
Role of Endothelial STAT3 in Cerebrovascular Function and Protection from Ischemic Brain Injury

 ,
,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Endotheilal STAT3 Activation Is Regulated by OGD
2.2. Inhibition of STAT3 Causes Endothelial Cell Death
2.3. Inhibition of STAT3 Abolishes Endothelial Response to Acetylcholine
2.4. Inhibition of STAT3 Reduces Barrier Integrity of Brain Microvascular Endothelial Cells
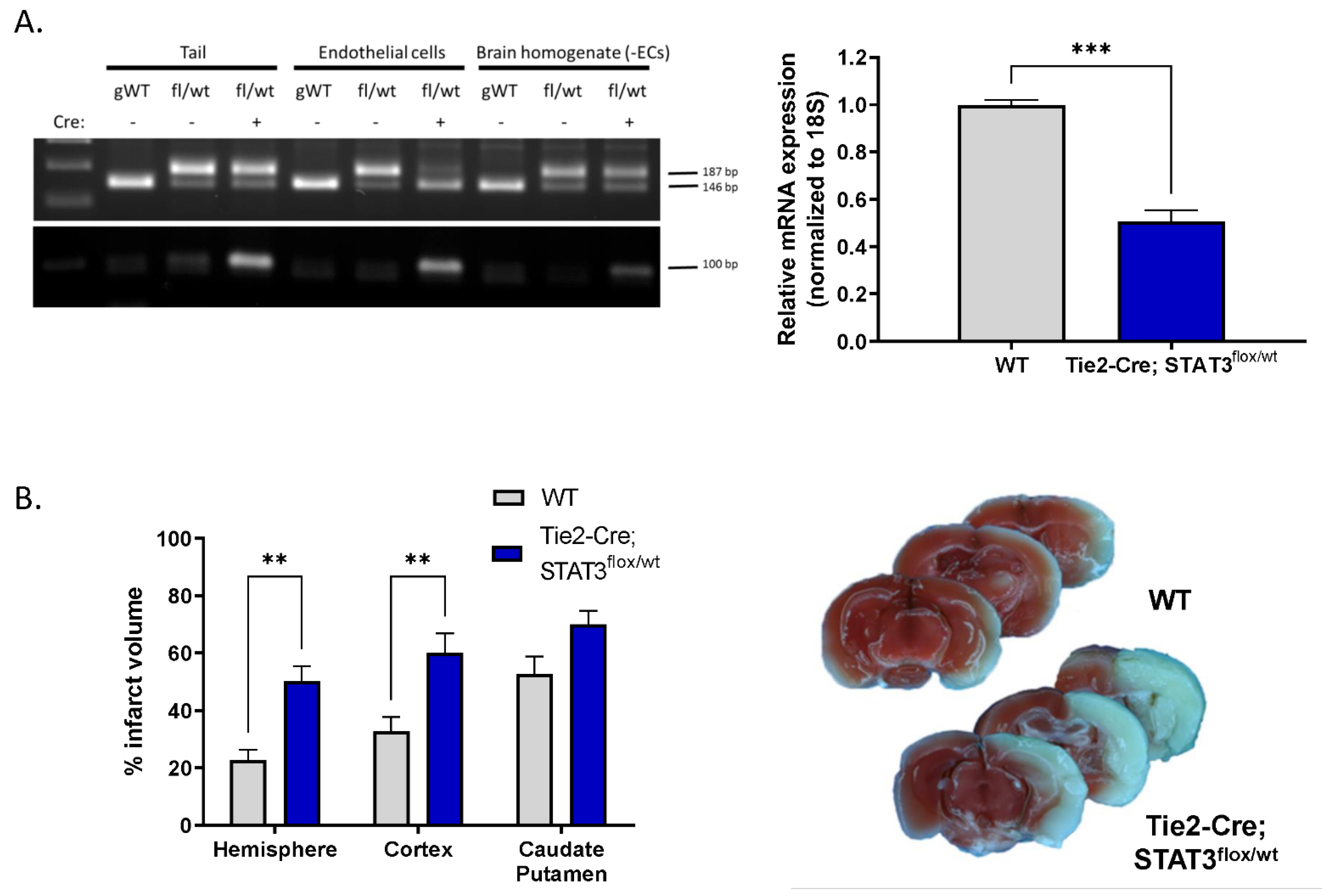
2.5. Increased Infarct in Mice with Attenuated Endothelial STAT3
3. Discussion
4. Materials and Methods
4.1. Primary Cerebral Microvessel Endothelial Cell Culture
4.2. Oxygen-Glucose Deprivation (OGD)
4.3. Western Blotting
4.4. Cell Death Assays
4.5. Immunocytochemistry
4.6. Transendothelial Electrical Resistance (TEER)
4.7. Permeability Assay
4.8. Nitrate/Nitrite Assay
4.9. Endothelial-Specific STAT3 Knock-Out Mice
4.10. Acute Isolation of Endothelial Cells
4.11. DNA Isolation
4.12. TaqMan Real-Time Quantitative Polymerase Chain Reaction
4.13. Middle Cerebral Artery Occlusion (MCAO)
4.14. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dziennis, S.; Alkayed, N.J. Role of signal transducer and activator of transcription 3 in neuronal survival and regeneration. Rev. Neurosci. 2008, 19, 341–361. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Tanaka, K.; Nogawa, S.; Dembo, T.; Kosakai, A.; Fukuuchi, Y. Phosphorylation of signal transducer and activator of transcription-3 (Stat3) after focal cerebral ischemia in rats. Exp. Neurol. 2001, 170, 63–71. [Google Scholar] [CrossRef]
- Amantea, D.; Tassorelli, C.; Russo, R.; Petrelli, F.; Morrone, A.L.; Bagetta, G.; Corasaniti, M.T. Neuroprotection by leptin in a rat model of permanent cerebral ischemia: Effects on STAT3 phosphorylation in discrete cells of the brain. Cell Death Dis. 2011, 2, e238. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Yan, Z.; Tao, K.; Li, Y.; Li, Y.; Li, J.; Dong, Y.; Feng, D.; Chen, H. Sinomenine activates astrocytic dopamine D2 receptors and alleviates neuroinflammatory injury via the CRYAB/STAT3 pathway after ischemic stroke in mice. J. Neuroinflamm. 2016, 13, 263. [Google Scholar] [CrossRef]
- Levy, D.E.; Darnell, J.E., Jr. Stats: Transcriptional control and biological impact. Nat. Rev. Mol Cell Biol 2002, 3, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Devgan, G.; Darnell, J.E., Jr.; Bromberg, J.F. Constitutively activated Stat3 protects fibroblasts from serum withdrawal and UV-induced apoptosis and antagonizes the proapoptotic effects of activated Stat1. Proc. Natl. Acad. Sci. USA 2001, 98, 1543–1548. [Google Scholar] [CrossRef] [PubMed]
- Lapp, D.W.; Zhang, S.S.; Barnstable, C.J. Stat3 mediates LIF-induced protection of astrocytes against toxic ROS by upregulating the UPC2 mRNA pool. Glia 2014, 62, 159–170. [Google Scholar] [CrossRef]
- Benito, C.; Davis, C.M.; Gomez-Sanchez, J.A.; Turmaine, M.; Meijer, D.; Poli, V.; Mirsky, R.; Jessen, K.R. STAT3 Controls the Long-Term Survival and Phenotype of Repair Schwann Cells during Nerve Regeneration. J. Neurosci. 2017, 37, 4255–4269. [Google Scholar] [CrossRef]
- Dziennis, S.; Jia, T.; Rønnekleiv, O.K.; Hurn, P.D.; Alkayed, N.J. Role of Signal Transducer and Activator of Transcription-3 in Estradiol-Mediated Neuroprotection. J. Neurosci. 2007, 27, 7268–7274. [Google Scholar] [CrossRef]
- Jung, J.E.; Kim, G.S.; Narasimhan, P.; Song, Y.S.; Chan, P.H. Regulation of Mn-superoxide dismutase activity and neuroprotection by STAT3 in mice after cerebral ischemia. J. Neurosci. 2009, 29, 7003–7014. [Google Scholar] [CrossRef]
- Sehara, Y.; Sawicka, K.; Hwang, J.Y.; Latuszek-Barrantes, A.; Etgen, A.M.; Zukin, R.S. Survivin Is a transcriptional target of STAT3 critical to estradiol neuroprotection in global ischemia. J. Neurosci. 2013, 33, 12364–12374. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, C.J.; Harms, U.; Rex, A.; Szulzewsky, F.; Wolf, S.A.; Grittner, U.; Lättig-Tünnemann, G.; Sendtner, M.; Kettenmann, H.; Dirnagl, U.; et al. Vascular signal transducer and activator of transcription-3 promotes angiogenesis and neuroplasticity long-term after stroke. Circulation 2015, 131, 1772–1782. [Google Scholar] [CrossRef]
- Li, L.; Li, H.; Li, M. Curcumin protects against cerebral ischemia-reperfusion injury by activating JAK2/STAT3 signaling pathway in rats. Int. J. Clin. Exp. Med. 2015, 8, 14985–14991. [Google Scholar]
- Sharma, S.; Yang, B.; Xi, X.; Grotta, J.C.; Aronowski, J.; Savitz, S.I. IL-10 directly protects cortical neurons by activating PI-3 kinase and STAT-3 pathways. Brain Res. 2011, 1373, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Dong, Z.; Cheng, M.; Zhao, Y.; Wang, M.; Sai, N.; Wang, X.; Liu, H.; Huang, G.; Zhang, X. Homocysteine exaggerates microglia activation and neuroinflammation through microglia localized STAT3 overactivation following ischemic stroke. J. Neuroinflamm. 2017, 14, 187. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Qian, J.; Li, H.; Shen, H.; Li, X.; Kong, Y.; Xu, Z.; Chen, G. Effects of SC99 on cerebral ischemia-perfusion injury in rats: Selective modulation of microglia polarization to M2 phenotype via inhibiting JAK2-STAT3 pathway. Neurosci. Res. 2019, 142, 58–68. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, J.; Yang, B.; Zheng, Y.; Yao, M.; Sun, M.; Xu, L.; Lin, C.; Chang, D.; Tian, F. Ginkgo biloba Extract Inhibits Astrocytic Lipocalin-2 Expression and Alleviates Neuroinflammatory Injury via the JAK2/STAT3 Pathway After Ischemic Brain Stroke. Front. Pharmacol. 2018, 9, 518. [Google Scholar] [CrossRef]
- Deli, M.A.; Abrahám, C.S.; Kataoka, Y.; Niwa, M. Permeability studies on in vitro blood-brain barrier models: Physiology, pathology, and pharmacology. Cell Mol. Neurobiol. 2005, 25, 59–127. [Google Scholar] [CrossRef] [PubMed]
- del Zoppo, G.J.; Milner, R. Integrin-matrix interactions in the cerebral microvasculature. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1966–1975. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Petito, C.K. Early and late mechanisms of increased vascular permeability following experimental cerebral infarction. J. Neuropathol. Exp. Neurol. 1979, 38, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, K.E.; Witt, K.A. Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiol. Dis. 2008, 32, 200–219. [Google Scholar] [CrossRef]
- Welte, T.; Zhang, S.S.; Wang, T.; Zhang, Z.; Hesslein, D.G.; Yin, Z.; Kano, A.; Iwamoto, Y.; Li, E.; Craft, J.E.; et al. STAT3 deletion during hematopoiesis causes Crohn’s disease-like pathogenesis and lethality: A critical role of STAT3 in innate immunity. Proc Natl. Acad. Sci. USA 2003, 100, 1879–1884. [Google Scholar] [CrossRef] [PubMed]
- Bailey, A.S.; Willenbring, H.; Jiang, S.; Anderson, D.A.; Schroeder, D.A.; Wong, M.H.; Grompe, M.; Fleming, W.H. Myeloid lineage progenitors give rise to vascular endothelium. Proc. Natl. Acad. Sci. USA 2006, 103, 13156–13161. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Alkayed, N.J. Hypoxic preconditioning and tolerance via hypoxia inducible factor (HIF) 1alpha-linked induction of P450 2C11 epoxygenase in astrocytes. J. Cereb. Blood Flow Metab. 2005, 25, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Mao, P.; Ardeshiri, A.; Jacks, R.; Yang, S.; Hurn, P.D.; Alkayed, N.J. Mitochondrial mechanism of neuroprotection by CART. Eur. J. Neurosci. 2007, 26, 624–632. [Google Scholar] [CrossRef]
- Avalle, L.; Poli, V. Nucleus, Mitochondrion, or Reticulum? STAT3 à La Carte. Int. J. Mol. Sci. 2018, 19, 2820. [Google Scholar] [CrossRef]
- Decker, T.; Kovarik, P. Serine phosphorylation of STATs. Oncogene 2000, 19, 2628–2637. [Google Scholar] [CrossRef]
- Yang, J.; Kunimoto, H.; Katayama, B.; Zhao, H.; Shiromizu, T.; Wang, L.; Ozawa, T.; Tomonaga, T.; Tsuruta, D.; Nakajima, K. Phospho-Ser727 triggers a multistep inactivation of STAT3 by rapid dissociation of pY705-SH2 through C-terminal tail modulation. Int. Immunol. 2020, 32, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.R.; Kim, H.J.; Kim, J.Y.; Hurt, E.M.; Klarmann, G.J.; Kawasaki, B.T.; Duhagon Serrat, M.A.; Farrar, W.L. Activation of signal transducer and activator of transcription 3 through a phosphomimetic serine 727 promotes prostate tumorigenesis independent of tyrosine 705 phosphorylation. Cancer Res. 2008, 68, 7736–7741. [Google Scholar] [CrossRef]
- Shi, X.; Zhang, H.; Paddon, H.; Lee, G.; Cao, X.; Pelech, S. Phosphorylation of STAT3 serine-727 by cyclin-dependent kinase 1 is critical for nocodazole-induced mitotic arrest. Biochemistry 2006, 45, 5857–5867. [Google Scholar] [CrossRef] [PubMed]
- Tammineni, P.; Anugula, C.; Mohammed, F.; Anjaneyulu, M.; Larner, A.C.; Sepuri, N.B. The import of the transcription factor STAT3 into mitochondria depends on GRIM-19, a component of the electron transport chain. J. Biol. Chem. 2013, 288, 4723–4732. [Google Scholar] [CrossRef]
- Garama, D.J.; White, C.L.; Balic, J.J.; Gough, D.J. Mitochondrial STAT3: Powering up a potent factor. Cytokine 2016, 87, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Wegrzyn, J.; Potla, R.; Chwae, Y.J.; Sepuri, N.B.; Zhang, Q.; Koeck, T.; Derecka, M.; Szczepanek, K.; Szelag, M.; Gornicka, A.; et al. Function of mitochondrial Stat3 in cellular respiration. Science 2009, 323, 793–797. [Google Scholar] [CrossRef]
- Doll, D.N.; Hu, H.; Sun, J.; Lewis, S.E.; Simpkins, J.W.; Ren, X. Mitochondrial crisis in cerebrovascular endothelial cells opens the blood-brain barrier. Stroke 2015, 46, 1681–1689. [Google Scholar] [CrossRef]
- Jiang, X.; Andjelkovic, A.V.; Zhu, L.; Yang, T.; Bennett, M.V.L.; Chen, J.; Keep, R.F.; Shi, Y. Blood-brain barrier dysfunction and recovery after ischemic stroke. Prog. Neurobiol. 2018, 163–164, 144–171. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Zhang, L.; Pu, H.; Mao, L.; Hu, X.; Jiang, X.; Xu, N.; Stetler, R.A.; Zhang, F.; Liu, X.; et al. Rapid endothelial cytoskeletal reorganization enables early blood-brain barrier disruption and long-term ischaemic reperfusion brain injury. Nat. Commun. 2016, 7, 10523. [Google Scholar] [CrossRef]
- Shi, Y.; Jiang, X.; Zhang, L.; Pu, H.; Hu, X.; Zhang, W.; Cai, W.; Gao, Y.; Leak, R.K.; Keep, R.F.; et al. Endothelium-targeted overexpression of heat shock protein 27 ameliorates blood-brain barrier disruption after ischemic brain injury. Proc. Natl. Acad. Sci. USA 2017, 114, E1243–E1252. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.; Le, X.; Zheng, L.; Wang, L.; Frey, J.A.; Gao, A.C.; Peng, Z.; Huang, S.; Xiong, H.Q.; Abbruzzese, J.L.; et al. Stat3 activation regulates the expression of vascular endothelial growth factor and human pancreatic cancer angiogenesis and metastasis. Oncogene 2003, 22, 319–329. [Google Scholar] [CrossRef]
- Chen, S.H.; Murphy, D.A.; Lassoued, W.; Thurston, G.; Feldman, M.D.; Lee, W.M. Activated STAT3 is a mediator and biomarker of VEGF endothelial activation. Cancer Biol. Ther. 2008, 7, 1994–2003. [Google Scholar] [CrossRef]
- Terpolilli, N.A.; Moskowitz, M.A.; Plesnila, N. Nitric oxide: Considerations for the treatment of ischemic stroke. J. Cereb. Blood Flow Metab. 2012, 32, 1332–1346. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Astone, M.; Alam, S.K.; Zhu, Z.; Pei, W.; Frank, D.A.; Burgess, S.M.; Hoeppner, L.H. Suppressing STAT3 activity protects the endothelial barrier from VEGF-mediated vascular permeability. Dis. Model. Mech. 2021, 14, 049029. [Google Scholar] [CrossRef]
- Zecchin, H.G.; Priviero, F.B.; Souza, C.T.; Zecchin, K.G.; Prada, P.O.; Carvalheira, J.B.; Velloso, L.A.; Antunes, E.; Saad, M.J. Defective insulin and acetylcholine induction of endothelial cell-nitric oxide synthase through insulin receptor substrate/Akt signaling pathway in aorta of obese rats. Diabetes 2007, 56, 1014–1024. [Google Scholar] [CrossRef] [PubMed]
- McCormick, M.E.; Goel, R.; Fulton, D.; Oess, S.; Newman, D.; Tzima, E. Platelet-endothelial cell adhesion molecule-1 regulates endothelial NO synthase activity and localization through signal transducers and activators of transcription 3-dependent NOSTRIN expression. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Saura, M.; Zaragoza, C.; Bao, C.; Herranz, B.; Rodriguez-Puyol, M.; Lowenstein, C.J. Stat3 mediates interleukin-6 [correction of interelukin-6] inhibition of human endothelial nitric-oxide synthase expression. J. Biol. Chem. 2006, 281, 30057–30062. [Google Scholar] [CrossRef] [PubMed]
- Sud, N.; Black, S.M. Endothelin-1 impairs nitric oxide signaling in endothelial cells through a protein kinase Cdelta-dependent activation of STAT3 and decreased endothelial nitric oxide synthase expression. DNA Cell Biol. 2009, 28, 543–553. [Google Scholar] [CrossRef] [PubMed]
- De-Fraja, C.; Conti, L.; Govoni, S.; Battaini, F.; Cattaneo, E. STAT signalling in the mature and aging brain. Int. J. Dev. Neurosci. 2000, 18, 439–446. [Google Scholar] [CrossRef]
- Percie du Sert, N.; Hurst, V.; Ahluwalia, A.; Alam, S.; Avey, M.T.; Baker, M.; Browne, W.J.; Clark, A.; Cuthill, I.C.; Dirnagl, U.; et al. The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. J. Cereb. Blood Flow Metab. 2020, 40, 1769–1777. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.C.; Davis, C.M.; Nelson, J.W.; Young, J.M.; Alkayed, N.J. Soluble epoxide hydrolase: Sex differences and role in endothelial cell survival. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1936–1942. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.M.; Ammi, A.Y.; Alkayed, N.J.; Kaul, S. Ultrasound stimulates formation and release of vasoactive compounds in brain endothelial cells. Am. J. Physiol. Circ. Physiol. 2015, 309, H583–H591. [Google Scholar] [CrossRef] [PubMed]
- Osada, T.; Gu, Y.H.; Kanazawa, M.; Tsubota, Y.; Hawkins, B.T.; Spatz, M.; Milner, R.; del Zoppo, G.J. Interendothelial claudin-5 expression depends on cerebral endothelial cell-matrix adhesion by β(1)-integrins. J. Cereb. Blood Flow Metab. 2011, 31, 1972–1985. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.M.; Zhang, W.H.; Allen, E.M.; Bah, T.M.; Shangraw, R.E.; Alkayed, N.J. Soluble Epoxide Hydrolase Blockade after Stroke Onset Protects Normal but Not Diabetic Mice. Int. J. Mol. Sci. 2021, 22, 5419. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Davis, C.M.; Lyon-Scott, K.; Varlamov, E.V.; Zhang, W.H.; Alkayed, N.J. Role of Endothelial STAT3 in Cerebrovascular Function and Protection from Ischemic Brain Injury. Int. J. Mol. Sci. 2022, 23, 12167. https://doi.org/10.3390/ijms232012167
Davis CM, Lyon-Scott K, Varlamov EV, Zhang WH, Alkayed NJ. Role of Endothelial STAT3 in Cerebrovascular Function and Protection from Ischemic Brain Injury. International Journal of Molecular Sciences. 2022; 23(20):12167. https://doi.org/10.3390/ijms232012167
Chicago/Turabian StyleDavis, Catherine M., Kristin Lyon-Scott, Elena V. Varlamov, Wenri H. Zhang, and Nabil J. Alkayed. 2022. "Role of Endothelial STAT3 in Cerebrovascular Function and Protection from Ischemic Brain Injury" International Journal of Molecular Sciences 23, no. 20: 12167. https://doi.org/10.3390/ijms232012167
APA StyleDavis, C. M., Lyon-Scott, K., Varlamov, E. V., Zhang, W. H., & Alkayed, N. J. (2022). Role of Endothelial STAT3 in Cerebrovascular Function and Protection from Ischemic Brain Injury. International Journal of Molecular Sciences, 23(20), 12167. https://doi.org/10.3390/ijms232012167