
Hydrogen Bonding Drives Helical Chirality via 10-Membered Rings in Dipeptide Conjugates of Ferrocene-1,1′-Diamine

 , , ,
, , ,  ,
,  , and
, and
Abstract
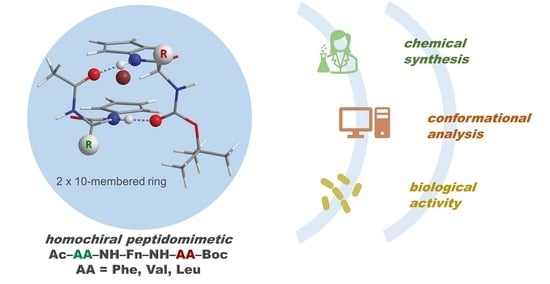
1. Introduction
2. Results and Discussion
2.1. Synthesis of Peptides 32–37
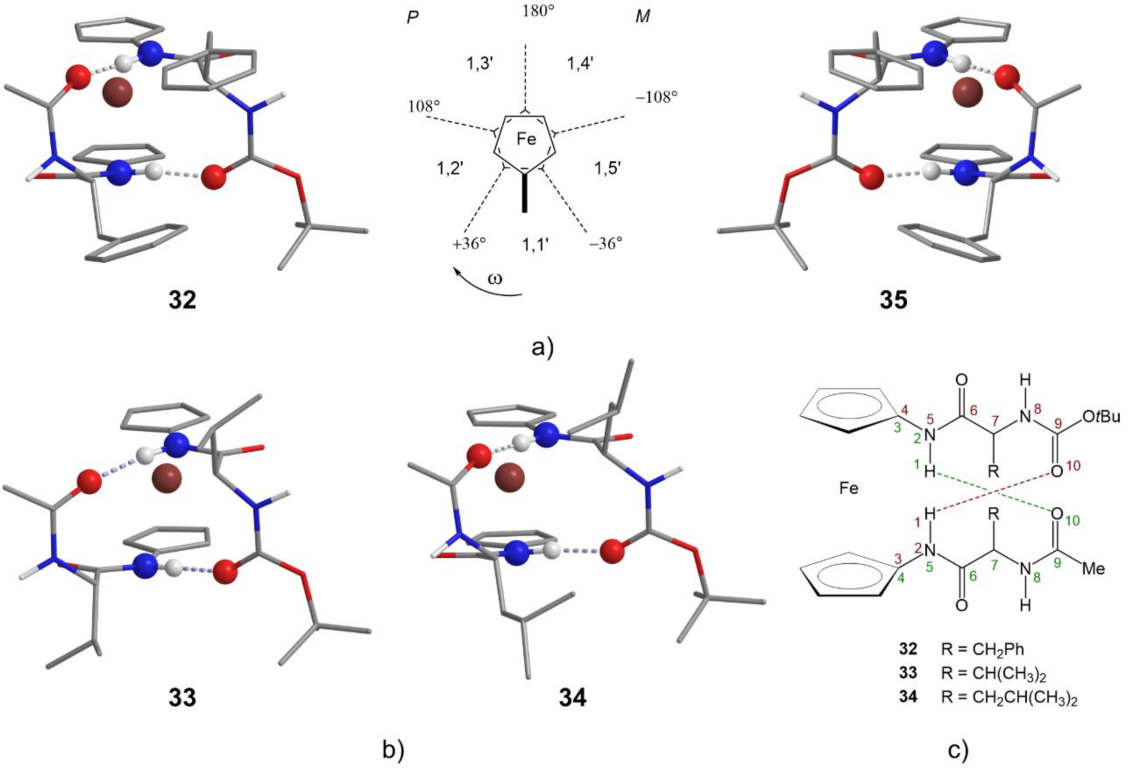
2.2. Computational Study
2.3. IR Spectroscopy
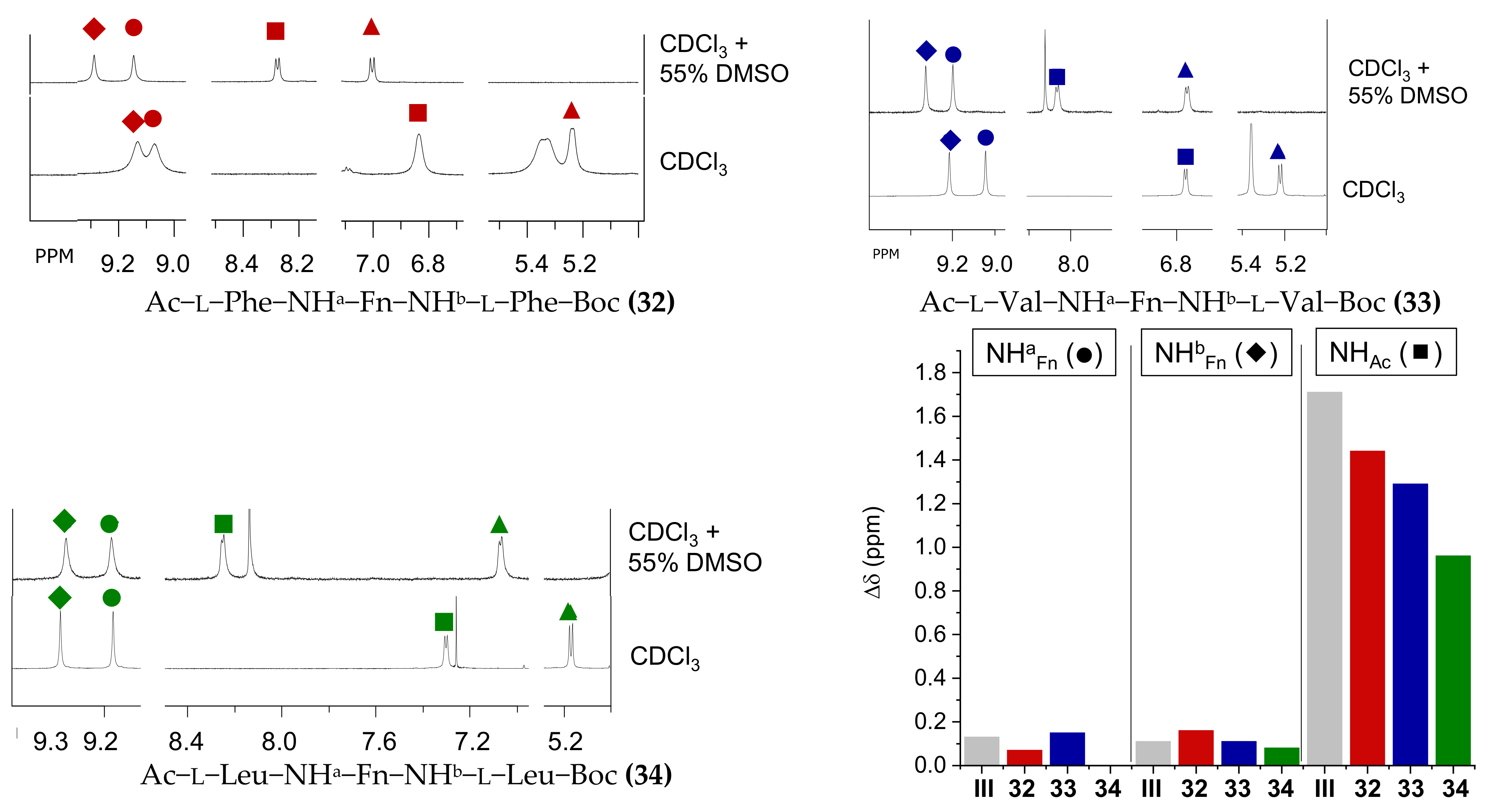
2.4. NMR Spectroscopy
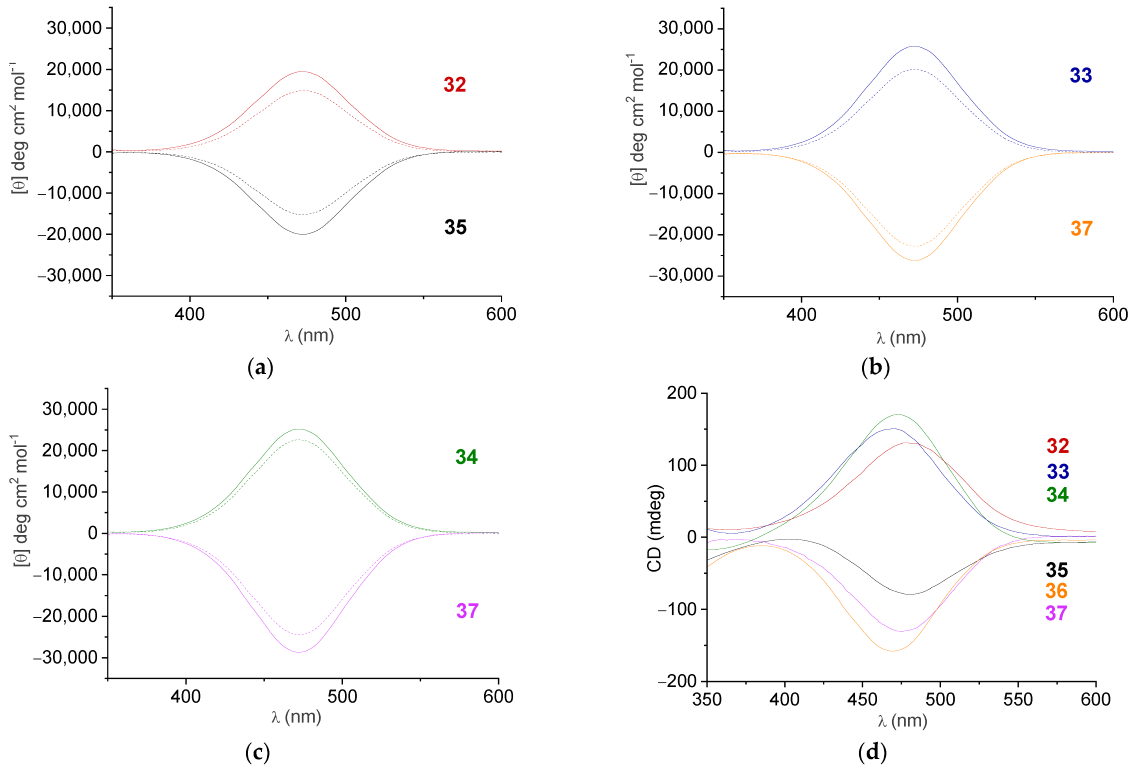
2.5. CD Spectroscopy
2.6. X-ray Crystal Structure Analysis
2.7. Biological Evaluation
2.7.1. Antitumor Activity
2.7.2. Antimicrobial Activity
2.7.3. Antioxidant Activity
3. Materials and Methods
3.1. General Procedure and Methods
3.1.1. Synthesis of Ac–l-AA–NH–Fn–NH–AA–Boc (32–37)
- Ac–l-Phe–NH–Fn–NH–l-Phe–Boc (32): Mp = 145.2 °C. IR (CH2Cl2) ῠmax/cm−1: 3430 m (NHfree), 3302, 3266, 3217 s (NHassoc.), 1731 s, 1706 s, 1683 s, 1668 s (C=OCONH), 1648 s 1575 s, 1541 s, 1498 s, 1467 s, 1457 s (amide II). IR (KBr) ῠmax/cm−1: 3294 m (NHassoc.), 1658 s, 1651 s (C=OCONH), 1568 s, 1533 s, 1499 (amide II). 1H-NMR (600 MHz, CDCl3) δ: 9.21 (1H, s, NHbFn), 9.15 (1H, s, NHaFn), 7.30–7.17 (11H, m, CHa and bphenyl and NHAc), 5.47–5.26 (3H, m, CH-7Fn, CH-10Fn and NHBoc), 4.84–4.76 (1H, s, CHaα-Phe), 4.67–4.59 (1H, s, CHbα-Phe), 4.09–3.88 (6H, m, CH-2Fn, CH-3Fn, CH-4Fn, CH-5Fn, CH-8Fn and CH-9Fn), 3.19 (1H, dd, J = 14.14; 4.46 Hz, CHaβ1-Phe), 3.14 (1H, dd, J = 14.89; 5.21 Hz, CHbβ1-Phe), 2.98 (1H, dd, J = 13.80; 10.42 Hz, CHaβ2-Phe), 2.89 (1H, dd, J = 14.14; 10.05 Hz, CHbβ2-Phe), 2.06 (s, 3H, CH3-Ac), 1.43 (9H, s, (CH3)3-Boc) ppm. 13C-NMR (150 MHz, CDCl3) δ: 171.8 (1C, COAc), 170.9 (1C, CObFn), 170.7 (1C, COaFn), 157.3 (1C, COBoc), 136.9 (1C, Cqa or bγ-Phe), 136.7 (1C, Cqa or bγ-Phe), 129.3 (2C, CHaor bδ-Phe), 129.2 (2C, CHaor bδ-Phe), 128.9 (2C, CHaor bε-Phe), 128.8 (2C, CHaor bε-Phe), 127.1 (1C, CHaor bζ-Phe), 127.0 (1C, CHaor bζ-Phe), 95.9 (1C, Cq-6Fn), 95.6 (1C, Cq-1Fn), 81.0 (1C, CqBoc), 65.9 (2C, CH-8 Fn and CH-9Fn), 65.0 (1C, CH-5Fn), 64.9 (1C, CH-4Fn), 63.1 (1C, CH-7Fn), 62.9 (1C, CH-10Fn), 61.8 (1C, CH-3Fn), 61.5 (1C, CH-2Fn), 56.6 (1C, CHbα-Phe), 56.3 (1C, CHaα-Phe), 38.1 (1C, CH2bβ-Phe), 37.7 (1C, CH2aβ-Phe), 28.7 (3C, (CH3)3-Boc), 23.1 (1C, CH3-Ac) ppm. ESI-MS (H2O:MeOH= 50:50): m/z 653.3 ((M+H)+). MALDI-HRMS m/z = 652.2337 (calculated for C35H40N4O5Fe = 652.2348).
- Ac–l-Val–NH–Fn–NH–l-Val–Boc (33): Mp = 182.4 °C. IR (CH2Cl2) ῠmax/cm−1: 3434 m (NHfree), 3305 s, 3249 m (NHassoc.), 1733 s, 1716 s, 1683 s, 166 s (C=OCONH), 1637 s, 1571 s, 1540 s, 1505 s, 1458 s (amide II). IR (KBr) ῠmax/cm−1: 3293 s (NHassoc.), 1672 s, 1652 s, 1578 s (C=OCONH), 1563 s, 1508 s, 1480 s, 1467 s (amide II). 1H-NMR (600 MHz, CDCl3) δ: 9.20 (1H, s, NHbFn), 9.02 (1H, s, NHaFn), 6.74 (1H, d, J = 7.26 Hz, NHAc), 5.34 (2H, s, CH-7Fn and CH-10Fn), 5.20 (1H, d, J = 8.38 Hz, NHBoc), 4.14 (1H, t, J = 7.95 Hz, CHaα-Val), 4.03 (1H, s, CH-2Fn), 3.98–3.94 (3H, m, CH-3Fn, CH-4Fn and CHbα-Val), 3.92 (1H, s, CH-5Fn), 3.88 (2H, s, CH-8Fn and CH-9Fn), 2.10 (3H, s, CH3-Ac), 2.05–1.98 (1H, m, CHaβ-Val), 1.98–1.91 (1H, m, CHbβ-Val), 1.45 (9H, s, (CH3)3-Boc), 1.04 (3H, d, J = 6.60 Hz, CH3aγ-Val), 1.02 (3H, d, J = 6.80 Hz, CH3bγ-Val), 0.98 (6H, d, J = 6.60 Hz, CH3aγ-Val and CH3bγ-Val) ppm. 13C-NMR (150 MHz, CDCl3) δ: 171.6 (1C, COAc), 171.0 (1C, CObFn), 170.7 (1C, COaFn), 157.3 (1C, COBoc), 95.9 (1C, Cq-6Fn), 95.6 (1C, Cq-1Fn), 80.6 (1C, CqBoc), 65.7 (2C, CH-8Fn and CH-9Fn), 65.1 (1C, CH-5Fn), 64.9 (1C, CH-4Fn), 62.9 (1C, CH-7Fn), 62.8 (1C, CH-10Fn), 61.9 (1C, CH-3Fn), 61.4 (1C, CH-2Fn), 61.3 (1C, CHbα-Val), 61.1 (1C, CHaα-Val), 30.5 (1C, CHbβ-Val), 30.3 (1C, CHaβ-Val), 28.7 (3C, (CH3)3-Boc), 23.4 (1C, CH3-Ac), 19.7 (1C, CH3a or bγ-Val), 19.44 (2C, CH3a and bγ-Val), 19.36 (1C, CH3a or bγ-Val) ppm. ESI-MS (H2O:MeOH= 50:50): m/z 579.3 ((M+Na)+). MALDI-HRMS m/z = 556.2339 (calculated for C27H40N4O5Fe = 556.2348).
- Ac–l-Leu–NH–Fn–NH–l-Leu–Boc (34): Mp = 206.6 °C. IR (CH2Cl2) ῠmax/cm−1: 3434 m (NHfree), 3301 s, 3253 s (NHassoc.), 1733 s, 1684 s, 1668 s (C=OCONH), 1653 s, 1573 s, 1540 s, 1506 s, 1457 s (amide II). IR (KBr) ῠmax/cm−1: 3281 s (NHassoc.), 1667 s, 1650 s (C=OCONH), 1568 s, 1530 s, 1484 s (amide II). 1H-NMR (600 MHz, CDCl3) δ: 9.36 (1H, s, NHbFn), 9.13 (1H, s, NHaFn), 7.28 (1H, d, J = 6.20 Hz, NHAc), 5.35 (1H, s, CH-7Fn), 5.34 (1H, s, CH-10Fn), 5.14 (1H, d, J = 8.02 Hz, NHBoc), 4.49–4.43 (1H, m, CHaα-Leu), 4.31–4.25 (1H, m, CHbα-Leu), 4.05 (1H, s, CH-3Fn), 3.98 (1H, s, CH-4Fn), 3.95 (1H, s, CH-2Fn), 3.90 (1H, s, CH-5Fn), 3.87 (2H, s, CH-8Fn and CH-9Fn), 2.09 (3H, s, CH3-Ac), 1.79–1.65 (3H, m, CHaβ1-Leu, CHaγ-Leu and CHbγ-Leu), 1.59–1.50 (3H, m, CHaβ2-Leu, CHbβ1-Leu and CHbβ2-Leu), 1.45 (9H, s, (CH3)3-Boc), 0.92 (6H, t, J = 6.80 Hz, CH3bδ1-Leu and CH3bδ2-Leu), 0.86 (6H, d, J = 6.58 Hz, CH3aδ1-Leu and CH3aδ2-Leu) ppm. 13C-NMR (150 MHz, CDCl3) δ: 172.0 (1C, COAc), 171.75 (1C, CObFn), 171.67 (1C, COaFn), 157.3 (1C, COBoc), 96.2 (1C, Cq-6Fn), 95.9 (1C, Cq-1Fn), 80.7 (1C, CqBoc), 65.7 (2C, CH-8Fn and CH-9Fn), 64.9 (1C, CH-5Fn), 64.7 (1C, CH-4Fn), 62.9 (1C, CH-7Fn), 62.7 (1C, CH-10Fn), 61.6 (1C, CH-3Fn), 61.3 (1C, CH-2Fn), 54.1 (1C, CHbα-Leu), 53.9 (1C, CHaα-Leu), 41.1 (1C, CH2bβ-Leu), 40.6 (1C, CH2aβ-Leu), 28.7 (3C, (CH3)3-Boc), 25.0 (1C, CHbγ-Leu), 24.9 (1C, CHaγ-Leu), 23.5 (1C, CH3-Ac), 23.1, 23.0, 21.8 and 21.7 (4C, CH3aδ1- and δ2-Leu and CH3b δ1- and δ2-Leu) ppm. ESI-MS (H2O:MeOH= 50:50): m/z 607.3 ((M+Na)+). MALDI-HRMS m/z = 584.2641 (calculated for C29H44N4O5Fe = 584.2661).
- Ac–d-Phe–NH–Fn–NH–d-Phe–Boc (35): Mp = 140.1 °C. IR (CH2Cl2) ῠmax/cm−1: 3432 m (NHfree), 3301 s (NHassoc.), 1731 s, 1682 s, 1668 s (C=OCONH), 1571 s, 1498 s, 1455 s, 1442 s (amide II). IR (KBr) ῠmax/cm−1: 3293 s (NHassoc.), 1657 s, 1651 s (C=OCONH), 1570 s, 1547 m, 1531 m, 1498 m, 1491 m, 1454 s (amide II). 1H-NMR (600 MHz, CDCl3) δ: 9.18 (1H, s, NHbFn), 9.12 (1H, s, NHaFn), 7.30–7.17 (10H, m, CHa and bphenyl), 7.07 (1H, s, NHAc), 5.36 (1H, s, CH-7Fn), 5.34 (1H, s, CH-10Fn), 5.30 (1H, s, J = 5.88 Hz, NHBoc), 4.79 (1H, s, CHaα-Phe), 4.62 (1H, s, CHbα-Phe), 4.08–3.89 (6H, m, CH-2Fn, CH-3Fn, CH-4Fn, CH-5Fn, CH-8Fn and CH-9Fn), 3.21–3.10 (2H, m, CHaβ1-Phe and CHbβ1-Phe), 2.97 (1H, t, J = 10.91 Hz, CHaβ2-Phe), 2.88 (1H, t, J = 10.91 Hz, CHaβ2-Phe), 2.05 (3H, s, CH3-Ac), 1.42 (9H, s, (CH3)3-Boc) ppm. 13C-NMR (150 MHz, CDCl3) δ: 171.8 (1C, COAc), 170.9 (1C, CObFn), 170.6 (1C, COaFn), 157.3 (1C, COBoc), 136.9 (1C, Cqa or bγ-Phe), 136.7 (1C, Cqa or bγ-Phe), 129.3 (2C, CHa or bδ-Phe), 129.2 (2C, CHa or bδ-Phe), 128.9 (2C, CHa or bε-Phe), 128.8 (2C, CHa or bε-Phe), 127.2 (1C, CHa or bζ-Phe), 127.1 (1C, CHa or bζ-Phe), 95.9 (1C, Cq-6Fn), 95.5 (1C, Cq-1Fn), 81.0 (1C, CqBoc), 65.9 (2C, CH-8 Fn and CH-9Fn), 65.0 (1C, CH-5Fn), 64.8 (1C, CH-4Fn), 63.1 (1C, CH-7Fn), 62.9 (1C, CH-10Fn), 61.9 (1C, CH-3Fn), 61.5 (1C, CH-2Fn), 56.6 (1C, CHbα-Phe), 56.3 (1C, CHaα-Phe), 38.1 (1C, CH2bβ-Phe), 37.7 (1C, CH2aβ-Phe), 28.7 (3C, (CH3)3-Boc), 23.1 (1C, CH3-Ac) ppm. ESI-MS (H2O:MeOH= 50:50): m/z 653.1 ((M+H)+), calculated for C35H40N4O5Fe = 652.2348.
- Ac–d-Val–NH–Fn–NH–d-Val–Boc (36): Mp = 201.4 °C. IR (CH2Cl2) ῠmax/cm−1: 3435 m (NHfree), 3306 s (NHassoc.), 1731 s, 1681 s, 1667 s (C=OCONH), 1571 s, 1503 s (amide II). IR (KBr) ῠmax/cm−1: 3287 s (NHassoc.), 1671 s, 1652 s (C=OCONH), 1577 s, 1564 s, 1508 s (amide II). 1H-NMR (600 MHz, CDCl3) δ: 9.21 (1H, s, NHbFn), 9.05 (1H, s, NHaFn), 6.78 (1H, d, J = 6.91 Hz, NHAc), 5.36 (2H, s, CH-7Fn and CH-10Fn), 5.21 (1H, d, J = 8.41 Hz, NHBoc), 4.16 (1H, t, J = 8.04 Hz, CHaα-Val), 4.04 (1H, s, CH-2Fn), 4.01–3.96 (3H, m, CH-3Fn, CH-4Fn and CHbα-Val), 3.94 (1H, s, CH-5Fn), 3.90 (2H, s, CH-8Fn and CH-9Fn), 2.11 (3H, s, CH3-Ac), 2.04 (1H, q, J = 6.96 Hz, CHaβ-Val), 1.97 (1H, q, J = 6.96 Hz, CHbβ-Val), 1.47 (9H, s, (CH3)3-Boc), 1.07 (3H, d, J = 6.69 Hz, CH3aγ-Val), 1.04 (3H, d, J = 6.69 Hz, CH3bγ-Val), 1.00 (6H, d, J = 6.69 Hz, CH3aγ-Val and CH3bγ-Val) ppm. 13C-NMR (150 MHz, CDCl3) δ: 171.5 (1C, COAc), 171.0 (1C, CObFn), 170.7 (1C, COaFn), 157.3 (1C, COBoc), 95.9 (1C, Cq-6Fn), 95.6 (1C, Cq-1Fn), 80.6 (1C, CqBoc), 65.7 (2C, CH-8Fn and CH-9Fn), 65.1 (1C, CH-5Fn), 64.9 (1C, CH-4Fn), 62.9 (1C, CH-7Fn), 62.8 (1C, CH-10Fn), 61.8 (1C, CH-3Fn), 61.4 (1C, CH-2Fn), 61.2 (1C, CHbα-Val), 61.1 (1C, CHaα-Val), 30.4 (1C, CHbβ-Val), 30.2 (1C, CHaβ-Val), 28.7 (3C, (CH3)3-Boc), 23.4 (1C, CH3-Ac), 19.7 (1C, CH3a or bγ-Val), 19.44 (2C, CH3a and bγ-Val), 19.38 (1C, CH3a or bγ-Val) ppm. ESI-MS (H2O:MeOH= 50:50): m/z 579.1 ((M+Na)+), calculated for C27H40N4O5Fe = 556.2348.
- Ac–d-Leu–NH–Fn–NH–d-Leu–Boc (37): Mp = 207.8 °C. IR (CH2Cl2) ῠmax/cm−1: 3434 m (NHfree), 3292 s (NHassoc.), 1733 w, 1663 s (C=OCONH), 1492 s (amide II). IR (KBr) ῠmax/cm−1: 3287 s (NHassoc.), 1683 s, 1667 s, 1651 s (C=OCONH), 1565 m, 1526 m, 1483 m (amide II). 1H-NMR (600 MHz, CDCl3) δ: 9.36 (1H, s, NHbFn), 9.14 (1H, s, NHaFn), 7.25 (1H, s, NHAc), 5.38 (2H, s, CH-7Fn and CH-10Fn), 5.15 (1H, d, J = 7.77 Hz, NHBoc), 4.51–4.41 (1H, m, CHaα-Leu), 4.32–4.24 (1H, m, CHbα-Leu), 4.13–3.83 (6H, m, CH-2Fn, CH-3Fn, CH-4Fn, CH-5Fn, CH-8Fn and CH-9Fn), 2.09 (3H, s, CH3-Ac), 1.79–1.64 (3H, m, CHaβ1-Leu, CHaγ-Leu and CHbγ-Leu), 1.60–1.49 (3H, m, CHaβ2-Leu, CHbβ1-Leu and CHbβ2-Leu), 1.45 (9H, s, (CH3)3-Boc), 0.92 (6H, t, J = 6.62 Hz, CH3bδ1-Leu and CH3bδ2-Leu), 0.86 (6H, d, J = 6.49 Hz, CH3aδ1-Leu CH3aδ2-Leu) ppm. 13C-NMR (150 MHz, CDCl3) δ: 171.9 (1C, COAc), 171.6 (2C, COaFn and CObFn), 157.2 (1C, COBoc), 96.2 (1C, Cq-6Fn), 95.9 (1C, Cq-1Fn), 80.7 (1C, CqBoc), 65.7 (2C, CH-8Fn and CH-9Fn), 65.0 (1C, CH-5Fn), 64.8 (1C, CH-4Fn), 62.9 (1C, CH-7Fn), 62.7 (1C, CH-10Fn), 61.5 (1C, CH-2Fn), 61.3 (1C, CH-3Fn), 54.0 (1C, CHbα-Leu), 53.9 (1C, CHaα-Leu), 41.0 (1C, CH2bβ-Leu), 40.5 (1C, CH2aβ-Leu), 28.7 (3C, (CH3)3-Boc), 25.0 (1C, CHbγ-Leu), 24.8 (1C, CHaγ-Leu), 23.4 (1C, CH3-Ac), 23.1, 23.0, 21.7 and 21.6 (4C, CH3aδ1- and δ2-Leu and CH3b δ1- and δ2-Leu) ppm. ESI-MS (H2O:MeOH = 50:50): m/z 607.2 ((M+Na)+), calculated for C29H44N4O5Fe = 584.2661.
3.1.2. Computational Details
3.1.3. Crystallographic Study
3.1.4. Biological Activity
- Antitumor activity
- Antimicrobial activity
- Antioxidant activity assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, X.; Ni, D.; Liu, Y.; Lu, S. Rational Design of Peptide-Based Inhibitors Disrupting Protein-Protein Interactions. Front. Chem. 2021, 9, 682675. [Google Scholar] [CrossRef] [PubMed]
- Seychell, B.C.; Beck, T. Molecular basis for protein-protein interactions. Beilstein J. Org. Chem. 2021, 17, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Zhou, Q.; He, J.; Jiang, Z.; Peng, C.; Tong, R.; Shi, J. Recent advances in the development of protein-protein interactions modulators: Mechanisms and clinical trials. Signal Transduct. Target. Ther. 2020, 5, 213–235. [Google Scholar] [CrossRef] [PubMed]
- Ovek, D.; Abali, Z.; Zeylan, M.E.; Keskin, O.; Gursoy, A.; Tuncbag, N. Artificial intelligence based methods for hot spot prediction. Curr. Opin. Struct. Biol. 2022, 72, 209–218. [Google Scholar] [CrossRef]
- Hoang, H.N.; Hill, T.A.; Ruiz-Gómez, G.; Diness, F.; Mason, J.M.; Wu, C.; Abbenante, G.; Shepherd, N.E.; Fairlie, D.P. Twists or turns: Stabilising alpha vs. beta turns in tetrapeptides. Chem. Sci. 2019, 10, 10595–10600. [Google Scholar] [CrossRef]
- Laxio Arenas, J.; Kaffy, J.; Ongeri, S. Peptides and peptidomimetics as inhibitors of protein-protein interactions involving β-sheet secondary structures. Curr. Opin. Chem. Biol. 2019, 52, 157–167. [Google Scholar] [CrossRef]
- Vagner, J.; Qu, H.; Hruby, V.J. Peptidomimetics, a synthetic tool of drug discovery. Curr. Opin. Chem. Biol. 2008, 12, 292–296. [Google Scholar] [CrossRef]
- Lenci, E.; Trabocchi, A. Peptidomimetic toolbox for drug discovery. Chem. Soc. Rev. 2020, 49, 3262–3277. [Google Scholar] [CrossRef]
- Huan, Y.; Kong, Q.; Mou, H.; Yi, H. Antimicrobial Peptides: Classification, Design, Application and Research Progress in Multiple Fields. Front. Microbiol. 2020, 11, 582779. [Google Scholar] [CrossRef]
- Kundu, R. Cationic Amphiphilic Peptides: Synthetic Antimicrobial Agents Inspired by Nature. ChemMedChem 2020, 15, 1887–1896. [Google Scholar] [CrossRef]
- Liscano, Y.; Oñate-Garzón, J.; Delgado, J.P. Peptides with Dual Antimicrobial–Anticancer Activity: Strategies to Overcome Peptide Limitations and Rational Design of Anticancer Peptides. Molecules 2020, 25, 4245. [Google Scholar] [CrossRef]
- Nair, R.V.; Baravkar, S.B.; Ingole, T.S.; Sanjayan, G.J. Synthetic turn mimetics and hairpin nucleators. Chem. Commun. 2014, 50, 13874–13884. [Google Scholar] [CrossRef]
- Herrick, R.S.; Jarret, R.M.; Curran, T.P.; Dragoli, D.R.; Flaherty, M.B.; Lindyberg, S.E.; Slate, R.A.; Thornton, L.C. Ordered conformations in bis(amino acid) derivatives of 1,1′-ferrocenedicarboxylic acid. Tetrahedron Lett. 1996, 37, 5289–5292. [Google Scholar] [CrossRef]
- Kirin, S.I.; Wissenbach, D.; Metzler-Nolte, N. Unsymmetrical 1,n′-disubstituted ferrocenoyl peptides: Convenient one pot synthesis and solution structures by CD and NMR spectroscopy. New J. Chem. 2005, 29, 1168–1173. [Google Scholar] [CrossRef]
- Van Staveren, D.R.; Weyhermüller, T.; Metzler-Nolte, N. Organometallic β-turn mimetics. A structural and spectroscopic study of inter-strand hydrogen bonding in ferrocene and cobaltocenium conjugates of amino acids and dipeptides. Dalton Trans. 2003, 210–220. [Google Scholar] [CrossRef]
- Barišić, L.; Čakić, M.; Mahmoud, K.A.; Liu, Y.-N.; Kraatz, H.-B.; Pritzkow, H.; Kirin, S.I.; Metzler-Nolte, N.; Rapić, V. Helically Chiral Ferrocene Peptides Containing 1′-Aminoferrocene-1-Carboxylic Acid Subunits as Turn Inducers. Chem. Eur. J. 2006, 12, 4965–4980. [Google Scholar] [CrossRef]
- Semencic, M.C.; Siebler, D.; Heinze, K.; Rapic, V. Bis- and Trisamides Derived From 1′-Aminoferrocene-1-carboxylic Acid and α-Amino Acids: Synthesis and Conformational Analysis. Organometallics 2009, 28, 2028–2037. [Google Scholar] [CrossRef]
- Chowdhury, S.; Mahmoud, K.A.; Schatte, G.; Kraatz, H.-B. Amino acid conjugates of 1,1′-diaminoferrocene. Synthesis and chiral organization. Org. Biomol. Chem. 2005, 3, 3018–3023. [Google Scholar] [CrossRef]
- Kovačević, M.; Kodrin, I.; Cetina, M.; Kmetič, I.; Murati, T.; Semenčić, M.Č.; Roca, S.; Barišić, L. The conjugates of ferrocene-1,1′-diamine and amino acids. A novel synthetic approach and conformational analysis. Dalton Trans. 2015, 44, 16405–16420. [Google Scholar] [CrossRef]
- Čakić Semenčić, M.; Kodrin, I.; Molčanov, K.; Kovačević, M.; Rapić, V. Novel ferrocene imide derivatives: Synthesis, conformational analysis and X-ray structure. Heliyon 2022, 8, e09470. [Google Scholar] [CrossRef]
- Nuskol, M.; Šutalo, P.; Kodrin, I.; Čakić Semenčić, M. Sensing of the Induced Helical Chirality by the Chiroptical Response of the Ferrocene Chromophore. Eur. J. Inorg. Chem. 2022, 2022, e202100880. [Google Scholar] [CrossRef]
- Nuskol, M.; Šutalo, P.; Đaković, M.; Kovačević, M.; Kodrin, I.; Čakić Semenčić, M. Testing the Potential of the Ferrocene Chromophore as a Circular Dichroism Probe for the Assignment of the Screw-Sense Preference of Tripeptides. Organometallics 2021, 40, 1351–1362. [Google Scholar] [CrossRef]
- Nuskol, M.; Studen, B.; Meden, A.; Kodrin, I.; Čakić Semenčić, M. Tight turn in dipeptide bridged ferrocenes: Synthesis, X-ray structural, theoretical and spectroscopic studies. Polyhedron 2019, 161, 137–144. [Google Scholar] [CrossRef]
- Čakić Semenčić, M.; Kodrin, I.; Barišić, L.; Nuskol, M.; Meden, A. Synthesis and Conformational Study of Monosubstituted Aminoferrocene-Based Peptides Bearing Homo- and Heterochiral Pro-Ala Sequences. Eur. J. Inorg. Chem. 2017, 2017, 306–307. [Google Scholar] [CrossRef]
- Moriuchi, T.; Ohmura, S.D.; Moriuchi-Kawakami, T. Chirality Induction in Bioorganometallic Conjugates. Inorganics 2018, 6, 111. [Google Scholar] [CrossRef]
- Kovačević, M.; Kodrin, I.; Roca, S.; Molčanov, K.; Shen, Y.; Adhikari, B.; Kraatz, H.B.; Barišić, L. Helically Chiral Peptides That Contain Ferrocene-1,1′-diamine Scaffolds as a Turn Inducer. Chem. Eur. J. 2017, 23, 10372–10395. [Google Scholar] [CrossRef]
- Kovačević, M.; Molčanov, K.; Radošević, K.; Srček Gaurina, V.; Roca, S.; Čače, A.; Barišić, L. Conjugates of 1′-aminoferrocene-1-carboxylic acid and proline: Synthesis, conformational analysis and biological evaluation. Molecules 2014, 21, 12852–12880. [Google Scholar] [CrossRef]
- Kovačević, M.; Čakić Semenčić, M.; Radošević, K.; Molčanov, K.; Roca, S.; Šimunović, L.; Kodrin, I.; Barišić, L. Conformational Preferences and Antiproliferative Activity of Peptidomimetics Containing Methyl 1′-Aminoferrocene-1-carboxylate and Turn-Forming Homo- and Heterochiral Pro-Ala Motifs. Int. J. Mol. Sci. 2021, 22, 13532. [Google Scholar] [CrossRef]
- Kovač, V.; Kodrin, I.; Radošević, K.; Molčanov, K.; Adhikari, B.; Kraatz, H.-B.; Barišić, L. Oxalamide-Bridged Ferrocenes: Conformational and Gelation Properties and In Vitro Antitumor Activity. Organometallics 2022, 41, 920–936. [Google Scholar] [CrossRef]
- Nanda, V.; Andrianarijaona, A.; Narayanan, C. The role of protein homochirality in shaping the energy landscape of folding. Protein Sci. 2007, 16, 1667–1675. [Google Scholar] [CrossRef]
- Zheng, Y.; Mao, K.; Chen, S.; Zhu, H. Chirality Effects in Peptide Assembly Structures. Front. Bioeng. Biotechnol. 2021, 22, 703004. [Google Scholar] [CrossRef] [PubMed]
- Krause, E.; Bienert, M.; Schmieder, P.; Wenschuh, H. The Helix-Destabilizing Propensity Scale of d-Amino Acids: The Influence of Side Chain Steric Effects. J. Am. Chem. Soc. 2000, 122, 4865–4870. [Google Scholar] [CrossRef]
- Sidorova, A.; Bystrov, V.; Lutsenko, A.; Shpigun, D.; Belova, E.; Likhachev, I. Quantitative Assessment of Chirality of Protein Secondary Structures and Phenylalanine Peptide Nanotubes. Nanomaterials 2021, 11, 3299. [Google Scholar] [CrossRef] [PubMed]
- Debnath, S.; Ghosh, S.; Pandit, G.; Satpati, P.; Chatterjee, S. Effect of Differential Geminal Substitution of γ Amino Acid Residues at the (i + 2) Position of αγ Turn Segments on the Conformation of Template β-Hairpin Peptides. J. Org. Chem. 2021, 86, 11310–11323. [Google Scholar] [CrossRef]
- Lesma, J.; Bizet, F.; Berardet, C.; Tonali, N.; Pellegrino, S.; Taverna, M.; Khemtemourian, L.; Soulier, J.L.; van Heijenoort, C.; Halgand, F.; et al. β-Hairpin Peptide Mimics Decrease Human Islet Amyloid Polypeptide (hIAPP) Aggregation. Front. Cell Dev. Biol. 2021, 9, 729001. [Google Scholar] [CrossRef]
- Schifano, N.P.; Caputo, G.A. Investigation of the Role of Hydrophobic Amino Acids on the Structure-Activity Relationship in the Antimicrobial Venom Peptide Ponericin L1. J. Membr. Biol. 2021. [Google Scholar] [CrossRef]
- Frederiksen, N.; Hansen, P.R.; Björkling, F.; Franzyk, H. Peptide/Peptoid Hybrid Oligomers: The Influence of Hydrophobicity and Relative Side-Chain Length on Antibacterial Activity and Cell Selectivity. Molecules 2019, 24, 4429. [Google Scholar] [CrossRef]
- Singh, A.; Lumb, I.; Mehra, V.; Kumar, V. Correction: Ferrocene-appended pharmacophores: An exciting approach for modulating the biological potential of organic scaffolds. Dalton Trans. 2019, 48, 3146. [Google Scholar] [CrossRef]
- Sharma, B.; Kumar, V. Has Ferrocene Really Delivered Its Role in Accentuating the Bioactivity of Organic Scaffolds? J. Med. Chem. 2021, 64, 16865–16921. [Google Scholar] [CrossRef]
- Ludwig, B.S.; Correia, J.D.G.; Kühn, F.E. Ferrocene derivatives as anti-infective agents. Coord. Chem. Rev. 2019, 396, 22–48. [Google Scholar] [CrossRef]
- Fiorina, V.J.; Dubois, R.J.; Brynes, S. Ferrocenyl polyamines as agents for the chemoimmunotherapy of cancer. J. Med. Chem. 1978, 21, 393–395. [Google Scholar] [CrossRef]
- Astruc, D. Why is Ferrocene so Exceptional? Eur. J. Inorg. Chem 2017, 6–29. [Google Scholar] [CrossRef]
- Patra, M.; Gasser, G. The medicinal chemistry of ferrocene and its derivatives. Nat. Rev. Chem. 2017, 1, 1–12. [Google Scholar] [CrossRef]
- Wang, R.; Chen, H.; Yan, W.; Zheng, M.; Zhang, T.; Zhang, Y. Ferrocene-containing hybrids as potential anticancer agents: Current developments, mechanisms of action and structure-activity relationships. Eur. J. Med. Chem. 2020, 190, 112109–112129. [Google Scholar] [CrossRef]
- Albada, B.; Metzler-Nolte, N. Highly Potent Antibacterial Organometallic Peptide Conjugates. Acc. Chem. Res. 2017, 50, 2510–2518. [Google Scholar] [CrossRef]
- Soulčre, L.; Bernard, J. Design, solid phase synthesis and evaluation of cationic ferrocenoyl peptide bioconjugates as potential antioxidant enzyme mimics. Bioorganic Med. Chem. Lett. 2009, 19, 1173–1176. [Google Scholar] [CrossRef]
- Xianjiao, M.; Shengling, L.; Wenbing, M.; Jianlong, W.; Zhiyong, H.; Duanlin, C. Synthesis and Antioxidant Activities of Ferrocenyl-containing Curcumin Analogues. Lett. Drug Des. Discov. 2018, 15, 1252–1258. [Google Scholar] [CrossRef]
- Barišić, L.; Rapić, V.; Kovač, V. Ferrocene Compounds. XXIX. Efficient Syntheses of 1′-Aminoferrocene-1-carboxylic Acid Derivatives. Croat. Chem. Acta 2002, 75, 199–210. [Google Scholar]
- Čakić Semenčić, M.; Kovač, V.; Kodrin, I.; Barišić, L.; Rapić, V. Synthesis and conformational study of bioconjugates derived from 1-acetyl-1′-aminoferrocene and α-amino acids. Eur. J. Inorg. Chem. 2015, 2015, 112–123. [Google Scholar] [CrossRef]
- Kovač, V.; Čakić Semenčić, M.; Kodrin, I.; Roca, S.; Rapić, V. Ferrocene-dipeptide conjugates derived from aminoferrocene and 1-acetyl-1′-aminoferrocene: Synthesis and conformational studies. Tetrahedron 2013, 69, 10497–10506. [Google Scholar] [CrossRef]
- Barišić, L.; Kovačević, M.; Mamić, M.; Kodrin, I.; Mihalić, Z.; Rapić, V. Synthesis and conformational analysis of methyl N-alanyl-1′- aminoferrocene-1-carboxylate. Eur. J. Inorg. Chem. 2012, 2012, 1810–1822. [Google Scholar] [CrossRef]
- Schrödinger, LLC. MacroModel; Schrödinger, LLC: New York, NY, USA, 2019. [Google Scholar]
- Mohamadi, F.; Richards, N.G.J.; Guida, W.C.; Liskamp, R.; Lipton, M.; Caufield, C.; Chang, G.; Hendrickson, T.; Still, W.C. Macromodel—An integrated software system for modeling organic and bioorganic molecules using molecular mechanics. J. Comput. Chem. 1990, 11, 440–467. [Google Scholar] [CrossRef]
- Koch, U.; Popelier, P.L.A.J. Characterization of C-H-O Hydrogen Bonds on the Basis of the Charge Density. J. Phys. Chem. 1995, 99, 9747–9754. [Google Scholar] [CrossRef]
- Mislow, K. Fuzzy restrictions and inherent uncertainties in chirality studies. In Fuzzy Logic in Chemistry; Rouvray, D.H., Ed.; Academic Press: New York, NY, USA, 1997; pp. 65–90. [Google Scholar] [CrossRef]
- Natarajan, R.; Basak, S.C.; Neumann, T.S. Novel approach for the numerical characterization of molecular chirality. J. Chem. Inf. Model. 2007, 47, 771–775. [Google Scholar] [CrossRef]
- Arnaboldi, S.; Benincori, T.; Cirilli, R.; Kutner, W.; Magni, M.; Mussini, P.R.; Noworyta, K.; Sannicolò, F. Inherently chiral electrodes: The tool for chiral voltammetry. Chem. Sci. 2015, 6, 1706–1711. [Google Scholar] [CrossRef]
- Vass, E.; Hollósi, M.; Besson, F.; Buchet, R. Vibrational spectroscopic detection of beta- and gamma-turns in synthetic and natural peptides and proteins. Chem. Rev. 2003, 103, 1917–1954. [Google Scholar] [CrossRef]
- Ramos, S.; Thielges, M.C. Site-Specific 1D and 2D IR Spectroscopy to Characterize the Conformations and Dynamics of Protein Molecular Recognition. J. Phys. Chem. B. 2019, 123, 3551–3566. [Google Scholar] [CrossRef]
- Jagesar, D.C.; Hartl, F.; Buma, W.J.; Brouwer, A.M. Infrared study of intercomponent interactions in a switchable hydrogen-bonded rotaxane. Chemistry 2008, 14, 1935–1946. [Google Scholar] [CrossRef]
- Joseph, J.; Jemmis, E.D. Red-, Blue-, or No-Shift in Hydrogen Bonds: A Unified Explanation. J. Am. Chem. Soc. 2007, 129, 4620–4632. [Google Scholar] [CrossRef]
- Arunan, E.; Desiraju, G.R.; Klein, R.A.; Sadlej, J.; Scheiner, S.; Alkorta, I.; Clary, D.C.; Crabtree, R.H.; Dannenberg, J.J.; Hobza, P.; et al. Defining the hydrogen bond: An account (IUPAC Technical Report). Pure Appl. Chem. 2011, 83, 1619–1636. [Google Scholar] [CrossRef]
- Fornaro, T.; Burini, D.; Biczysko, M.; Barone, V. Hydrogen-bonding effects on infrared spectra from anharmonic computations: Uracil-water complexes and uracil dimers. J. Phys. Chem. A 2015, 119, 4224–4236. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, S.; Jayakumar, R. Role of N-t-Boc group in helix initiation in a novel tetrapeptide. J. Pept. Res. 2002, 59, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Ananthananarayanan, V.; Cameron, T. Proline-containing β-turns. Int. J. Pept. Protein Res. 1998, 31, 399–411. [Google Scholar] [CrossRef]
- Lapić, J.; Siebler, D.; Heinze, K.; Rapić, V. Conformational analysis of heteroannularly substituted ferrocene oligoamides. Eur. J. Inorg. Chem. 2007, 2014–2024. [Google Scholar] [CrossRef]
- Nerli, S.; McShan, A.C.; Sgourakis, N.G. Chemical shift-based methods in NMR structure determination. Prog. Nucl. Magn. Reson. Spectrosc. 2018, 106–107, 1–25. [Google Scholar] [CrossRef]
- Vincenzi, M.; Mercurio, F.A.; Leone, M. NMR Spectroscopy in the Conformational Analysis of Peptides: An Overview. Curr. Med. Chem. 2021, 28, 2729–2782. [Google Scholar] [CrossRef]
- Pardi, A.; Wagner, G.; Wuthrich, K. Protein conformation and proton nuclear-magnetic-resonance chemical shifts. Eur. J. Biochem. 1983, 137, 445–454. [Google Scholar] [CrossRef]
- Gellman, S.H.; Dado, G.P.; Liang, G.-B.; Adams, B.R. Conformation-directing effects of a single intramolecular amide-amide hydrogen bond: Variable-temperature NMR and IR studies on a homologous diamide series. J. Am. Chem. Soc. 1991, 113, 1164–1173. [Google Scholar] [CrossRef]
- Wagner, G.; Pardi, A.; Wuethrich, K. Hydrogen bond length and proton NMR chemical shifts in proteins. J. Am. Chem. Soc. 1983, 105, 5948–5949. [Google Scholar] [CrossRef]
- Baxter, N.J.; Williamson, M.P. Temperature dependence of 1H chemical shifts in proteins. J. Biomol. NMR 1997, 9, 359–369. [Google Scholar] [CrossRef]
- Liu, J.-Y.; Sun, X.-Y.; Tang, Q.; Song, J.-J.; Li, X.-Q.; Gong, B.; Liu, R.; Lu, Z.-L. An unnatural tripeptide structure containing intramolecular double H-bond mimics a turn-hairpin conformation. Org. Biomol. Chem. 2021, 19, 4359–4363. [Google Scholar] [CrossRef]
- Berger, N.; Wollny, L.J.B.; Sokkar, P.; Mittal, S.; Mieres-Perez, J.; Stoll, R.; Sander, W.; Sanchez-Garcia, E. Solvent-Enhanced Conformational Flexibility of Cyclic Tetrapeptides. ChemPhysChem 2019, 20, 1664–1670. [Google Scholar] [CrossRef]
- Stevens, E.S.; Sugawara, N.; Bonora, G.M.; Toniolo, C. Conformational analysis of linear peptides. 3. Temperature dependence of NH chemical shifts in chloroform. J. Am. Chem. Soc. 1980, 102, 7048–7050. [Google Scholar] [CrossRef]
- Kessler, H. Conformation and Biological Activity of cyclic Peptides. Angew. Chem. Int. Ed. Engl. 1982, 21, 512–523. [Google Scholar] [CrossRef]
- Vijayakumar, E.K.S.; Balaram, P. Stereochemistry of a-aminoisobutyric acid peptides in solution. Helical conformations of protected decapeptides with repeating Aib-L-Ala and Aib-L-Val sequences. Biopolymers 1983, 22, 2133–2140. [Google Scholar] [CrossRef]
- Andersen, N.H.; Neidigh, J.W.; Harris, S.M.; Lee, G.M.; Liu, Z.; Tong, H. Extracting Information from the Temperature Gradients of Polypeptide NH Chemical Shifts. 1. The Importance of Conformational Averaging. J. Am. Chem. Soc. 1997, 119, 8547–8561. [Google Scholar] [CrossRef]
- Newberry, R.W.; Raines, R.T. Secondary Forces in Protein Folding. ACS Chem. Biol. 2019, 14, 1677–1686. [Google Scholar] [CrossRef]
- Pignataro, M.F.; Herrera, M.G.; Dodero, V.I. Evaluation of Peptide/Protein Self-Assembly and Aggregation by Spectroscopic Methods. Molecules 2020, 25, 4854. [Google Scholar] [CrossRef]
- Rogers, D.M.; Jasim, S.B.; Dyer, N.T.; Auvray, F.; Réfrégiers, M.; Hirst, J.D. Electronic Circular Dichroism Spectroscopy of Proteins. Chem 2019, 5, 2751–2774. [Google Scholar] [CrossRef]
- Moriuchi, T. Helical Chirality of Ferrocene Moieties in Cyclic Ferrocene-Peptide Conjugates. Chem. Eur. J. 2022, e202100902. [Google Scholar] [CrossRef]
- Giordanetto, F.; Tyrchan, C.; Ulander, J. Intramolecular Hydrogen Bond Expectations in Medicinal Chemistry. ACS Med. Chem. Lett. 2017, 8, 139–142. [Google Scholar] [CrossRef]
- Boyd, M.R.; Kenneth, D.P. Some practical considerations and applications of the national cancer institute in vitro anticancer drug discovery screen. Drug Dev. Res. 1995, 34, 91–109. [Google Scholar] [CrossRef]
- Genomics of Drug Sensitivity in Cancer. Drug: Cisplatin—Cancerrxgene—Genomics of Drug Sensi-tivity in Cancer. Available online: https://www.cancerrxgene.org/compound/Cisplatin/1005/overview/ic50 (accessed on 11 February 2022).
- Pascal, F.; Bedouet, L.; Baylatry, M.; Namur, J.; Laurent, A. Comparative Chemosensitivity of VX2 and HCC Cell Lines to Drugs Used in TACE. Anticancer Res. 2015, 35, 6497–6503. [Google Scholar]
- Petrucci, E.; Pasquini, L.; Bernabei, M.; Saulle, E.; Biffoni, M.; Accarpio, F.; Sibio, S.; Di Giorgio, A.; Di Donato, V.; Casorelli, A.; et al. A Small Molecule SMAC Mimic LBW242 Potentiates TRAIL- and Anticancer Drug-Mediated Cell Death of Ovarian Cancer Cells. PLoS ONE 2012, 7, e35073. [Google Scholar] [CrossRef]
- Chantson, J.T.; Verga Falzacappa, M.V.; Crovella, S.; Metzler-Nolte, N. Solid-Phase Synthesis, Characterization, and Antibacterial Activities of Metallocene–Peptide Bioconjugates. Chem. Med. Chem. 2006, 1, 1268–1274. [Google Scholar] [CrossRef]
- Albada, H.B.; Chiriac, A.I.; Wenzel, M.; Penkova, M.; Bandow, J.E.; Sahl, H.G.; Metzler-Nolte, N. Modulating the activity of short arginine-tryptophan containing antibacterial peptides with N-terminal metallocenoyl groups. Beilstein J. Org. Chem. 2012, 8, 1753–1764. [Google Scholar] [CrossRef]
- Zou, T.B.; He, T.P.; Li, H.B.; Tang, H.W.; En-Qin Xia, E.Q. The Structure-Activity Relationship of the Antioxidant Peptides from Natural Proteins. Molecules 2016, 21, 72. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision, C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1998, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parameterization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Keith, T.A. AIMAll, Version 19.02.13; TK Gristmill Software: Overland Park, KS, USA, 2017. [Google Scholar]
- Farrugia, L.J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Cryst. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Cryst. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Rigaku Corporation; Rigaku, P.R.O. CrysAlis; Version: 1.171.39.46; Rigaku Oxford Diffraction Ltd.: Yarnton, UK, 2018. [Google Scholar]
- Sheldrick, G.M. SHELXT-integrated space-group and crystal-structure determination. Acta Crystallogr. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Spek, A.L. CheckCIF validation ALERTS: What they mean and how to respond. Acta Crystallogr. 2020, 76, 1–11. [Google Scholar] [CrossRef]
- Farrugia, L.J. ORTEP-3 for Windows-a version of ORTEP-III with a Graphical User Interface (GUI). J. Appl. Cryst. 1997, 30, 565. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Brand-Williams, W.; Cuvelier, M.E.; Berset, C. Use of a freeradical method to evaluate antioxidant activity. Food Sci. Technol. 1995, 28, 25–30. [Google Scholar] [CrossRef]
- Benzie, F.F.; Strain, J.J. Ferric reducing/antioxidant power assay: Direct measure of total antioxidant activity of biological fluids and modified version for simultaneous measurement of total antioxidant power and ascorbic acid concentration. Meth. Enzymol. 1999, 299, 15–23. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| D–H/Å | H···A/Å | D···A/Å | D–H···A/° | Symm. op. on A | |
|---|---|---|---|---|---|
| 33 | |||||
| N1–H1···O5 | 0.86 | 2.12 | 2.940(5) | 160 | x, y, z |
| N2–H2···O4 | 0.86 | 2.21 | 3.011(4) | 154 | −1/2 + x, 1/2–y, –z |
| N3–H3···O2 | 0.86 | 2.20 | 2.980(5) | 150 | x, y, z |
| N4–H4···O1 | 0.86 | 2.18 | 2.995(5) | 158 | 1/2 + x, 1/2–y, –z |
| C5–H5···O1 | 0.93 | 2.52 | 2.940(6) | 108 | x, y, z |
| C7–H7···O4 | 0.93 | 2.52 | 2.925(6) | 107 | x, y, z |
| C16–H16C···O2 | 0.96 | 2.44 | 3.041(7) | 120 | x, y, z |
| C17–H17B···O2 | 0.96 | 2.37 | 2.984(7) | 121 | x, y, z |
| C18–H18···O4 | 0.98 | 2.58 | 3.241(6) | 125 | −1/2 + x, 1/2–y, –z |
| C20–H20A···N2 | 0.96 | 2.57 | 2.925(7) | 102 | x, y, z |
| C27–H27A···N4 | 0.96 | 2.53 | 2.895(8) | 103 | x, y, z |
| Ferrocene Peptidomimetics | HeLa | HepG2 | MCF-7 |
|---|---|---|---|
| Ac–l-Phe–NH–Fn–NH–l-Phe–Boc (32) | >350 μM | >350 μM | 53.1 ± 23 μM |
| Ac–d-Phe–NH–Fn–NH–d-Phe–Boc (35) | >350 μM | >350 μM | 32.7 ± 6.89 μM |
| Ac–l-Val–NH–Fn–NH–l-Val–Boc (33) | >350 μM | >350 μM | >350 μM |
| Ac–d-Val–NH–Fn–NH–d-Val–Boc (36) | >350 μM | >350 μM | >350 μM |
| Ac–l-Leu–NH–Fn–NH–l-Leu–Boc (34) | 331 ± 39 μM | >350 μM | 261 ± 97 μM |
| Ac–d-Leu–NH–Fn–NH–d-Leu–Boc (37) | 80.8 ± 15 μM | >350 μM | >350 μM |
| IIIa | N.T. | 259.33 | N.T. |
| Cisplatin | 46.14 b | 15.9 c | 97.86 b |
| Ferrocene Peptidomimetics (c = 1 mM) | FRAP (mM Trolox) | DPPH (% Inhibition of 0.1 mM Trolox) |
|---|---|---|
| Ac–l-Phe–NH–Fn–NH–l-Phe–Boc (32) | 0.63 ± 0.21 | 113.77 ± 4.02 |
| Ac–d-Phe–NH–Fn–NH–d-Phe–Boc (35) | 0.58 ± 0.20 | 116.80 ± 18.19 |
| Ac–l-Val–NH–Fn–NH–l-Val–Boc (33) | 0.52 ± 0.06 | 102.62 ± 20.66 |
| Ac–d-Val–NH–Fn–NH–d-Val–Boc (36) | 0.62 ± 0.15 | 88.68 ± 24.52 |
| Ac–l-Leu–NH–Fn–NH–l-Leu–Boc (34) | 0.56 ± 0.17 | 81.98 ± 7.04 |
| Ac–d-Leu–NH–Fn–NH–d-Leu–Boc (37) | 0.60 ± 0.10 | 122.75 ± 2.17 |
| Trolox 0.1 mM | - | 100.00 ± 0.0 |
| Compound | 33 | 36 |
|---|---|---|
| Empirical formula | C27H40FeN4O5 | C27H40FeN4O5 |
| Formula wt./g mol−1 | 556.48 | 556.48 |
| Color | Yellow | yellow |
| Crystal dimensions/mm | 0.35 × 0.25 × 0.15 | 0.28 × 0.16 × 0.14 |
| Space group | P 212121 | P 212121 |
| a/Å | 17.196(2) | 17.0774(4) |
| b/Å | 9.239(3) | 9.2182(2) |
| c/Å | 18.716(6) | 18.6791(5) |
| α/° | 90 | 90 |
| β/° | 90 | 90 |
| γ/° | 90 | 90 |
| Z | 4 | 4 |
| V/Å3 | 2973.6(14) | 2940.52(12) |
| Dcalc/g cm−3 | 1.243 | 1.257 |
| λ/Å | 0.71073 (MoKα) | 1.54179 (CuKα) |
| μ/mm−1 | 0.547 | 4.444 |
| Θ range/° | 1.61–24.95 | 3.51–79.42 |
| T/K | 293(2) | 293(2) |
| Diffractometer type | CAD4 | Synergy S |
| Range of h, k, l | 0 < h < 19; −10 < k < 0; −21 < l < 21 | −11 < h < 11; −21 < k < 20; −23 < l < 23 |
| Reflections collected | 5477 | 14,271 |
| Independent reflections | 4981 | 5644 |
| Observed reflections (I ≥ 2σ) | 4351 | 5127 |
| Absorption correction | None | Multi-scan |
| Tmin, Tmax | - | 0.4351; 1.0000 |
| Rint | 0.0170 | 0.0477 |
| R (F) | 0.0354 | 0.0542 |
| Rw (F2) | 0.0983 | 0.1429 |
| Goodness of fit | 1.145 | 1.122 |
| H atom treatment | Constrained | Constrained |
| No. of parameters | 334 | 334 |
| No. of restraints | 0 | 0 |
| Δρmax, Δρmin (eÅ−3) | 0.291–0.215 | 0.409; –0.773 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kovačević, M.; Markulin, D.; Zelenika, M.; Marjanović, M.; Lovrić, M.; Polančec, D.; Ivančić, M.; Mrvčić, J.; Molčanov, K.; Milašinović, V.; et al. Hydrogen Bonding Drives Helical Chirality via 10-Membered Rings in Dipeptide Conjugates of Ferrocene-1,1′-Diamine. Int. J. Mol. Sci. 2022, 23, 12233. https://doi.org/10.3390/ijms232012233
Kovačević M, Markulin D, Zelenika M, Marjanović M, Lovrić M, Polančec D, Ivančić M, Mrvčić J, Molčanov K, Milašinović V, et al. Hydrogen Bonding Drives Helical Chirality via 10-Membered Rings in Dipeptide Conjugates of Ferrocene-1,1′-Diamine. International Journal of Molecular Sciences. 2022; 23(20):12233. https://doi.org/10.3390/ijms232012233
Chicago/Turabian StyleKovačević, Monika, Dora Markulin, Matea Zelenika, Marko Marjanović, Marija Lovrić, Denis Polančec, Marina Ivančić, Jasna Mrvčić, Krešimir Molčanov, Valentina Milašinović, and et al. 2022. "Hydrogen Bonding Drives Helical Chirality via 10-Membered Rings in Dipeptide Conjugates of Ferrocene-1,1′-Diamine" International Journal of Molecular Sciences 23, no. 20: 12233. https://doi.org/10.3390/ijms232012233
APA StyleKovačević, M., Markulin, D., Zelenika, M., Marjanović, M., Lovrić, M., Polančec, D., Ivančić, M., Mrvčić, J., Molčanov, K., Milašinović, V., Roca, S., Kodrin, I., & Barišić, L. (2022). Hydrogen Bonding Drives Helical Chirality via 10-Membered Rings in Dipeptide Conjugates of Ferrocene-1,1′-Diamine. International Journal of Molecular Sciences, 23(20), 12233. https://doi.org/10.3390/ijms232012233