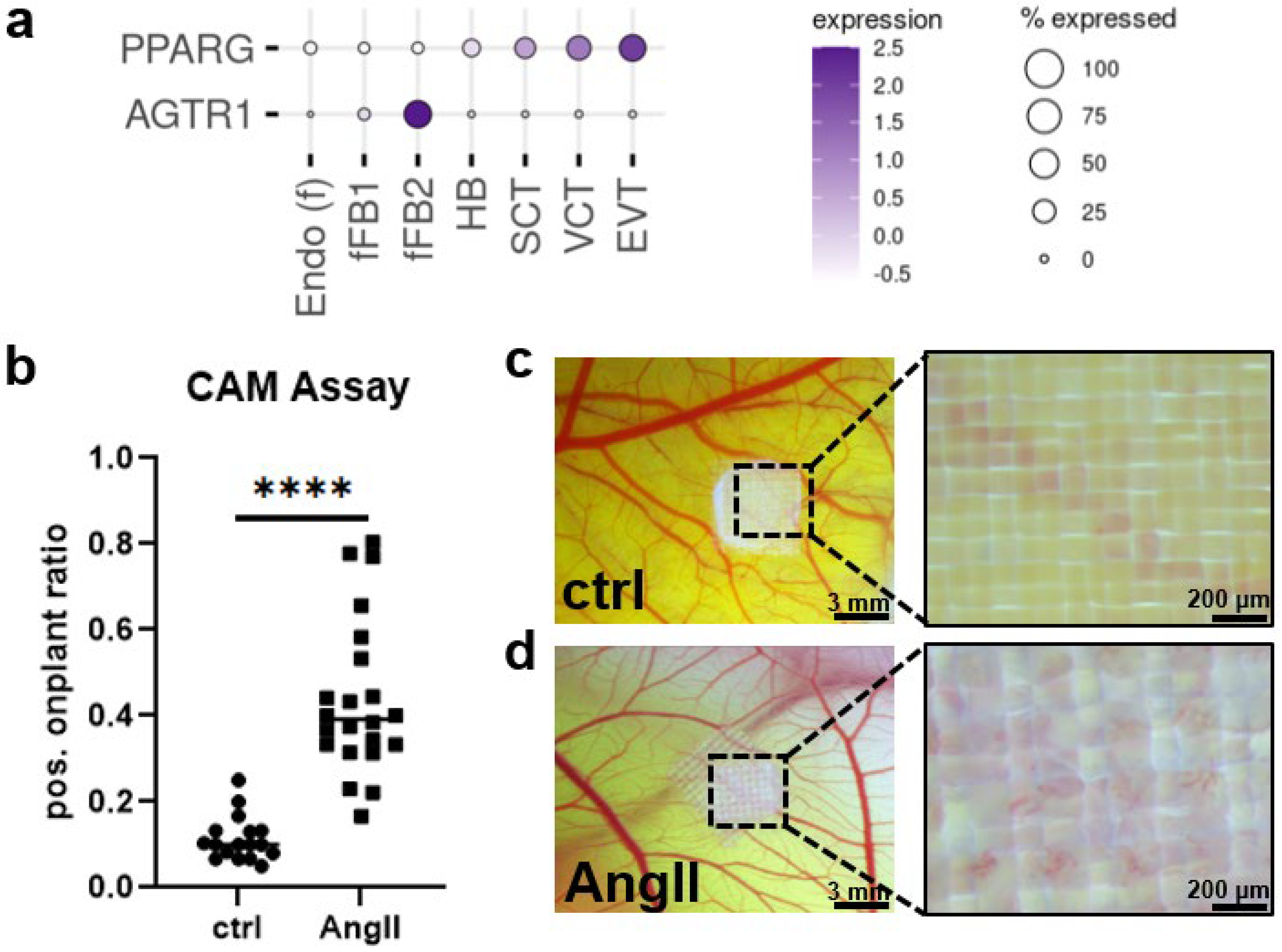
2. Results
3. Discussion
4. Methods and Materials
4.1. Cell Culture
4.2. Explant Culture
4.3. Protein Analysis
4.4. RNA Analysis
4.5. Immunofluorescence Staining and Imaging
4.6. Chicken Chorioallantoic Membrane (CAM) Assay
4.7. LDH Assay
4.8. Leptin ELISA
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Elliott, W.J. Systemic Hypertension. Curr. Probl. Cardiol. 2007, 32, 201–259. Available online: https://www.sciencedirect.com/science/article/pii/S0146280607000035 (accessed on 1 January 2020). [CrossRef] [PubMed]
- Mann, J.F. Choice of Drug Therapy in Primary (essential) Hypertension: Recommendations. Up-To-Date. 2015, pp. 1–29. Available online: http://www.uptodate.com/contents/choice-of-drug-therapy-in-primary-essential-hypertension-recommendations?source=see_link (accessed on 1 January 2020).
- Statistik Austria. Österreichische Gesundheitsbefragung 2019. 2019. Available online: https://www.statistik.at/web_de/statistiken/menschen_und_gesellschaft/gesundheit/gesundheitsdeterminanten/rauchen/index.html (accessed on 1 January 2020).
- Simonetti, G.D.; Baumann, T.; Pachlopnik, J.M.; Von Vigier, R.O.; Bianchetti, M.G. Non-lethal fetal toxicity of the angiotensin receptor blocker candesartan. Pediatr. Nephrol. 2006, 21, 1329–1330. [Google Scholar] [CrossRef] [PubMed]
- Karthikeyan, V.J.; E Ferner, R.; Baghdadi, S.; A Lane, D.; Lip, G.Y.; Beevers, D.G. Are angiotensin-converting enzyme inhibitors and angiotensin receptor blockers safe in pregnancy: A report of ninety-one pregnancies. J. Hypertens. 2011, 29, 396–399. [Google Scholar] [CrossRef] [PubMed]
- Bullo, M.; Tschumi, S.; Bucher, B.S.; Bianchetti, M.G.; Simonetti, G.D. Pregnancy Outcome Following Exposure to Angioten-sin-Converting Enzyme Inhibitors or Angiotensin: A systematic review. Hypertension 2012, 60, 444–450. [Google Scholar] [CrossRef]
- Bald, M.; Holder, M.; Zieger, M.; Vochem, M.; Leichter, H.E. Increased renal echogenicity in a preterm neonate: Question. Pediatr. Nephrol. 2005, 20, 1664–1665. [Google Scholar] [CrossRef]
- Schaefer, C. Angiotensin II-receptor-antagonists: Further evidence of fetotoxicity but not teratogenicity. Birth Defects Res. Part A: Clin. Mol. Teratol. 2003, 67, 591–594. [Google Scholar] [CrossRef]
- Porta, M.; Hainer, J.W.; Jansson, S.-O.; Malm, A.; Bilous, R.; Chaturvedi, N.; Fuller, J.H.; Klein, R.; Orchard, T.; Parving, H.-H.; et al. Exposure to candesartan during the first trimester of pregnancy in type 1 diabetes: Experience from the placebo-controlled diabetic retinopathy candesartan trials. Diabetologia 2011, 54, 1298–1303. [Google Scholar] [CrossRef][Green Version]
- Polifka, J.E. Is there an embryopathy associated with first-trimester exposure to angiotensin-converting enzyme inhibitors and angiotensin receptor antagonists? A critical review of the evidence. Birth Defects Res. Part A: Clin. Mol. Teratol. 2012, 94, 576–598. [Google Scholar] [CrossRef]
- Lu, Y.; Chen, R.; Cai, J.; Huang, Z.; Yuan, H. The management of hypertension in women planning for pregnancy. Br. Med Bull. 2018, 128, 75–84. [Google Scholar] [CrossRef]
- Agrawal, A.; Wenger, N.K. Hypertension During Pregnancy. Curr. Hypertens. Rep. 2020, 22, 1–9. [Google Scholar] [CrossRef]
- Lo, J.O.; Mission, J.F.; Caughey, A.B. Hypertensive disease of pregnancy and maternal mortality. Curr. Opin. Obstet. Gynecol. 2013, 25, 124–132. [Google Scholar] [CrossRef]
- Vestgaard, M.; Sommer, M.C.; Ringholm, L.; Damm, P.; Mathiesen, E. Prediction of preeclampsia in type 1 diabetes in early pregnancy by clinical predictors: A systematic review. J. Matern. Neonatal Med. 2017, 31, 1933–1939. [Google Scholar] [CrossRef]
- Roberts, J.M.; Redman, C.W. Global Pregnancy Collaboration symposium: Prepregnancy and very early pregnancy antecedents of adverse pregnancy outcomes: Overview and recommendations. Placenta 2017, 60, 103–109. [Google Scholar] [CrossRef]
- August, A.P.; Sibai, B.M. Preeclampsia: Clinical features and diagnosis. Up-To-Date. 2022, pp. 1–57. Available online: https://www.uptodate.com/contents/preeclampsia-clinical-features-and-diagnosis (accessed on 9 October 2022).
- Neuhaus, S.; Neuhaus, C.; Weigand, M.A.; Bremerich, D. Spezielle intensivmedizinische Krankheitsbilder der schwangeren Patientin. Der Anaesthesist 2021, 70, 717–730. [Google Scholar] [CrossRef]
- Sedgh, G.; Singh, S.; Hussain, R. Intended and Unintended Pregnancies Worldwide in 2012 and Recent Trends. Stud. Fam. Plan. 2014, 45, 301–314. [Google Scholar] [CrossRef]
- Huppertz, B.; Schleußner, E. Die Plazenta: Grundlagen und Klinische Bedeutung; Springer: Berlin/Heidelberg, Germany, 2018. [Google Scholar]
- Fournier, T.; Handschuh, K.; Tsatsaris, V.; Evain-Brion, D. Involvement of PPARγ in Human Trophoblast Invasion. Placenta 2007, 28, S76–S81. [Google Scholar] [CrossRef]
- Pham, J.; Rajan, K.A.N.; Li, P.; Parast, M.M. The role of Sirtuin1–PPARγ axis in placental development and function. J. Mol. Endo-Crinol. 2018, 60, R201–R212. [Google Scholar] [CrossRef]
- Janani, C.; Kumari, B.R. PPAR gamma gene—A review. Diabetes Metab. Syndr. Clin. Res. Rev. 2015, 9, 46–50. [Google Scholar] [CrossRef]
- Mukherjee, R.; Jow, L.; Croston, G.E.; Paterniti, J.R. Identification, characterization, and tissue distribution of human peroxisome proliferator-activated receptor (PPAR) isoforms PPARγ2 versus PPARγ1 and activation with retinoid X receptor agonists and antagonists. J. Biol. Chem. 1997, 272, 8071–8076. [Google Scholar] [CrossRef]
- Gleiter, C.H.; E Mörike, K. Clinical Pharmacokinetics of Candesartan. Clin. Pharmacokinet. 2002, 41, 7–17. [Google Scholar] [CrossRef]
- Yan, W.-H.; Pan, C.-Y.; Dou, J.-T.; Meng, J.-H.; Wang, B.-A.; Mu, Y.-M. Candesartan cilexetil prevents diet-induced insulin resistance via peroxisome proliferator-activated receptor-γ activation in an obese rat model. Exp. Ther. Med. 2016, 12, 272–278. [Google Scholar] [CrossRef]
- Chmiel-Perzyńska, I.; Perzyński, A.; Olajossy, B.; Gil-Kulik, P.; Kocki, J.; Urbańska, E.M. Losartan Reverses Hippocampal Increase of Kynurenic Acid in Type 1 Diabetic Rats: A Novel Procognitive Aspect of Sartan Action. J. Diabetes Res. 2019, 2019, 4957879. [Google Scholar] [CrossRef]
- Wang, S.; Dougherty, E.J.R.L.D. Effects of telmisartan on TNFα induced PPARγ phosphorylation and insulin resistance in adipocytes. Pharm. Res. 2016, 111, 3044–3049. [Google Scholar]
- Villapol, S.; Balarezo, M.G.; Affram, K.; Saavedra, J.M.; Symes, A. Neurorestoration after traumatic brain injury through angiotensin II receptor blockage. Brain 2015, 138, 3299–3315. [Google Scholar] [CrossRef] [PubMed]
- Haaland, K. Angiotensin II receptor antagonists against migraine in pregnancy: Fatal outcome. J. Headache Pain 2010, 11, 167–169. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Serreau, R.; Luton, D.; Macher, M.-A.; Delezoide, A.-L.; Garel, C.; Jacqz-Aigrain, E. Developmental toxicity of the angiotensin II type 1 receptor antagonists during human pregnancy: A report of 10 cases. BJOG: Int. J. Obstet. Gynaecol. 2005, 112, 710–712. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Okuda, M.; Ishikawa, H.; Takahashi, T.; Hirahara, F. Oligohydramnios and pulmonary hypoplasia: A case in which in-volvement of an angiotensin II receptor antagonist was suspected. J. Obstet. Gynaecol. Res. 2008, 34, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Nonn, O.; Fischer, C.; Geisberger, S.; El-Heliebi, A.; Kroneis, T.; Forstner, D.; Desoye, G.; Staff, A.C.; Sugulle, M.; Dechend, R.; et al. Maternal Angiotensin Increases Placental Leptin in Early Gestation via an Alternative Renin-Angiotensin System Pathway: Suggesting a Link to Preeclampsia. Hypertension 2021, 77, 1723–1736. [Google Scholar] [CrossRef]
- Vento-Tormo, R.; Efremova, M.; Botting, R.A.; Turco, M.Y.; Vento-Tormo, M.; Meyer, K.B.; Park, J.-E.; Stephenson, E.; Polański, K.; Goncalves, A.; et al. Single-cell reconstruction of the early maternal–fetal interface in humans. Nature 2018, 563, 347–353. [Google Scholar] [CrossRef]
- Pastuschek, J.; Nonn, O.; Gutiérrez-Samudio, R.N.; Murrieta-Coxca, J.M.; Müller, J.; Sanft, J.; Huppertz, B.; Markert, U.R.; Groten, T.; Morales-Prieto, D.M. Molecular characteristics of established trophoblast-derived cell lines. Placenta 2021, 108, 122–133. [Google Scholar] [CrossRef]
- Choy, M.; Manyonda, I. The phagocytic activity of human first trimester extravillous trophoblast. Hum. Reprod. 1998, 13, 2941–2949. [Google Scholar] [CrossRef]
- Nonn, O.; Güttler, J.; Forstner, D.; Maninger, S.; Zadora, J.; Balogh, A.; Frolova, A.; Glasner, A.; Herse, F.; Gauster, M. Placental CX3CL1 is Deregulated by Angiotensin II and Contributes to a Pro-Inflammatory Trophoblast-Monocyte Interaction. Int. J. Mol. Sci. 2019, 20, 641. [Google Scholar] [CrossRef]
- Ino, K.; Uehara, C.; Kikkawa, F.; Kajiyama, H.; Shibata, K.; Suzuki, T.; Khin, E.E.; Ito, M.; Takeuchi, M.; Itakura, A.; et al. Enhancement of aminopeptidase A expression during Angiotensin II-induced choriocarcinoma cell proliferation through AT1 receptor involving protein kinase C- and mitogen-activated protein kinase-dependent signaling pathway. J. Clin. Endocrinol. Metab. 2003, 88, 3973–3982. [Google Scholar] [CrossRef][Green Version]
- Qie, S.; Ran, Y.; Lu, X.; Su, W.; Li, W.; Xi, J.; Gong, W.; Liu, Z. Candesartan modulates microglia activation and polarization via NF-κB signaling pathway. Int. J. Immunopathol. Pharmacol. 2020, 34, 2058738420974900. [Google Scholar] [CrossRef]
- Alhusban, A.; Al-Azayzih, A.; Goc, A.; Gao, F.; Fagan, S.C.; Somanath, P.R. Clinically Relevant Doses of Candesartan Inhibit Growth of Prostate Tumor Xenografts In Vivo through Modulation of Tumor Angiogenesis. J. Pharmacol. Exp. Ther. 2014, 350, 635–645. [Google Scholar] [CrossRef]
- Levytska, K.; Drewlo, S.; Baczyk, D.; Kingdom, J. PPAR-γ Regulates Trophoblast Differentiation in the BeWo Cell Model. PPAR Res. 2014, 2014, 637251. [Google Scholar] [CrossRef]
- Kohan-Ghadr, H.-R.; A Kilburn, B.; Kadam, L.; Johnson, E.; Kolb, B.L.; Rodriguez-Kovacs, J.; Hertz, M.; Armant, D.R.; Drewlo, S. Rosiglitazone augments antioxidant response in the human trophoblast and prevents apoptosis. Biol. Reprod. 2018, 100, 479–494. [Google Scholar] [CrossRef]
- Kim, T.H.; Kim, M.G.; Shin, S.; Chi, Y.-H.; Paik, S.-H.; Lee, J.-H.; Yoo, S.D.; Youn, Y.S.; Bulitta, J.B.; Joo, S.H.; et al. Placental transfer and mammary excretion of a novel angiotensin receptor blocker fimasartan in rats. BMC Pharmacol. Toxicol. 2016, 17, 35. [Google Scholar] [CrossRef]
- Barak, Y.; Nelson, M.C.; Ong, E.S.; Jones, Y.Z.; Ruiz-Lozano, P.; Chien, K.R.; Koder, A.; Evans, R.M. PPARγ is required for placental, cardiac, and adipose tissue development. Mol Cell. 1999, 4, 585–595. [Google Scholar] [CrossRef]
- Tarrade, A.; Schoonjans, K.; Pavan, L.; Auwerx, J.; Rochette-Egly, C.; Evain-Brion, D.; Fournier, T. PPARγ/RXRα heterodimers control human trophoblast invasion. J. Clin. Endocrinol. Metab. 2001, 86, 5017–5024. [Google Scholar] [CrossRef][Green Version]
- Roberti, S.L.; Higa, R.; White, V.; Powell, T.L.; Jansson, T.; Jawerbaum, A. Critical role of mTOR, PPARγ and PPARδ signaling in regulating early pregnancy decidual function, embryo viability and feto-placental growth. Mol. Hum. Reprod. 2018, 24, 327–340. [Google Scholar] [CrossRef]
- Schaiff, W.T.; Bildirici, I.; Cheong, M.; Chern, P.L.; Nelson, D.M. Peroxisome Proliferator-Activated Receptor-γ and Retinoid X Re-ceptor Signaling Regulate Fatty Acid Uptake by Primary Human Placental Trophoblasts. J. Clin. Endocrinol. Metab. 2005, 90, 4267–4275. [Google Scholar] [CrossRef]
- Schaiff, W.T.; Knapp, F.F.; Barak, Y.; Biron-Shental, T.; Nelson, D.M.; Sadovsky, Y. Ligand-Activated Peroxisome Proliferator Activated Receptor γ Alters Placental Morphology and Placental Fatty Acid Uptake in Mice. Endocrinology 2007, 148, 3625–3634. [Google Scholar] [CrossRef]
- Perucca, E.; Crema, A. Plasma Protein Binding of Drugs in Pregnancy. Clin. Pharmacokinet. 1982, 7, 336–352. [Google Scholar] [CrossRef] [PubMed]
- Notarianni, L.J. Plasma Protein Binding of Drugs in Pregnancy and in Neonates. Clin. Pharmacokinet. 1990, 18, 20–36. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.I.; Green, D.J.; van den Anker, J.N.; Rakhmanina, N.Y.; Ahmadzia, H.K.; Momper, J.D.; Momper, J.D.; Park, K.; Burckart, G.J.; Dallmann, A.; et al. Mechanistic Modeling of Placental Drug Transfer in Humans: How Do Differences in Maternal/Fetal Fraction of Unbound Drug and Placental Influx/Efflux Transfer Rates Affect Fetal Pharmacokinetics? Front. Pediatr. 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Handschuh, K.; Guibourdenche, J.; Cocquebert, M.; Tsatsaris, V.; Vidaud, M.; Evain-Brion, D.; Fournier, T. Expression and Regulation by PPARγ of hCG α- and β-subunits: Comparison between Villous and Invasive Extravillous Trophoblastic Cells. Placenta 2009, 30, 1016–1022. [Google Scholar] [CrossRef]
- Elchalal, U.; Humphrey, R.G.; Smith, S.D.; Hu, C.; Sadovsky, Y.; Nelson, D.M. Troglitazone attenuates hypoxia-induced injury in cultured term human trophoblasts. Am. J. Obstet. Gynecol. 2004, 191, 2154–2159. [Google Scholar] [CrossRef] [PubMed]
- Schaiff, W.T.; Carlson, M.G.; Smith, S.D.; Levy, R.; Nelson, D.M.; Sadovsky, Y. Peroxisome Proliferator-Activated Receptor-g Modulates Differentiation of Human Trophoblast in a Ligand-Specific Manner. J. Clin. Endocrinol. Metab. 2000, 85, 3874–3881. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yun, Z.; Maecker, H.; Johnson, R.; Giaccia, A. Inhibition of PPAR gamma 2 gene expression by the HIF-1-regulated gene DEC1/Stra13: A mechanism for regulation of adipogenesis by hypoxia. Dev. Cell. 2002, 2, 331–341. [Google Scholar] [CrossRef]
- He, P.; Chen, Z.; Sun, Q.; Li, Y.; Gu, H.; Ni, X. Reduced Expression of 11β-Hydroxysteroid Dehydrogenase Type 2 in Preeclamptic Placentas Is Associated With Decreased PPARγ but Increased PPARα Expression. Endocrinology 2014, 155, 299–309. [Google Scholar] [CrossRef]
- Alwan, S.; Polifka, J.; Friedman, J. Angiotensin II receptor antagonist treatment during pregnancy. Birth Defects Res. Part A: Clin. Mol. Teratol. 2005, 73, 123–130. [Google Scholar] [CrossRef]
- Shanab, A.Y.; Elshaer, S.L.; El-Azab, M.F.; Soliman, S.; Sabbineni, H.; Matragoon, S.; Fagan, S.C.; El-Remessy, A.B. Candesartan stimulates reparative angio-genesis in ischemic retinopathy model: Role of hemeoxygenase-1 (HO-1). Angiogenesis 2015, 18, 137–150. [Google Scholar] [CrossRef]
- Lee, S.; Chae, I.; Kim, H.; Kang, D.; Kim, S.-H.; Kim, H.-S. Predictors of candesartan’s effect on vascular reactivity in patients with coronary artery disease. Cardiovasc. Ther. 2017, 35, e12291. [Google Scholar] [CrossRef]
- Andone, S.; Bajko, Z.; Motataianu, A.; Maier, S.; Barcutean, L.; Balasa, R. Neuroprotection in Stroke—Focus on the Renin-Angiotensin System: A Systematic Review. Int. J. Mol. Sci. 2022, 23, 3876. [Google Scholar] [CrossRef]
- Nishimura, Y.; Ito, T.; Saavedra, J.M. Angiotensin II AT 1 Blockade Normalizes Cerebrovascular Autoregulation and Reduces Cerebral Ischemia in Spontaneously Hypertensive Rats. Stroke 2000, 31, 2478–2486. [Google Scholar] [CrossRef]
- Alhusban, A.; Kozak, A.; Ergul, A.; Fagan, S.C. AT1 Receptor Antagonism Is Proangiogenic in the Brain: BDNF a Novel Mediator. J. Pharmacol. Exp. Ther. 2012, 344, 348–359. [Google Scholar] [CrossRef]
- Schaefer, C.; Hannemann, D.; Meister, R. Post-marketing surveillance system for drugs in pregnancy—15 years experience of ENTIS. Reprod. Toxicol. 2005, 20, 331–343. [Google Scholar] [CrossRef]
- Deryugina, E.I.; Quigley, J.P. Chapter 2 Chick Embryo Chorioallantoic Membrane Models to Quantify Angiogenesis Induced by Inflammatory and Tumor Cells or Purified Effector Molecules. Methods Enzymol. 2008, 444, 21–41. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence |
|---|---|
| TBP | F: 5‘-TGA CCC AGC ATC ACT GTT TC-3’ R: 5’-CCA GCA CAC TCT TCT CAG CA-3’ |
| ACTB | F: 5’-AAA GAC CTG TAC GCC AAC AC-3’ R: 5’-GTC ATA CTC CTG CCT GCT GAT-3’ |
| B2M | F: 5’-GAT GAG TAT GCC TGC CGT GT-3’ R: 5‘-TGT CTC GAT CCC ACT TAA CTA TCT-3’ |
| HPRT1 | F: 5’-GAA AGG GTG TTT ATT CCT CAT GAA-3’ R: 5’-CAA GCA GGT CAG CAA AGA ATT T-3’ |
| YWHAZ | F: 5’-GGT GGC CAA TAT GGG GAT GT-3’ R: 5‘-TCC CTT TTA TTC CCC GCC AG-3’ |
| GAPDH | F: 5’-ACC CAC TCC TCC ACC TTT-3’ R: 5’-CTG TTG CTG TAG CCA AAT-3’ |
| LEP | F: 5’-GTG CGG ATT CTT GTG GCT TT-3’ R: 5’-AGG AGA CTG ACT GCG TGT GT-3’ |
| FABP4 | F: 5’-AAG GCG TCA CTT CCA CGA GAG-3’ R: 5‘-AAT GCG AAC TTC AGT CCA GGT-3‘ |
| CEBPα | F: 5’-CTT GTG CCT TGG AA-3’ R: 5’-GCT GTA GCC TCG GAA AGG A-3’ |
| CD34 | F: 5’-CGG CCA TTC AGC AAG ACA AC-3’ R: 5’-TAG CAC GTG GTC AGA TGC AG-3‘ |
| PPARG1 | F: 5’-AAG GCC ATT TTC TCA AAC GA-3’ R: 5‘-AGG AGT GGG AGT GGT CTT CC-3‘ |
| PPARG2 | F: 5’-CCA TGC TGT TAT GGG TGA AA-3’ R: 5‘-TCA AAG GAG TGG GAG TGG TC-3‘ |
| hMMP1 | F: 5‘-CTC TGG AGT AAT GTC ACA CCT CT-3’ R: 5‘-TGT TGG TCC ACC TTT CAT CTT C-3’ |
| AGTR1 | F: 5‘-GCG CGG GTT TGA TAT TTG ACA-3’ R: 5‘-TCA AAT ACA CCT GGT GCC GA-3’ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neuper, L.; Kummer, D.; Forstner, D.; Guettler, J.; Ghaffari-Tabrizi-Wizsy, N.; Fischer, C.; Juch, H.; Nonn, O.; Gauster, M. Candesartan Does Not Activate PPARγ and Its Target Genes in Early Gestation Trophoblasts. Int. J. Mol. Sci. 2022, 23, 12326. https://doi.org/10.3390/ijms232012326
Neuper L, Kummer D, Forstner D, Guettler J, Ghaffari-Tabrizi-Wizsy N, Fischer C, Juch H, Nonn O, Gauster M. Candesartan Does Not Activate PPARγ and Its Target Genes in Early Gestation Trophoblasts. International Journal of Molecular Sciences. 2022; 23(20):12326. https://doi.org/10.3390/ijms232012326
Chicago/Turabian StyleNeuper, Lena, Daniel Kummer, Désirée Forstner, Jacqueline Guettler, Nassim Ghaffari-Tabrizi-Wizsy, Cornelius Fischer, Herbert Juch, Olivia Nonn, and Martin Gauster. 2022. "Candesartan Does Not Activate PPARγ and Its Target Genes in Early Gestation Trophoblasts" International Journal of Molecular Sciences 23, no. 20: 12326. https://doi.org/10.3390/ijms232012326
APA StyleNeuper, L., Kummer, D., Forstner, D., Guettler, J., Ghaffari-Tabrizi-Wizsy, N., Fischer, C., Juch, H., Nonn, O., & Gauster, M. (2022). Candesartan Does Not Activate PPARγ and Its Target Genes in Early Gestation Trophoblasts. International Journal of Molecular Sciences, 23(20), 12326. https://doi.org/10.3390/ijms232012326