1. Introduction
Alcohol dehydrogenases (ADHs) are ubiquitous enzymes with a wide variety of substrate specificities, which are involved in several physiological processes that are essential for life and cell function. Moreover, ADHs, frequently indicated as ketoreductases (KREDs), represent one of the most important enzyme classes for the sustainable industrial preparation of optically pure
sec-alcohols, e.g., active pharmaceutical ingredient (APIs) intermediates, from the corresponding ketones in stereoselective biocatalyzed processes [
1,
2].
Among others, nicotinamide (NAD(P)(H)) cofactors are the most used coenzymes in ADH-catalyzed oxidation/reduction of alcohol/ketone substrates [
1,
3,
4]. Depending on the possible presence of metal ions in their active site and on the protein subunits’ length, NAD(P)(H)-dependent ADHs are mainly classified into two major groups: the zinc-dependent medium-chain dehydrogenases/reductases (MDRs) superfamily and the short-chain dehydrogenases/reductases (SDRs) superfamily, whose members do not require the presence of metallic ions for activity [
5]. Other enzymes capable of performing carbonyl reduction reactions belong to the aldo–keto reductases (AKRs) superfamily, and are distinguishable from MDRs and SDRs by a (α/β)
8-barrel fold and the lack of a typical Rossmann fold NAD(P)(H)-binding motif [
6].
Various ADHs from extremophilic, e.g., (hyper)thermophilic, microorganisms have been discovered and characterized to some extent [
7]. These enzymes show great potential for possible industrial applications since they frequently possess a superior stability at high temperatures, as well as in the presence of organic solvents and other protein denaturants [
8].
Among the different possible approaches leading to the discovery of novel useful biocatalysts, metagenomics, i.e., the application of culture-independent methods for the identification of target sequences coding for the enzymes of interest in environmental DNA, is currently considered one of the most promising, especially when dealing with samples from extreme environments [
9,
10,
11]. During the last years, the exploitation of metagenomics has indeed permitted the finding of thermostable and synthetically valuable biocatalysts from different enzyme classes, e.g., hydrolases [
12,
13,
14], oxidoreductases [
15,
16] and amine transferases [
17,
18].
Recently, while searching for novel hydroxysteroid dehydrogenases (HSDHs), NAD(P)(H)-dependent oxidoreductases applicable in the selective oxidation/reduction of the hydroxyl/keto groups of steroids and bile acids [
19], a new SDR sequence, named Is2-SDR (GenBank: QNN88924.1) and showing some phylogenetic relationship with known 7α- and 12α-HSDHs (
Figure 1a), was identified in a metagenome prepared from an Icelandic hot spring sample [
20,
21]. However, when analyzing the sequence alignment results, the percent identity values of Is2-SDR with different HSDHs were modest (<40%), among the highest those obtained with a 7α-HSDH from
Bosea sp. and with a 12α-HSDH from
Lysinibacillus sphaericus (38% and 36% identity, respectively,
Figure 1b). Nonetheless, other HSDH sequences, e.g., 3α-HSDHs and 3β-HSDHs, aligned with relatively close identity values, thus, making a direct inference of Is2-SDR substrate specificity from sequence analysis quite difficult (see
Table S1, supplementary materials for the full set of data).
Subsequently, the preliminary characterization of Is2-SDR after recombinant production in
Escherichia coli demonstrated that, although showing NADPH-dependent reductase activity toward different ketones, this new enzyme could not be classified as a HSDH since it did not accept any of the tested bile acids as substrates [
20].
Nonetheless, very interesting results were obtained when using Is2-SDR in the biocatalyzed reduction of selected bulky substrates. Specifically, this novel biocatalyst permitted a kinetic resolution of the so-called Wieland–Miescher ketone, a useful intermediate in the synthesis of various complex natural products [
22], as well as the stereoselective reduction of the bulky–bulky ketone 6-gingerol [
23] (see the results and discussion sections for further details).
In this work, we report about the functional characterization of Is2-SDR, which permitted us to further highlight the broad substrate scope and outstanding stability of this new biocatalyst. Moreover, a promiscuous reductive activity toward oxidized steroid derivatives was demonstrated and rationalized by docking analysis on the protein 3D structural model.
3. Discussion
The discovery of new extremozymes by exploiting metagenomics approaches paves the way to the development of sustainable biocatalytic industrial processes, which are highly foreseen in a green chemistry and circular economy view.
However, the lack of defined information about the microbial source and physiological role of metagenome-derived enzymes frequently poses some difficulties in envisaging their natural substrate specificity, as well as the possible influence of different reaction parameters on their catalytic performances.
These limitations are further amplified in the case of oxidoreductases belonging to the SDR superfamily, since automatic functional annotation of these biocatalysts is very often challenging due to the generally low sequence identity and high divergency within this protein family [
30]. Moreover, some useful enzymatic features, e.g., tolerance to high temperatures and organic solvents, are still hardly predictable starting from the corresponding protein sequences.
In the case of Is2-SDR, the functional role and microbial origin of this enzyme could not be defined by simple databases search, the closest homologue (GenBank: MBC7327662.1; 90% identity at the amino acid level) being an uncharacterized oxidoreductase found in the metagenome-assembled genome of a bacterium isolated in an oil production facility [
31]. It is worth mentioning that most of the (hyper)thermophilic ADHs characterized so far belong to the zinc-containing MDRs or the aldo–keto reductase (AKRs) superfamily [
32,
33]. A highly thermostable ADH from the SDR superfamily, possibly involved in carbohydrate metabolism, was found some years ago in
Pyrococcus furiosus [
34]; however, the similarity to Is2-SDR was not significative (about 33% sequence identity).
The first part of this work was, therefore, devoted to completing the functional characterization of Is2-SDR by extending the information about its substrate scope and selectivity, as well as about its performances under different reaction conditions.
A preliminary spectrophotometric screening of various alcohol and ketone substrates showed us a clear preference of Is2-SDR for reduction reactions instead of oxidations, along with a quite broad substrate scope, this fact confirming its applicative potential.
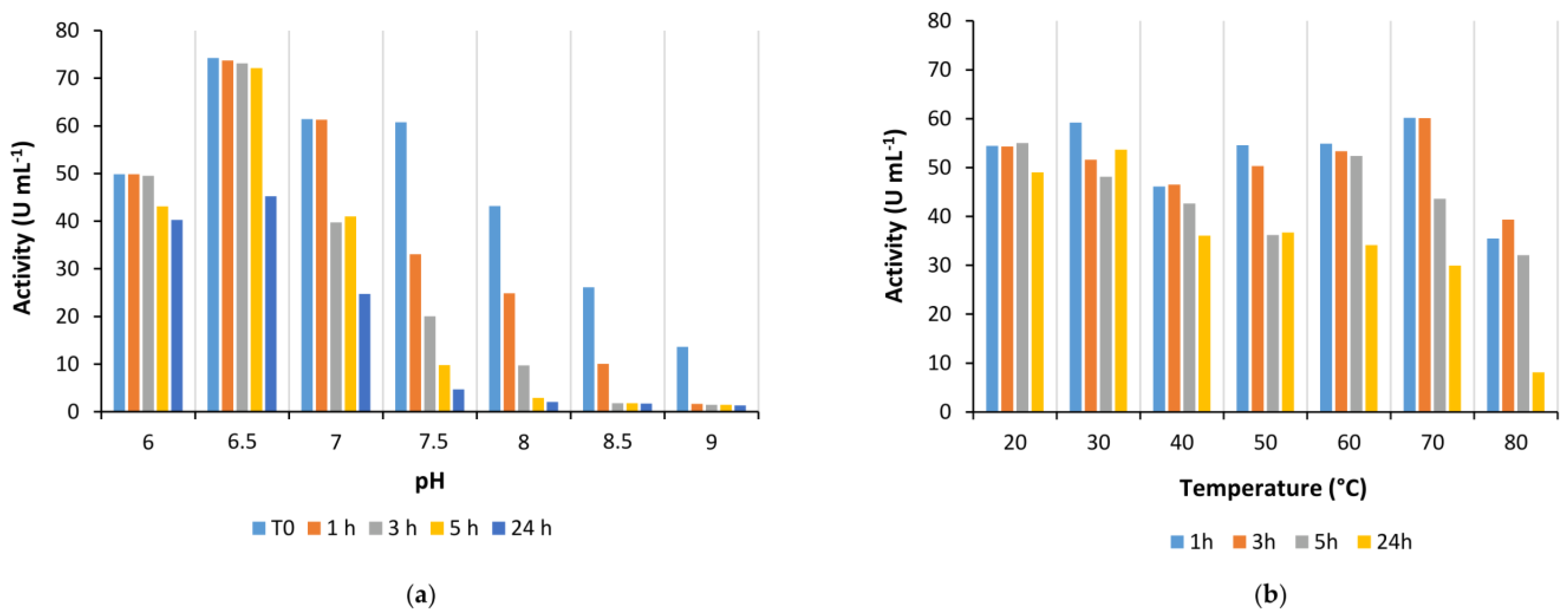
Is2-SDR also showed good activity in the quite broad pH range between 6.0 to 9.5, whereas its stability was generally higher at slightly acidic and neutral pHs when compared to alkaline pHs.
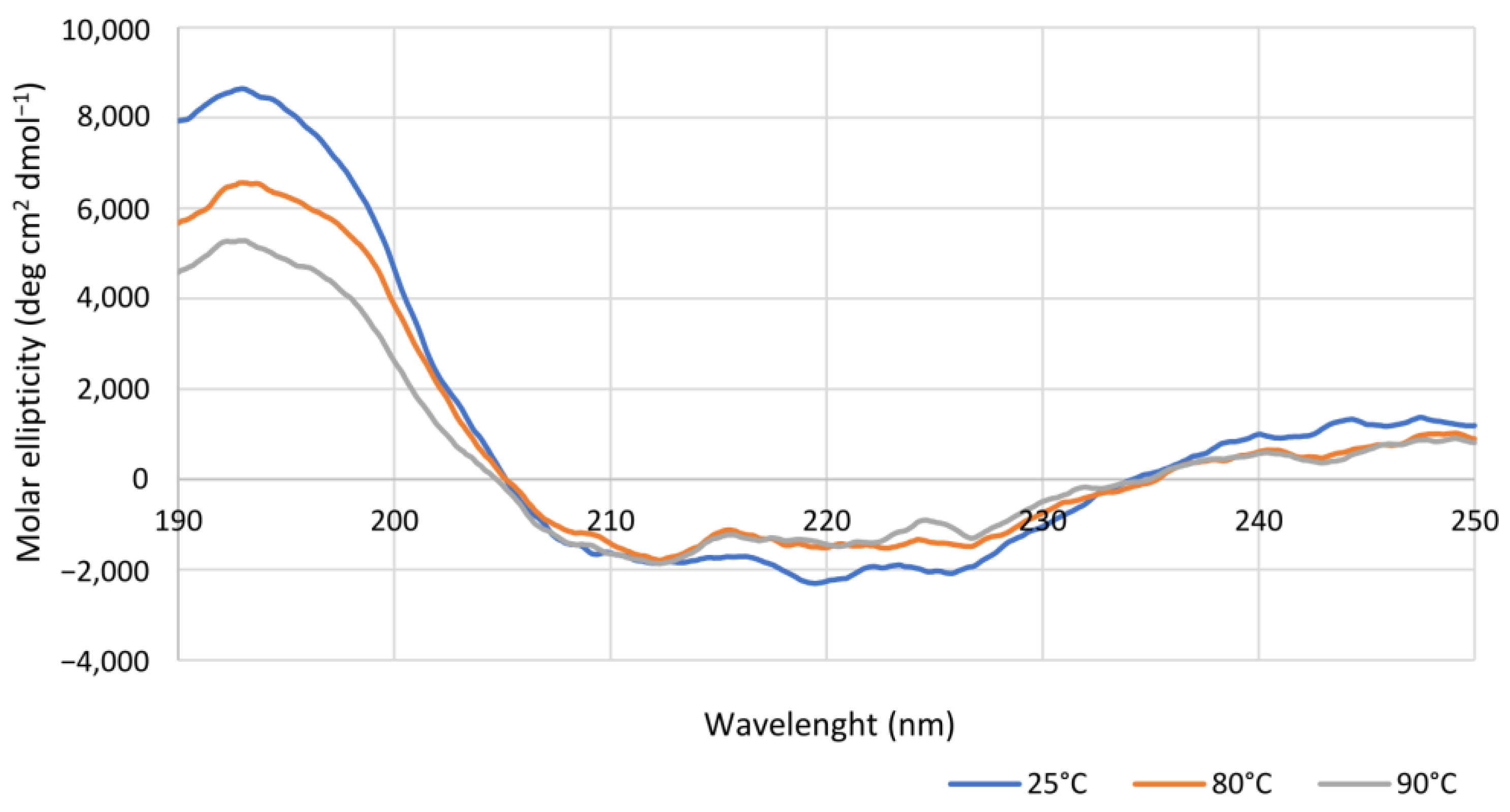
Coming to the effect of temperature on enzyme performances, Is2-SDR showed both a high thermophilicity (T
opt = 70 °C) and thermostability (T
M = 75 °C), these data being consistent with the environmental conditions of collection of the starting metagenomic DNA (hot spring, 85–90 °C, pH 5.0) [
21]. An overall good stability was also observed when exposing Is2-SDR to both water-miscible and water-immiscible organic solvents.
The extended investigation about the synthetic performances of Is2-SDR in the biocatalyzed reduction of a set of different carbonyl compounds (
Table 1) depicted a profile of a highly promiscuous ketoreductase showing interesting features in terms of substrate scope. Entries
17–
19 highlight how the typical pattern of carbonyl groups possessing a sterically large and a small Cα groups is preferred by Is2-SDR, as both benzaldehyde (
17) and acetophenone (
18, [
20]) were efficiently converted (with high e.e. in the case of the prochiral substrate
18), while the symmetrically substituted, bulky–bulky ketone
19 resulted in a low conversion (25%) under the same reaction conditions.
Is2-SDR was found able to convert 1,2-ketoesters (entries
20 and
21) with excellent conversions and e.e.s [
20], while the enzymatic reduction of the 1,2-diketone 1-phenylpropane-1,2-dione (
22) resulted in a complex mixture of regioisomeric alcohols, as this enzyme showed no specific regioselectivity for the 1 or 2 position of the substrate. No indications on the stereoselectivity in the reduction of
22 with Is2-SDR could be obtained as the outcome of the reaction could be analyzed only by the
1H NMR of the products mixture (see the material and methods section and the representative NMR spectra in the
supplementary materials).
Carbonyl group position resulted as crucial for Is2-SDR in the reduction of polycyclic compounds: α-tetralone (
23) was in fact efficiently reduced (conversion and e.e. of 93% and 98%), whereas β-tetralone (
24) reduction resulted in a very low conversion and e.e.. Further exploring polycyclic compounds with a higher degree of conformation freedom when compared to aryl–alkyl condensed systems, racemic
trans-decalone (
25) was found to be a good substrate for Is2-SDR, obtaining two different diastereomers with modest to very good e.e.s. [
20]. Is2-SDR was also able to reduce the Wieland–Miescher ketone, a structurally complex aliphatic bicyclic ketone (
26), catalyzing its kinetic resolution (see ref. [
22] for details).
The substrate scope of Is2-SDR was further investigated with a focus on bulky or bulky–bulky ketones [
35]. Zingerone (
27), whose structural features could be at first regarded as a “small–large” case ketone, represents a peculiar substrate for Is2-SDR given the fact the “large” group—the substituted phenyl ring—is connected to the carbonyl group by a flexible ethylene chain. Is2-SDR reduced
27 with a low conversion and a modest e.e. [
23]. Nevertheless, this enzyme was capable of converting the natural compound 6
S-gingerol (
28), a bulky–bulky substrate, which presents the same “large” group of
27 but is characterized by the presence of 1,3-hydroxyketone installed on the 6 and 8 positions (by traditional nomenclature) of a highly flexible decane chain, resulting in a modest conversion (50%) but in an excellent e.e. in favor of the 8
R,6
S diol [
23]. Interestingly, substrate
29, a 1,2-ketoester (formal) derivative of
27, was efficiently reduced by Is2-SDR (conversion and e.e. >99%) highlighting, as demonstrated also by entries
20 and
21, a preference towards this class of compounds.
In light of the broad substrate scope and the prevalent reductase activity shown by Is2-SDR, oxidized steroidal compounds were tested. Interestingly, a 3β/17β ketoreductase activity was demonstrated, thus, confirming the capability of this new biocatalyst of accepting sterically hindered substrates. This outcome could not be envisioned given the low sequence similarity with enzymes showing the same activity, i.e., 3β/17β-HSDHs (
Table S1, supplementary materials).
Moreover, the specific activities of Is2-SDR toward the accepted steroids and bile acids were about three orders of magnitude lower than those usually observed with “true” HSDHs [
20]; this finding further supporting the idea that these compounds are most likely reduced on the basis of Is2-SDR promiscuity. Additionally, at variance with the behavior of HSDHs in biphasic media [
36], it is worth noting that Is2-SDR, although quite stable when incubated in the presence of different water-immiscible organic solvents, was incapable of reducing the tested steroids in such heterogeneous systems, thus, possibly suggesting a very low affinity toward these compounds [
37].
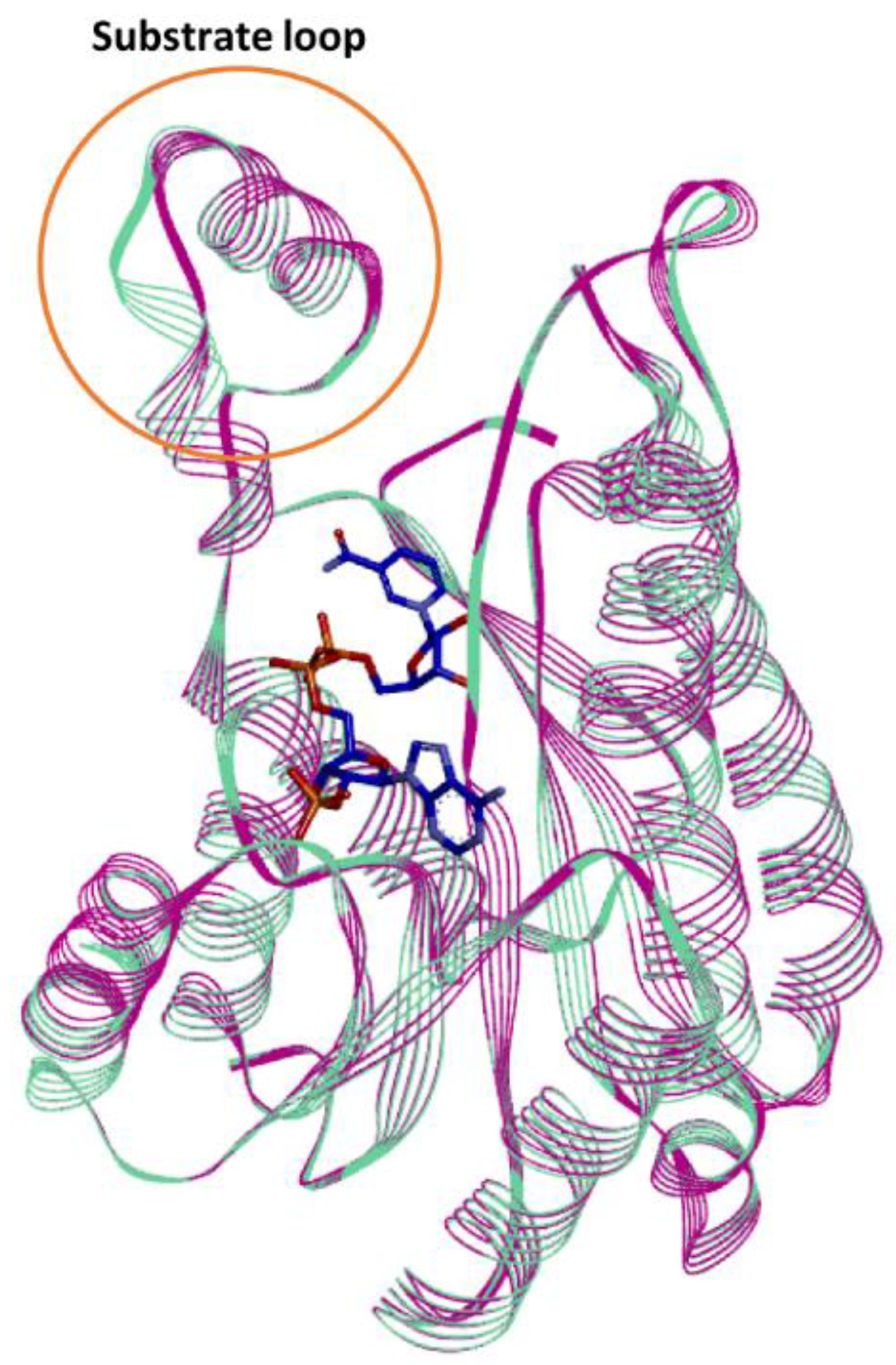
In silico studies of the Is2-SDR structural model built using the Lm-FabG structure revealed a substantial similarity between Is2-SDR and the proteins of the SDR superfamily in terms of overall folding and conserved residues involved in cofactor binding and catalytic activity. The substrate-binding loop, that allows the correct substrate recognition, shows a helix–loop–helix structure, which is typical of the SDR superfamily.
The structure similarity between Lm-FabG and Is2-SDR suggests that these two enzymes could partially share substrate specificity. In fact, Lm-FabG is a β-ketoacyl-acyl carrier protein reductase that reduces 1,3-keto-thioesters and it is involved in lipid metabolism being required for elongation of C4 to C16 acyl chains [
27]. Accordingly, Is2-SDR demonstrated to be active toward 1,2-keto esters, as stated above, and toward substrates containing long acyl chains, such as
28 and
29.
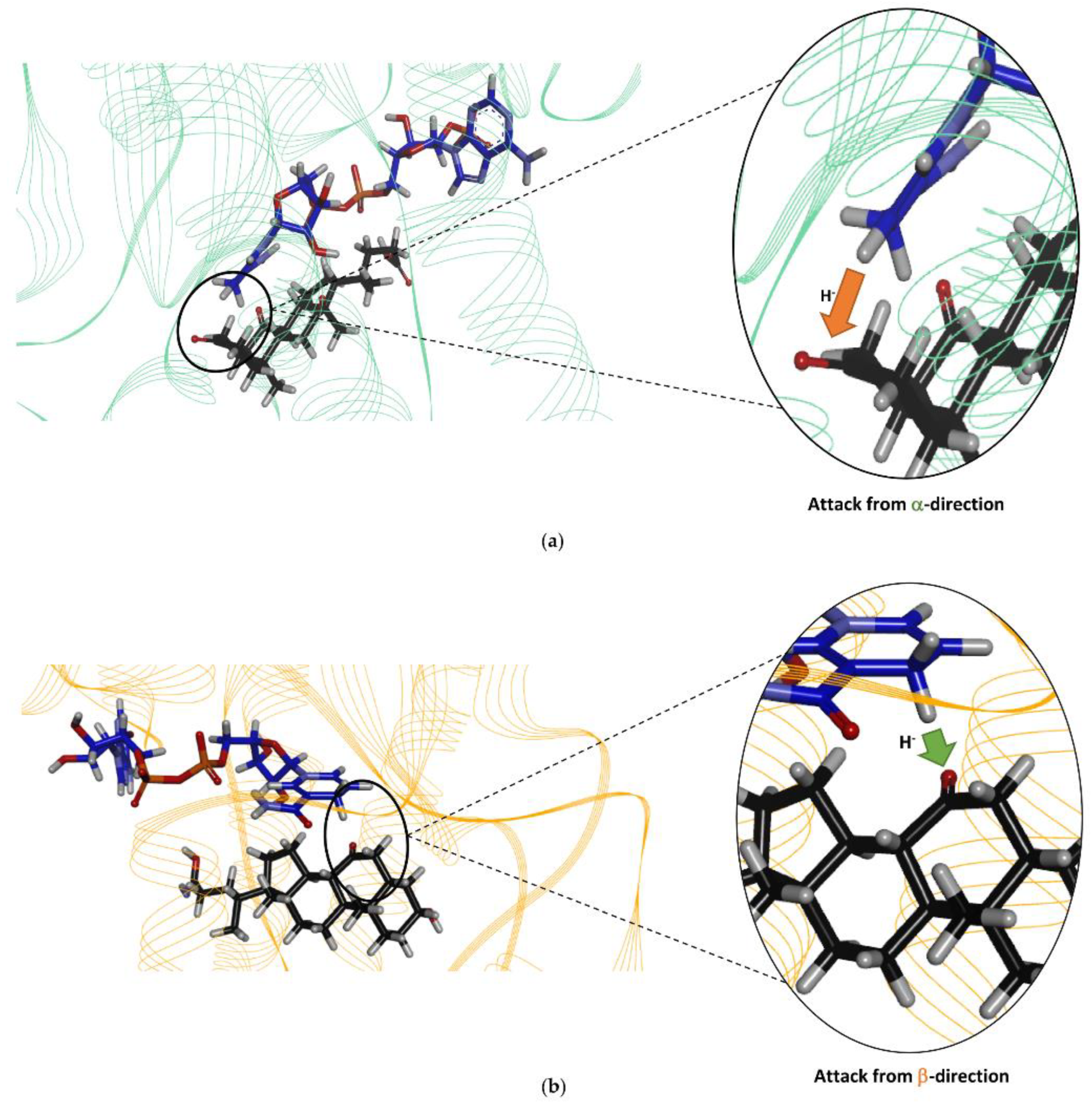
Finally, the docking of the bile acid DHCA in the Is2-SDR active site clearly showed that this substrate, as experimentally observed, can be suitably positioned for the reduction of the 3-keto group to 3β-OH.
In conclusion, the extensive characterization of Is2-SDR carried out in this work highlighted the synthetic potential and the broad substrate scope of a novel, thermostable ketoreductase that also showed an interesting promiscuous activity towards steroidal compounds.
4. Materials and Methods
4.1. General
All reagents and solvents were purchased from either Merck (Darmstadt, Germany), TCI (Zwijndrecht, Belgium), or Fluorochem (Hadfield, UK) and used without further purification, unless otherwise stated. The glucose dehydrogenase from
Bacillus megaterium (Bm-GDH) was recombinantly produced from
E. coli BL21(DE3) cells harboring the plasmid pKTS-GDH, as previously described [
38]. The NADP(H)-dependent FDH from
Pseudomonas sp. 101 (Ps-FDH) was a kind gift from Prof. Tishkov (M.V. Lomonosov Moscow State University). The recombinant production of Ps-FDH was carried out by inoculating an overnight preculture (100 mL) in 1 L of LB
KAN30 medium. The culture was maintained at 37 °C and 220 rpm up to an OD
600 of 0.4–0.6; then, the expression was induced by the addition of 0.5 M IPTG (final concentration: 1 mM). After incubation at 17 °C for 72 h, the cells were harvested by centrifugation, and enzyme purification was carried out by immobilized metal affinity chromatography (IMAC), as described for Is2-SDR in the following.
4.2. Is2-SDR Expression and Purification
E. coli BL21(DE3) cells harboring plasmid pETite Is2-SDR were plated on a Petri dish containing LBKAN30 agar medium and incubated at 37 °C o/n. Then, a single E. coli colony was picked, pre-inoculated in 100 mL of LBKAN30 and incubated for 24 h at 37 °C and 220 rpm. After that, 20 mL of preculture was inoculated and grown in 1 L of LBKAN30 at 37 °C and 220 rpm up to an OD600 of 0.4–0.6. Once the desired OD600 value was reached, the expression was induced by the addition of 0.5 M IPTG to a final concentration of 1 mM, and the bacterial culture was incubated at 200 rpm and 30 °C for 24 h.
The cells were harvested by centrifugation (30 min, 4 °C, 5000 rpm) and resuspended in 20 mL of wash buffer (20 mM potassium phosphate buffer (PB), pH 7.0, 500 mM NaCl, 20 mM imidazole). Then, the lysis of the bacterial cells was conducted by ultrasonication (5 cycles, 30 s each at 40% of maximum power, Omni Ruptor 250-Watt Ultrasonic Cell Distrupter), followed by centrifugation (30 min, 4 °C, 10,000 rpm). Protein purification was performed using immobilized metal affinity chromatography (IMAC) with Ni-NTA Sepharose 6 Fast Flow (GE Healthcare, Milan, Italy) as stationary phase. Protein-soluble fraction was incubated on ice with Ni-NTA Sepharose resin (10 mL) for 90 min, and then loaded into a glass column (10 × 110 mm) connected to a Gilson Minipuls 3 peristaltic pump (flow rate: 1 mL min−1). After washing the resin with wash buffer, protein elution was carried out by increasing the concentration of imidazole, up to 300 mM.
Protein fractions were quantified by using the Bradford method, collected, and dialyzed o/n in 20 mM PB, pH 7.0, at 4 °C overnight. The presence of the target protein in the eluted fractions was verified by SDS-PAGE analysis.
4.3. Enzyme Assays and Is2-SDR Characterization
Is2-SDR activity was determined spectrophotometrically by monitoring the oxidation of NADPH or reduction of NADP+ at 340 nm (ε: 6.22 mM−1 cm−1) in the presence of a suitable substrate at room temperature using disposable plastic cuvettes. A total of 2–10 µL of 1:10 diluted enzyme solution was added to the assay solution containing 2 mM substrate and 0.2 mM cofactor in 50 mM PB, pH 7.0 (1 mL final volume), before measuring absorbance. The enzymatic unit is defined as the amount of enzyme that catalyses the conversion of 1 μmole of substrate in one minute under defined assay conditions.
Is2-SDR optimum pH, optimum temperature, stability at different pHs, temperatures, and cosolvents have been evaluated by the above-described assay using ethyl-3-methyl-2-oxobutyrate (EMOB,
5,
Figure 2) as a substrate. Assays were performed at least in duplicate, and results were compared to blanks.
To determine enzyme pH optimum, activity assays were performed in PB in a range of pHs between 6.0 to 9.0. Enzyme stability at different pH values was estimated by diluting 1:50 purified enzyme solutions in PB at different pHs (6.0–9.0) and incubating the diluted enzyme at room temperature. Activity assays were performed immediately after the dilution and at scheduled times (1, 3, 5, and 24 h).
To determine enzyme temperature optimum, activity assays were performed in a thermostatic spectrophotometer at temperatures between 20–90 °C. Once the instrument was set up at the desired temperature, a cuvette containing the substrate solution (2 mM in 50 mM PB, pH 7.0) was incubated in a thermostatic bath at the same temperature for 10 min. Then, 20 μL of 10 mM cofactor solution (NADPH in water) and 2–50 µL of 1:10 diluted enzyme solution were quickly added, and the cuvette was transferred to the instrument for the analysis. Enzyme stability at different temperatures was evaluated by diluting 1:50 the Is2-SDR purified solution in 50 mM PB, pH 7.0, in 1.5 mL tubes and incubating the solution at the desired temperature in a thermomixer. Activity assays were performed immediately after the dilution and at scheduled times (1, 3, 5, and 24 h).
The stability of Is2-SDR was evaluated in the presence of various organic solvents (methanol, ethanol, dimethyl sulfoxide, and acetonitrile) at different concentrations (5, 10, and 20 % (v/v)) and in biphasic systems in the presence of water-immiscible organic solvents (toluene, ethyl acetate, petroleum ether, tert-butylmethylether (tBME), and cyclopentyl methyl ether (CPME)). Specifically, Is2-SDR was diluted 1:50 with 50 mM PB, pH 7.0, and the organic solvent was added to reach the desired concentration, or an equivalent volume of a water-immiscible solvent was added to the diluted enzyme. The mixtures were incubated in a thermoshaker at 25 °C and 100 rpm, and activity assays were performed immediately and at scheduled times (1, 3, 5, and 24 h).
Bm-GDH and Ps-FDH activity assays were performed following the above-described assay procedure. For GDH, assay mixture was 50 mM glucose, 50 mM PB, pH 7.0, 0.2 mM NAD(P)+, while for FDH, assay mixture was 20 mM ammonium formate, 50 mM PB, pH 7.0, 0.2 mM NAD(P)+.
4.4. Bioinformatic and Protein Structure Analysis
Sequence alignments were carried out using standard protein Blast tool (
https://blast.ncbi.nlm.nih.gov/, accessed on 1 April 2022). Phylogenetic trees were created using the Clustal Omega tool (
http://www.ebi.ac.uk/Tools/msa/clustalo/, accessed on 1 April 2022) [
39] and visualized with iTOL webserver (
http://itol.embl.de/, accessed on 1 April 2022) [
40]. Is2-SDR homology model was generated through the SWISS MODEL web server [
26]. 4JRO corresponding to the 3-oxoacyl-[acyl-carrier protein]reductase (FabG) from
Listeria monocytogenes was selected as the template to build up the homology model. The quality of the model was assessed by using PROCHECK [
41] and Verify 3D tools [
42] available on the UCLA-DOE LAB platform (
https://saves.mbi.ucla.edu/, accessed on 1 May 2022). Protein visualization and ligand optimization were carried out using the Discovery Studio package 2021 (version 21.1.0 BIOVIA, San Diego, CA, USA). The conformers of the optimized ligand were generated using the Mercury module of the CCDC suite (
https://www.ccdc.cam.ac.uk, accessed on 1 May 2022) and docked into the Is2-SDR active site using the GOLD module of the Hermes CCDC-suite [
43].
4.5. Analytical Methods
CD spectra were recorded on a nitrogen-flushed Jasco J-1100 spectropolarimeter (Easton, MD, USA) interfaced with a thermostatically controlled cell holder. CD analyses were carried out in quartz cuvettes with 0.1 cm path length using purified Is2-SDR diluted in degassed water to a concentration of 0.15 mg mL−1. CD spectra were recorded in the range between 190 and 250 nm at 25, 80, or 90 °C, while for the determination of apparent melting temperature (TM), CD signal variations were detected at 220 nm using the following temperature program: 20 up to 65 °C at 5 °C min−1, data pitch each 2 °C, hold 30 s; 65 up to 90 °C at 2.5 °C min−1, data pitch each 0.5 °C, hold 30 s.
The NMR spectra were acquired in CDCl3, DMSO-d6, or in CD3OD, at room temperature (rt) on a Bruker AV 400 MHz spectrometer with a z gradient at 400 MHz for 1H-NMR analysis and 101 MHz for 13C-NMR.
Reactions were monitored via TLC (thin-layer chromatography) on pre-coated glass plates silica gel 60 with fluorescent indicator UV254 and treated with oxidizing solutions (phosphomolybdic reagent: (NH4)6MoO4 42 g, Ce(SO4)2 2 g, H2SO4 62 mL, H2O 1 L; Komarowski reagent: 4-hydroxybenzaldehyde, 3 g; MeOH, 200 mL; H2SO4 50%, 20 mL). At scheduled times, reaction samples were extracted with EtOAc and dried over Na2SO4, then submitted to chiral HPLC, GC, GC-MS, or NMR.
HPLC analyses were conducted using a Shimadzu LC-20AD high-performance liquid chromatography system equipped with a Shimadzu SPD-20 A UV detector. The samples, as EtOAc solutions, were analyzed on a Chiralcel OD (10 µm, 250 × 4.6 mm) through an isocratic elution (petroleum ether/isopropanol = 98:2), with detection at 210 nm. Retention times of ethyl 2-oxo-4-phenylbutanoate and
R- and
S-ethyl 2-hydroxy-4-phenylbutanoate were: 8.71 min, 16.02 min, and 10.45 min, which is in agreement with the literature reference data [
44].
GC analyses were performed on an AGILENT 6850 (Net-work GC System) gas chromatograph equipped with a chiral capillary column (MEGA DEX DAC-BETA, Legnano, Italy; 25 m × 0.25 mm × 0.25 μm) and a flame ionization detector. At scheduled times, reactions samples (100 µL) were extracted with an equal volume of EtOAc and injected into the GC system. Different temperature gradients were applied to analyze the different compounds (according to the details reported below) using a general instrument setup: Tinjector = 200 °C, Tdetector = 250 °C, flow = 1 mL min−1, p= 100 kPa.
GC-MS analyses were performed using an Agilent HP-5MS column (30 m × 0.25 mm × 0.25 μm) on a Finnigan TRACE DSQ GC/MS instrument (Thermo Quest, San Jose, CA, USA). Inlet temperature: 280 °C; ion source temperature: 280 °C; MS transferline temperature: 280 °C.
4.6. Preparation of Standard Racemates
The chemical reductions of the substrates were performed following a standard protocol using NaBH4 as reducing agent. To a stirred solution of 0.15 M substrate (1 eq) in EtOH at 0 °C, NaBH4 (0.25 to 1 eq) was added. The reaction was maintained at 0 °C until the complete dissolution of NaBH4, then at rt for 4 h. After the indicated time, the reaction was quenched with a saturated solution of NH4Cl, then extracted with EtOAc (3×). Combined organic layers were dried over Na2SO4, concentrated in vacuo, affording the desired racemic alcohols that were used without any further purification. The racemic products were characterized by GC, HPLC, or NMR.
4.7. Analytical Scale, Biocatalyzed Reduction of Ketone Substrates with Is2-SDR
General screening protocol (final volume = 1–2 mL): BmGDH, NADP+, Is2-SDR and substrate (variable amounts depending on reduced compound, see details below), and 50 mM glucose were dissolved in a 10% (v/v) solution of DMSO in 50 mM PB, pH 7.0. Reaction mixtures were incubated at 25 °C, 180 rpm for 24–48 h. Conversions and enantiomeric excesses were determined by GC, HPLC, and/or NMR analyses after extraction with EtOAc.
4.7.1. Enzymatic Reduction of 17
Reaction mixture: 10 mM
17, 2.0 U mL
−1 BmGDH, 4.0 U mL
−1 Is2-SDR, and 0.4 mM NADP
+. TLC = DCM, phosphomolybdic reagent; GC = 100 °C for 30 min, T
injector = T
detector = 220 °C, flow = 1 mL min
−1, p= 103 kPa
Rt: 17, 8.62 min; benzaldehyde, 3.47 min (
Figure S8, supplementary materials), in agreement with the literature reference data [
45].
4.7.2. Enzymatic Reduction of 18
Reaction mixture: 10 mM
18, 1.0 U mL
−1 BmGDH, 4.0 U mL
−1 Is2-SDR, and 0.4 mM NADP
+. TLC = petroleum ether: EtOAc 9:1 (
v/v), UV
254nm. GC = 70 °C for 5 min, 2 °C/min until 120 °C, hold 8 min, 20 °C/min until 180 °C, hold 1 min,
Rt:
18 13.98 min,
R-1-phenylethan-1-ol 20.84 min,
S-1-phenylethan-1-ol 21.34 min (
Figure S9, supplementary materials).
4.7.3. Enzymatic Reduction of 19
Reaction mixture: 10 mM
19, 2.0 U mL
−1 BmGDH, 4.0 U mL
−1 Is2-SDR, and 0.4 mM NADP
+. TLC = DCM: EtOAc 9:1 (
v/v), permanganate solution. GC-MS = 60 °C, hold 1 min, 6 °C min
−1 until 150 °C, hold 1 min, 12 °C min
−1 until 280 °C, hold 5 min,
Rt:
19 20.58 min, diphenylmethanol 20.64 min (
Figure S10, supplementary materials).
4.7.4. Enzymatic Reduction of 22
Reaction mixture: 10 mM
22, 1.0 U mL
−1 BmGDH, 1.0 U mL
−1 Is2-SDR, and 0.4 mM NADP
+. TLC = DCM:MeOH 98:2 (
v/
v), phosphomolybdic reagent.
1H-NMR = 1-phenylpropane-1,2-dione ppm, singlet at 2.00 ppm PhCOCO
CH3; 1-hydroxy-1-phenylpropan-2-one, quartet centered at 5.25 ppm PhC
H(OH)COCH
3; 2-hydroxy-1-phenylpropan-1-one, singlet at 5.17 ppm PhCOC
H(OH)CH
3; 1-phenylpropane-1,2-diol, broad doublet centered at 4.36 ppm PhC
H(OH)CH(OH)CH
3 (
Figure S11, supplementary materials).
4.7.5. Enzymatic Reduction of 23
Reaction mixture: 10 mM
23, 1.0 U mL
−1 BmGDH, 4.0 U mL
−1 Is2-SDR, and 0.2 mM NADP
+. TLC = petroleum ether: EtOAc 9:1 (
v/
v), UV
254nm. GC = 120 °C for 20 min,
Rt:
23 9.28 min,
R-α-tetralol 10.49,
S-α-tetralol 12.17 min (
Figure S12, supplementary materials), in agreement with the literature reference data [
46].
4.7.6. Enzymatic Reduction of 24
Reaction mixture: 10 mM
24, 1.0 U mL
−1 BmGDH, 4.0 U mL
−1 Is2-SDR, and 0.4 mM NADP
+. TLC = petroleum ether: EtOAc 9:1 (
v/
v), UV
254nm. GC = after derivatization as acetates (Ac
2O, Py in EtOAc) 110 °C for 40 min,
Rt:
24 31.96 min,
R-β-tetralol acetate 27.71 min,
S-β-tetralol acetate diphenylmethanol 28.64 min (
Figure S13, supplementary materials), in agreement with the literature reference data [
47].
4.7.7. Enzymatic Reduction of 29
Reaction mixture: 20 mM
29, 1.0 U mL
−1 BmGDH, 4.0 U mL
−1 Is2-SDR, and 0.2 mM NADP
+. TLC = DCM, phosphomolybdic reagent, UV
254nm. HPLC = method described in material and methods,
Rt:
29 8.71 min,
R-ethyl 2-hydroxy-4-phenylbutanoate 16.02 min,
S-ethyl 2-hydroxy-4-phenylbutanoate 10.45 min (
Figure S14, supplementary materials), in agreement with the literature reference data [
44].
4.8. Is2-SDR Biocatalyzed Reduction of Bile Acids and Steroids
4.8.1. Enzymatic Reduction of Dehydrocholic Acid (30)
Dehydrocholic acid (30, 25 mg, 0.01 mmol) was dissolved (5 mM) in 2.4 mL of 100 mM PB, pH 8.0. To this solution, glucose (25 mM), NADP+ (0.4 mM), BmGDH (2.0 U mL−1), and Is2-SDR (3.0 U mL−1) were added. The obtained mixture was incubated at 25 °C for 24 h and monitored by TLC analysis (DCM: MeOH 9:1 (v/v), Komarovsky reagent). After the complete conversion of the starting material, the reaction mixture was taken to pH 4, and it was freeze-dried. Reduction product was recovered by taking up the solid residue with EtOAc and by in vacuo concentration. The sample was dissolved in deuterated DMSO, and the NMR spectra were acquired on a Bruker AV 400 instrument at 302 K. Since the spectrum is quite complex, only a partial analysis was possible. The proton and carbon assignments were based on the proton homonuclear experiment COSY and on the proton–carbon heteronuclear experiments HSQC and HMBC. The multiplicity of the carbon nuclei was determined by the DEPT 135 experiment, which allows to distinguish among quaternary (s), methyne (d), methylene (t), and methyl (q) carbons. 1H NMR (DMSO-D6), δ: 3.79 (1H, t br, J = 2.7 Hz, H-3), 2.98 (1H, dd, J6ax,5 = 9.3, J6ax.6eq = 12.6, H-6ax), 2.96 (1H, t, J = 13.0, J = 13.0, H-8), 2.75 (t, J = 12.7, J = 12.7 Hz, H-11ax), 2.18 (1H, m, H-5), 2.06 (1H, td, J11ax,9 = 12.6, J8,9 = 12.6 and J11eq,9 = 5.1 Hz, H-9), 1.88 (1H, J = 12.4 and J = 5.1 Hz, H-11eq), 1.58 (1H, m, H-14), 1.68 (1H, dd, J = 12.8 and 2.4 Hz, H-6eq), 1.26 (3H, s, CH3-19), 0.98 (3H, s, CH3-18), 0.75(3H, d, J = 6.3 Hz, CH3-21); 13C NMR (DMSO-D6), δ: 212.5 (1C, s. CO-12), 210.4 (1C, s, CO-7), 174.8 (1C, s, COO-24), 63.8 (1C, d, C-3), 56.3 (1C, s, C-13), 51.8 (1C, d, C-14), 48.0 (1C, d, C-8), 45.3 (1C, d, C-17), 44.8 (1C, t, C-6), 44.5 (1C, d, C-9), 39.9 (1C, d, C-5), 38.4 (1C, t, C-11), 35.9 (1C, s, C-10), 35.0 (1C, d, C-20), 34.5 (1C, t), 31.1 (1C, t), 30.4 (1C, t, C-22), 28.7 (1C, t), 27.2 (1C, t), 26.9 (1C, t), 24.6 (1C, t), 22.7 (1C, q, CH3-19), 18.6 (1C, q, CH3-21), 11.4 (1C, q, CH3-18).
4.8.2. Enzymatic Reduction of 5α-Dihydrotestosterone (31)
5α-Dihydrotestosterone (31, 10 mg, 0.03 mmol) was dissolved (5 mM) in a 10% (v/v) solution of DMSO in 6.8 mL of 50 mM PB, pH 7.0. To this solution, glucose (25 mM), NADP+ (0.4 mM), BmGDH (2.0 U mL−1), and Is2-SDR (3.0 U mL−1) were added. The obtained suspension was incubated at 25 °C for 96 h and monitored by TLC analysis (DCM: MeOH 95:5 (v/v), Komarovsky reagent). After extraction with EtOAc and anhydrification over Na2SO4, the crude product was purified by flash column chromatography [DCM: MeOH 95:5 (v/v)] to yield a reduction product whose structure was attributed to 5α-androstane-3β,17β-diol (5.2 mg, 0.02 mmol, 60%).
1H-NMR (CDCl
3, 400 MHz) δ 3.56 (t,
J = 8.6 Hz, 1H), 3.53–3.46 (m, 1H), 2.03–1.90 (m, 1H), 1.87–1.65 (m, 4H), 1.64–0.98 (m, 17H), 0.85 (s, 3H), 0.73 (s, 3H). Signals in accordance with the literature data [
48].
4.8.3. Enzymatic Reduction of Androsterone (32)
Androsterone (32, 20 mg, 0.07 mmol) was dissolved (5 mM) in a 40% (v/v) solution of DMSO in 14 mL of 50 mM PB, pH 7.0. To this solution, glucose (25 mM), NADP+ (0.4 mM), BmGDH (2.0 U/mL), and Is2-SDR (3.0 U mL−1) were added. The obtained mixture was incubated at 25 °C for 96 h and monitored by TLC analysis (DCM: MeOH 95:5 (v/v), Komarovsky reagent). After extraction with EtOAc and anhydrification over Na2SO4, the crude product was purified by preparative TLC [DCM: MeOH 95:5 (v/v)] to yield a reduction product whose structure was attributed to 5α-androstane-3α,17β-diol (8.4 mg, 0.03 mmol, 42.8%).
1H-NMR (CDCl
3, 400 MHz) δ 3.97 (q,
J = 2.4 Hz, 1H), 3.56 (t,
J = 8.6 Hz, 1H), 1.77–1.68 (m, 1H), 1.64–1.26 (m, 21H), 0.72 (s, 3H), 0.66 (s, 3H). Signals in accordance with literature data [
48].


 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}












