
A Mitophagy-Related Gene Signature for Subtype Identification and Prognosis Prediction of Hepatocellular Carcinoma


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
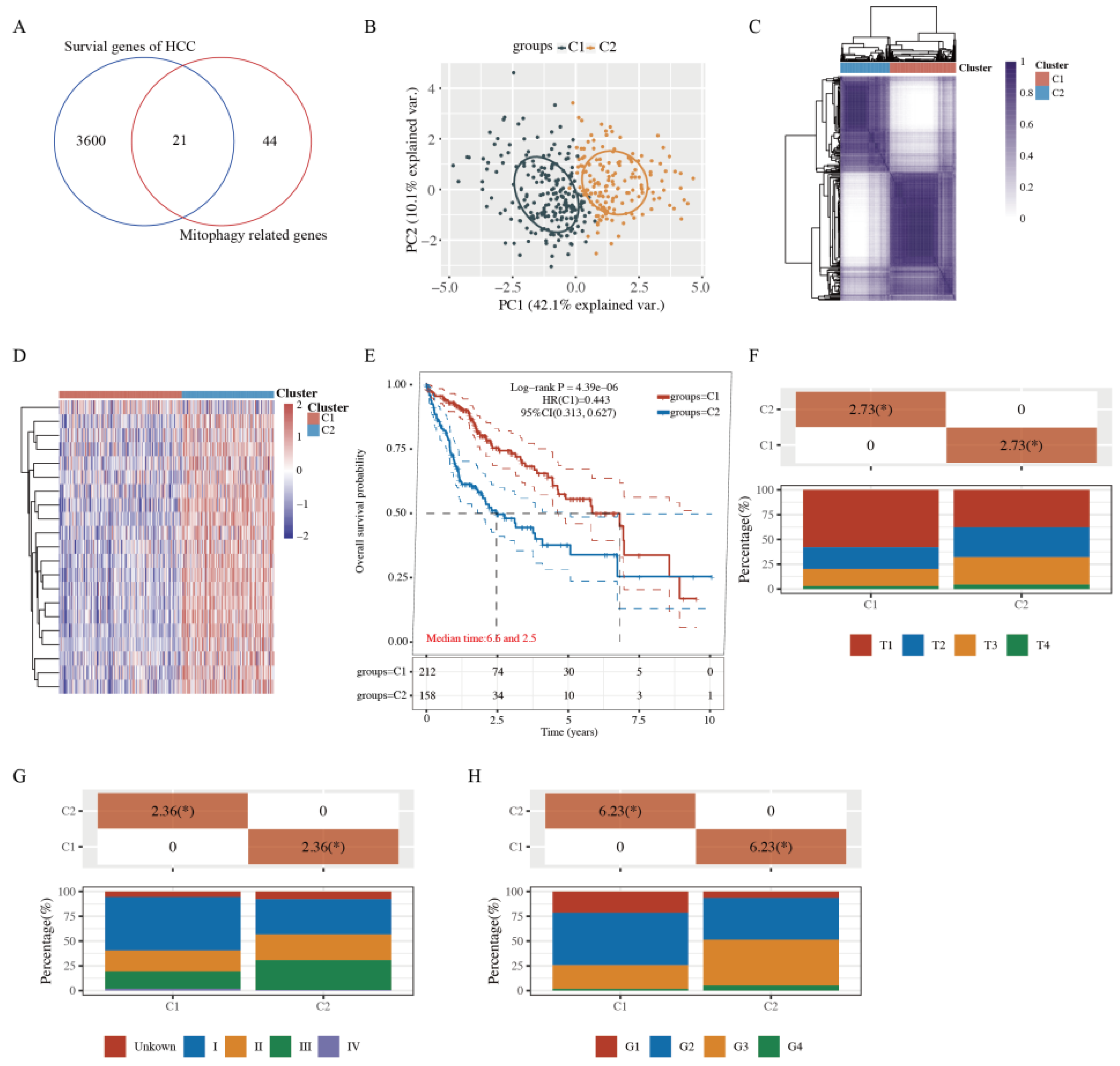
2.1. Tumor Classification Based on MRGs
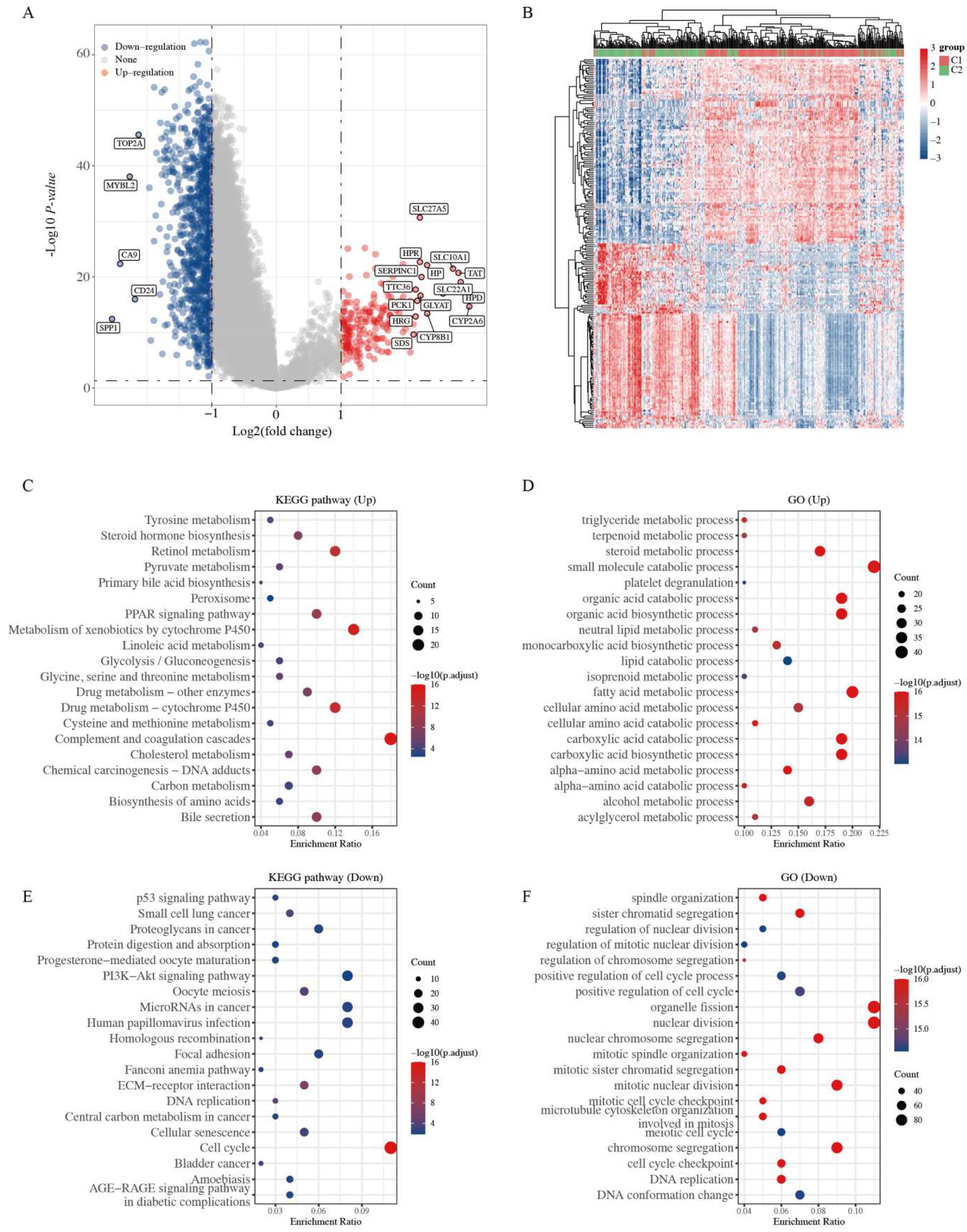
2.2. Functional Enrichment Analysis of Two Clusters
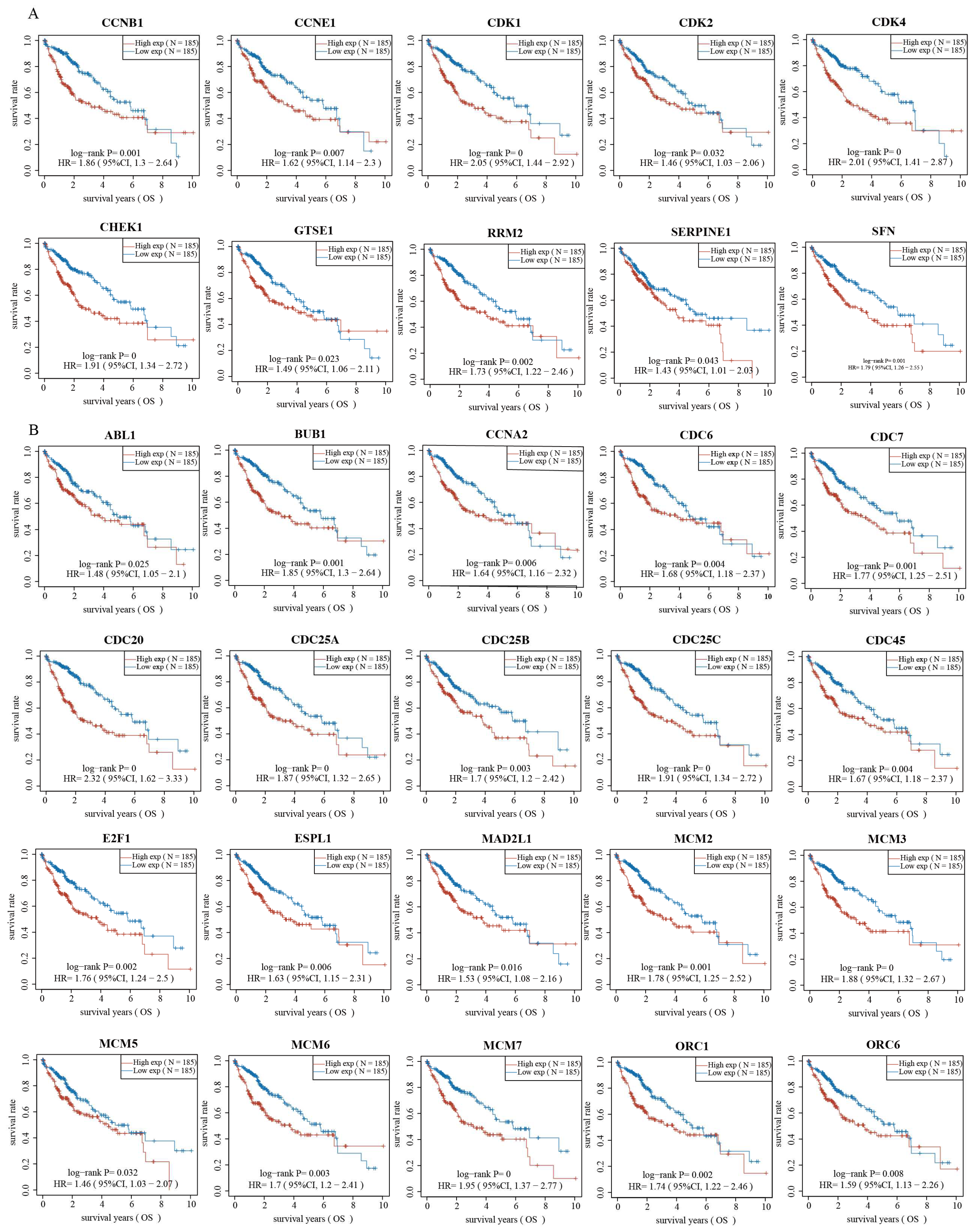
2.3. Validation of Genes Associated with the p53 Signaling Pathway and Cell Cycle
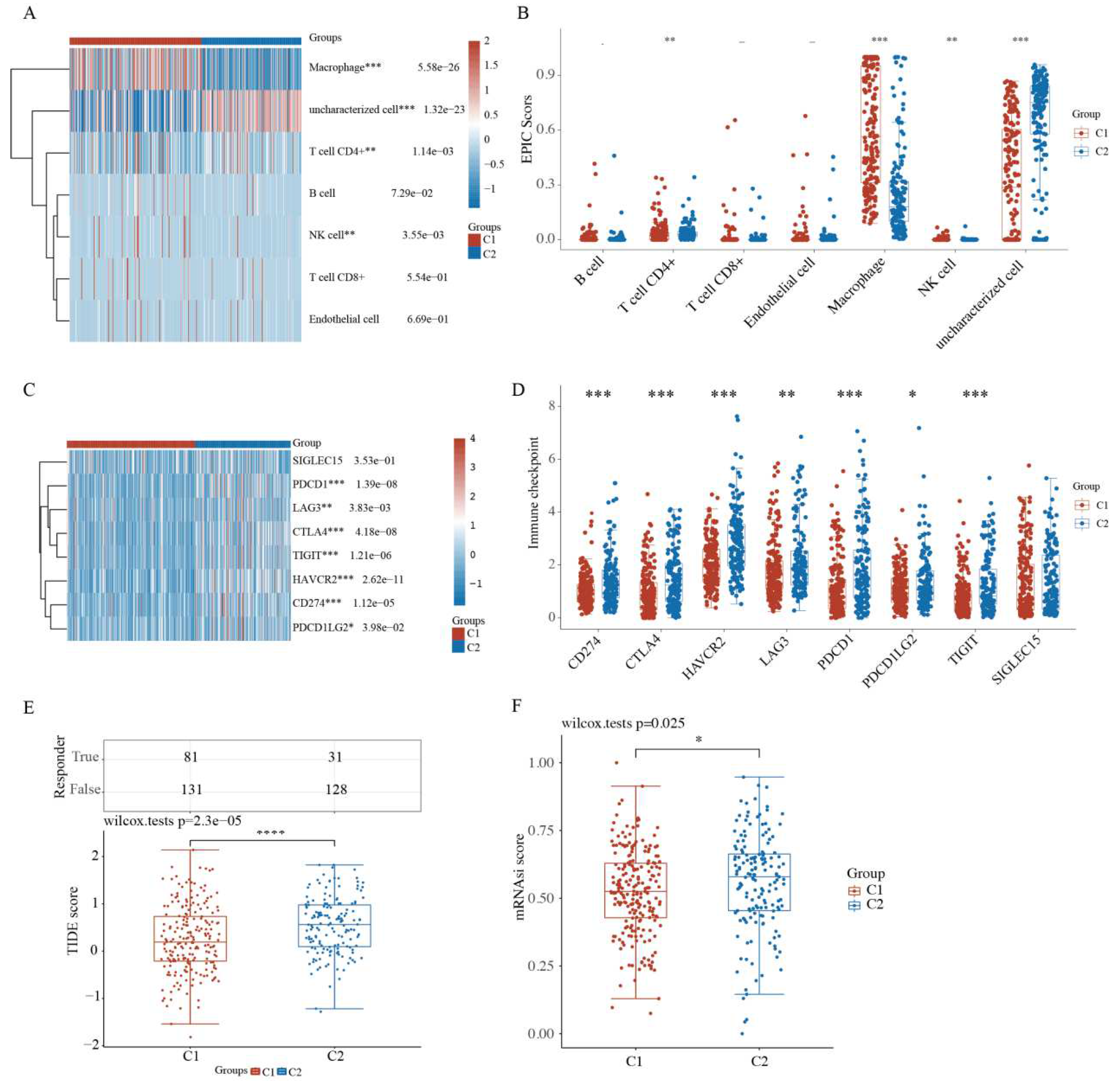
2.4. Comparison of Immune Activity and Tumor Stemness between HCC Subtypes
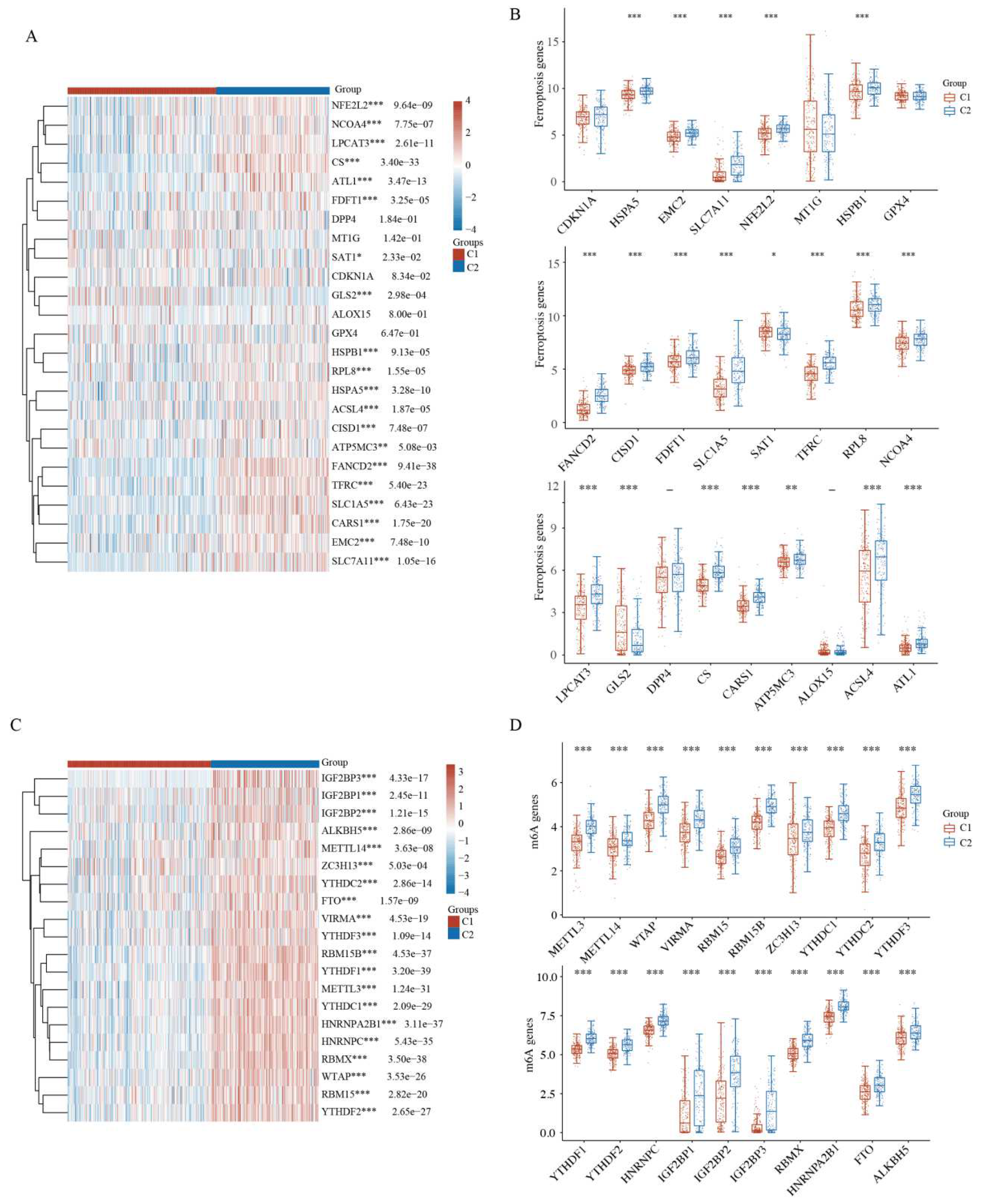
2.5. Comparison of Ferroptosis and N6-Methyladenosine between HCC Subtypes
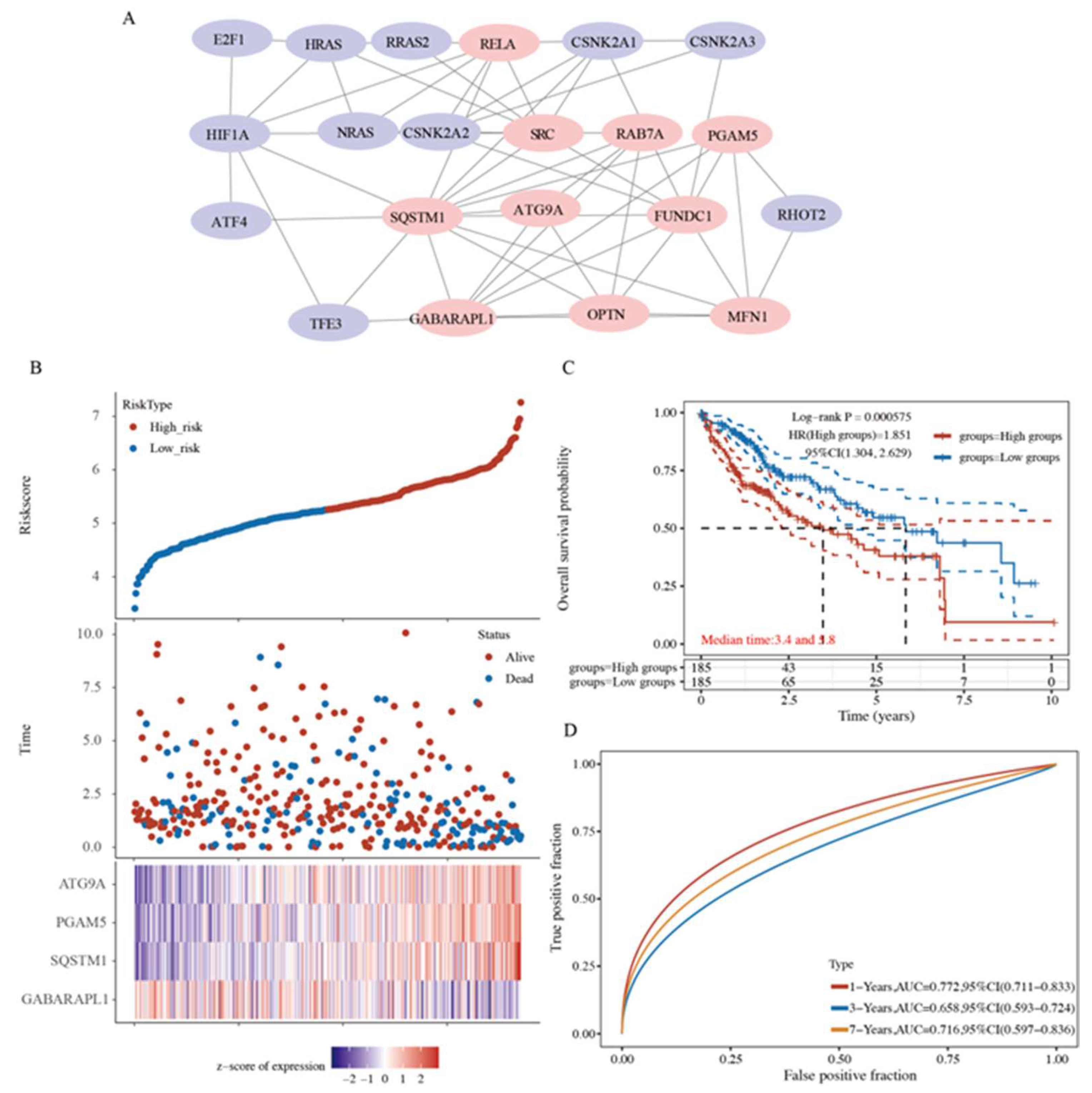
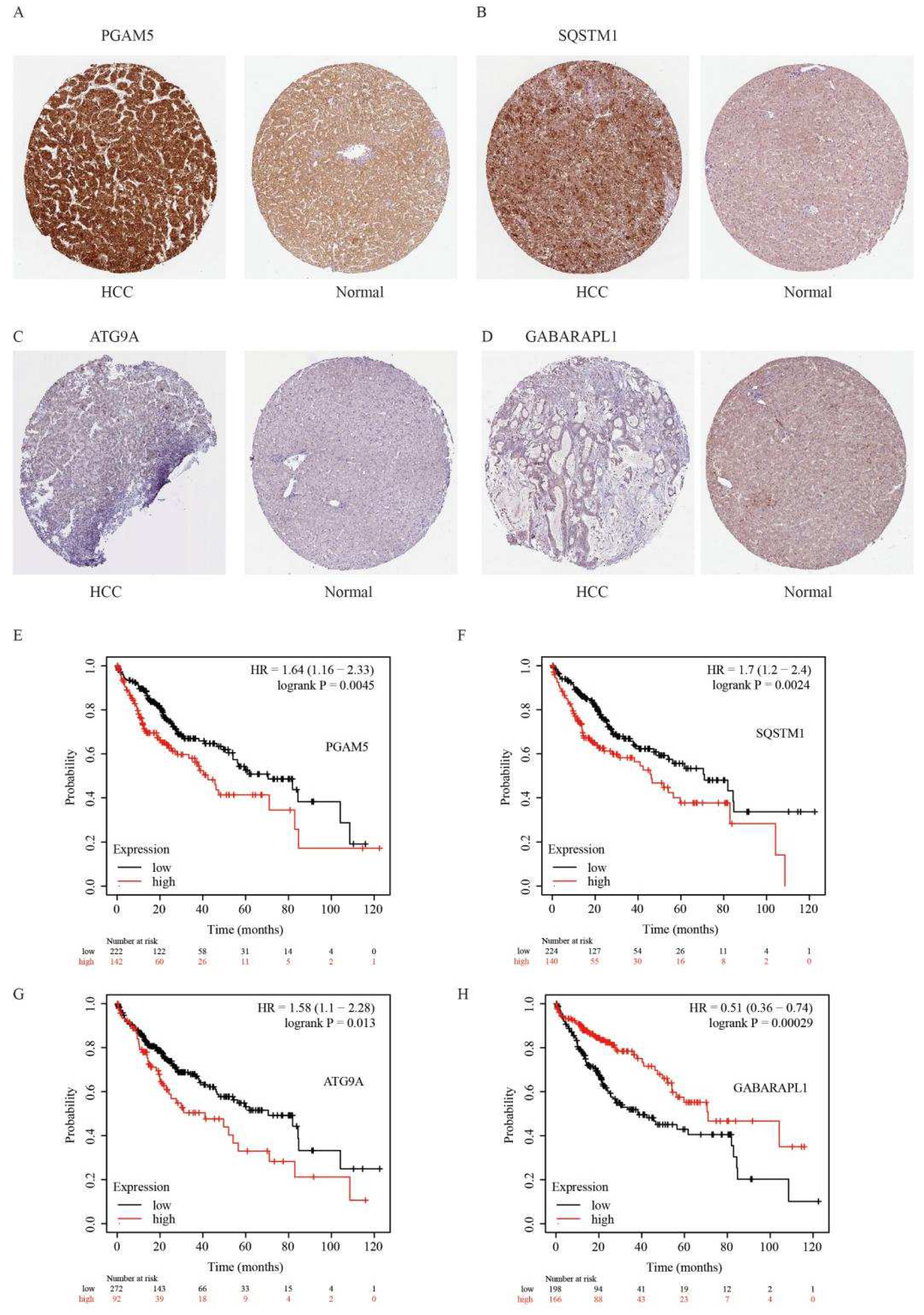
2.6. A Prognostic Model Construction in the TCGA-HCC Cohort
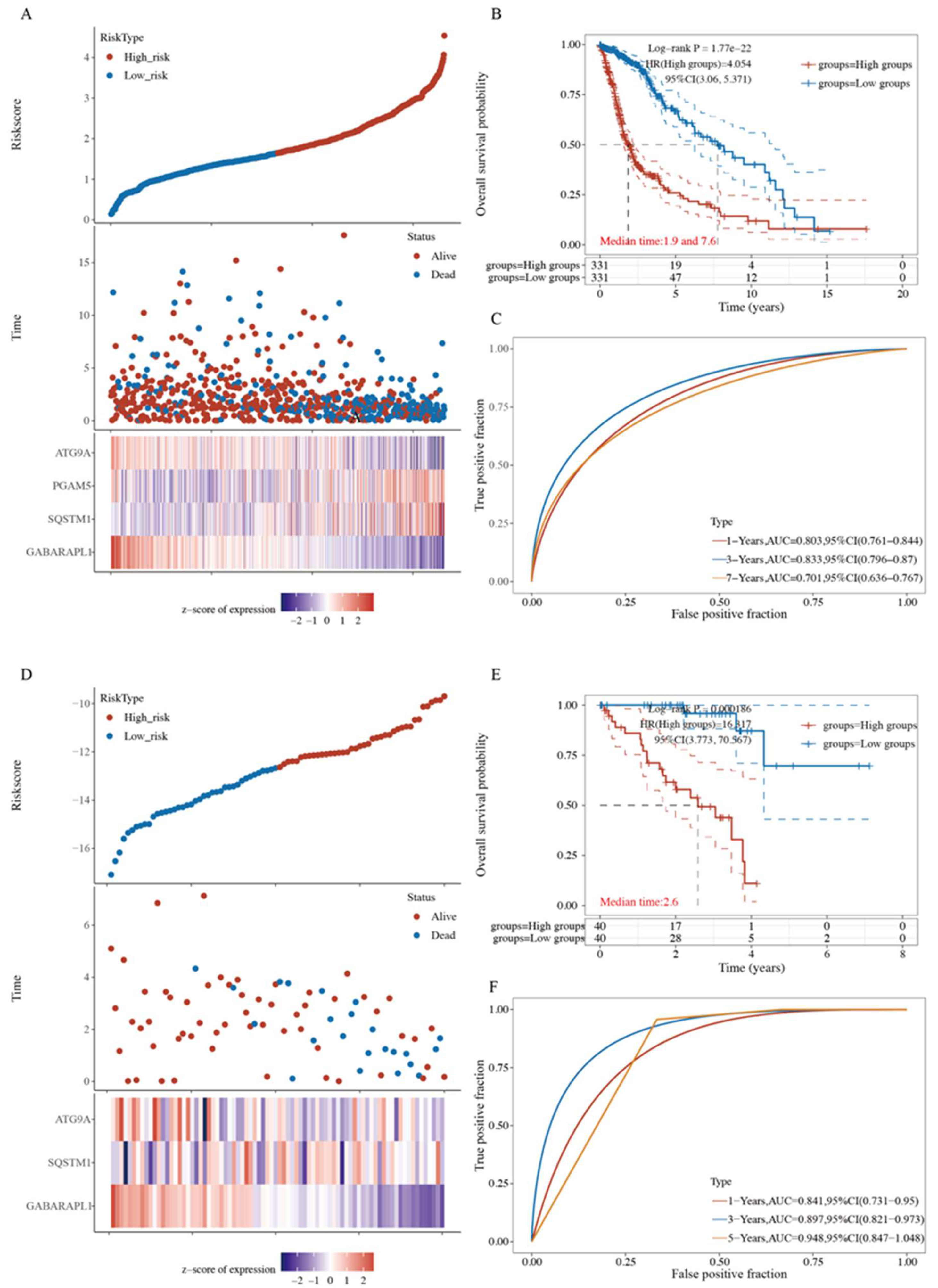
2.7. Four-Gene Signature Validation in Pan-Cancer TCGA Cohorts
3. Discussion
4. Materials and Methods
4.1. Data Collection Information of HCC Patients
4.2. Selection of MRGs and Consensus Clustering Analysis
4.3. Identification of Differentially Expressed Genes and Enrichment Analysis
4.4. Immune Infiltration and Immune Checkpoints Analysis
4.5. Aberrancies and Functional Implications of Ferroptosis and N6-methyladenosine [m6A)
4.6. Gene Signature Identification
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hepatocellular Carcinoma. Nat. Rev. Dis. Primers 2021, 7, 7. [CrossRef] [PubMed]
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.X.; Yang, T.W.; Yin, J.M.; Yang, P.X.; Kou, B.X.; Chai, M.Y.; Liu, X.; Chen, D. Progress and Prospects of Biomarkers in Primary Liver Cancer (Review). Int. J. Oncol. 2020, 57, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: Globocan Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Gera, S.; Ettel, M.; Acosta-Gonzalez, G.; Xu, R. Clinical Features, Histology, and Histogenesis of Combined Hepatocellular-Cholangiocarcinoma. World J. Hepatol. 2017, 9, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Anwanwan, D.; Singh, S.K.; Singh, S.; Saikam, V.; Singh, R. Challenges in Liver Cancer and Possible Treatment Approaches. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188314. [Google Scholar] [CrossRef] [PubMed]
- Renne, S.L.; Sarcognato, S.; Sacchi, D.; Guido, M.; Roncalli, M.; Terracciano, L.; Di Tommaso, L. Hepatocellular Carcinoma: A Clinical and Pathological Overview. Pathologica 2021, 113, 203–217. [Google Scholar] [CrossRef] [PubMed]
- Tsilimigras, D.I.; Pawlik, T.M. Prognostication in Hepatocellular Carcinoma: Is It a Burden or a Ticket? Br. J. Surg. 2021, 108, 337–339. [Google Scholar] [CrossRef]
- Vyas, M.; Zhang, X. Hepatocellular Carcinoma: Role of Pathology in the Era of Precision Medicine. Clin. Liver Dis. 2020, 24, 591–610. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, X.; Li, X.; Bao, H.; Li, G.; Li, N.; Li, H.; Dou, J. Connection between Cdc20 Expression and Hepatocellular Carcinoma Prognosis. Med. Sci. Monit. 2021, 27, e926760. [Google Scholar] [CrossRef]
- Hartke, J.; Johnson, M.; Ghabril, M. The Diagnosis and Treatment of Hepatocellular Carcinoma. Semin. Diagn. Pathol. 2017, 34, 153–159. [Google Scholar] [CrossRef]
- Pinero, F.; Dirchwolf, M.; Pessoa, M.G. Biomarkers in Hepatocellular Carcinoma: Diagnosis, Prognosis and Treatment Response Assessment. Cells 2020, 9, 1370. [Google Scholar] [CrossRef]
- Onishi, M.; Yamano, K.; Sato, M.; Matsuda, N.; Okamoto, K. Molecular Mechanisms and Physiological Functions of Mitophagy. EMBO J. 2021, 40, e104705. [Google Scholar] [CrossRef]
- Poole, L.P.; Macleod, K.F. Mitophagy in Tumorigenesis and Metastasis. Cell Mol. Life Sci. 2021, 78, 3817–3851. [Google Scholar] [CrossRef]
- Panigrahi, D.P.; Praharaj, P.P.; Bhol, C.S.; Mahapatra, K.K.; Patra, S.; Behera, B.P.; Mishra, S.R.; Bhutia, S.K. The Emerging, Multifaceted Role of Mitophagy in Cancer and Cancer Therapeutics. Semin. Cancer Biol. 2020, 66, 45–58. [Google Scholar] [CrossRef]
- Ma, X.; McKeen, T.; Zhang, J.; Ding, W.X. Role and Mechanisms of Mitophagy in Liver Diseases. Cells 2020, 9, 837. [Google Scholar] [CrossRef]
- Thangaraj, A.; Periyasamy, P.; Guo, M.L.; Chivero, E.T.; Callen, S.; Buch, S. Mitigation of Cocaine-Mediated Mitochondrial Damage, Defective Mitophagy and Microglial Activation by Superoxide Dismutase Mimetics. Autophagy 2020, 16, 289–312. [Google Scholar] [CrossRef]
- Han, S.; Jeong, Y.Y.; Sheshadri, P.; Cai, Q. Mitophagy Coordination with Retrograde Transport Ensures the Integrity of Synaptic Mitochondria. Autophagy 2020, 16, 1925–1927. [Google Scholar] [CrossRef]
- Elswood, J.; Pearson, S.J.; Payne, H.R.; Barhoumi, R.; Rijnkels, M.; Porter, W.W. Autophagy Regulates Functional Differentiation of Mammary Epithelial Cells. Autophagy 2021, 17, 420–438. [Google Scholar] [CrossRef]
- Lou, G.; Palikaras, K.; Lautrup, S.; Scheibye-Knudsen, M.; Tavernarakis, N.; Fang, E.F. Mitophagy and Neuroprotection. Trends Mol. Med. 2020, 26, 8–20. [Google Scholar] [CrossRef]
- Praharaj, P.P.; Panigrahi, D.P.; Bhol, C.S.; Patra, S.; Mishra, S.R.; Mahapatra, K.K.; Behera, B.P.; Singh, A.; Patil, S.; Bhutia, S.K. Mitochondrial Rewiring through Mitophagy and Mitochondrial Biogenesis in Cancer Stem Cells: A Potential Target for Anti-Csc Cancer Therapy. Cancer Lett. 2021, 498, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Vara-Perez, M.; Felipe-Abrio, B.; Agostinis, P. Mitophagy in Cancer: A Tale of Adaptation. Cells 2019, 8, 493. [Google Scholar]
- Kulikov, A.V.; Luchkina, E.A.; Gogvadze, V.; Zhivotovsky, B. Mitophagy: Link to Cancer Development and Therapy. Biochem. Biophys. Res. Commun. 2017, 482, 432–439. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of Cancer Metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef]
- Bernardini, J.P.; Lazarou, M.; Dewson, G. Parkin and Mitophagy in Cancer. Oncogene 2017, 36, 1315–1327. [Google Scholar] [CrossRef]
- Veeriah, S.; Taylor, B.S.; Meng, S.; Fang, F.; Yilmaz, E.; Vivanco, I.; Janakiraman, M.; Schultz, N.; Hanrahan, A.J.; Pao, W.; et al. Somatic Mutations of the Parkinson’s Disease-Associated Gene Park2 in Glioblastoma and Other Human Malignancies. Nat. Genet. 2010, 42, 77–82. [Google Scholar]
- Moossavi, M.; Parsamanesh, N.; Bahrami, A.; Atkin, S.L.; Sahebkar, A. Role of the Nlrp3 Inflammasome in Cancer. Mol. Cancer 2018, 17, 158. [Google Scholar] [CrossRef]
- O’Sullivan, T.E.; Johnson, L.R.; Kang, H.H.; Sun, J.C. Bnip3- and Bnip3l-Mediated Mitophagy Promotes the Generation of Natural Killer Cell Memory. Immunity 2015, 43, 331–342. [Google Scholar] [CrossRef]
- Donisi, C.; Puzzoni, M.; Ziranu, P.; Lai, E.; Mariani, S.; Saba, G.; Impera, V.; Dubois, M.; Persano, M.; Migliari, M.; et al. Immune Checkpoint Inhibitors in the Treatment of Hcc. Front. Oncol. 2020, 10, 601240. [Google Scholar]
- Wallace, D.F. The Regulation of Iron Absorption and Homeostasis. Clin. Biochem. Rev. 2016, 37, 51–62. [Google Scholar]
- Li, C.; Zhang, Y.; Cheng, X.; Yuan, H.; Zhu, S.; Liu, J.; Kroemer, G. Pink1 and Park2 Suppress Pancreatic Tumorigenesis through Control of Mitochondrial Iron-Mediated Immunometabolism. Dev. Cell 2018, 46, 441–455.e8. [Google Scholar] [CrossRef]
- Chen, M.; Wei, L.; Law, C.T.; Tsang, F.H.; Shen, J.; Cheng, C.L.; Tsang, L.-H.; Ho, D.W.H.; Chiu, D.K.-C.; Lee, J.M.-F.; et al. Rna N6-Methyladenosine Methyltransferase-Like 3 Promotes Liver Cancer Progression through Ythdf2-Dependent Posttranscriptional Silencing of Socs2. Hepatology 2018, 67, 2254–2270. [Google Scholar] [CrossRef]
- Ma, J.Z.; Yang, F.; Zhou, C.C.; Liu, F.; Yuan, J.H.; Wang, F.; Wang, T.; Xu, Q.; Zhou, W.; Sun, S. Mettl14 Suppresses the Metastatic Potential of Hepatocellular Carcinoma by Modulating N(6) -Methyladenosine-Dependent Primary Microrna Processing. Hepatology 2017, 65, 529–543. [Google Scholar] [CrossRef]
- Sun, T.; Wu, R.; Ming, L. The Role of M6a Rna Methylation in Cancer. BioMed. Pharm. 2019, 112, 108613. [Google Scholar] [CrossRef]
- Chen, L.Y.; Yang, B.; Zhou, L.; Ren, F.; Duan, Z.P.; Ma, Y.J. Promotion of Mitochondrial Energy Metabolism During Hepatocyte Apoptosis in a Rat Model of Acute Liver Failure. Mol. Med. Rep. 2015, 12, 5035–5041. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy Proteins Regulate Innate Immune Responses by Inhibiting the Release of Mitochondrial DNA Mediated by the Nalp3 Inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef]
- Grazioli, S.; Pugin, J. Mitochondrial Damage-Associated Molecular Patterns: From Inflammatory Signaling to Human Diseases. Front. Immunol. 2018, 9, 832. [Google Scholar] [CrossRef]
- Liu, Z.; Ren, B.; Wang, Y.; Zou, C.; Qiao, Q.; Diao, Z.; Mi, Y.; Zhu, D.; Liu, X. Sesamol Induces Human Hepatocellular Carcinoma Cells Apoptosis by Impairing Mitochondrial Function and Suppressing Autophagy. Sci. Rep. 2017, 7, 45728. [Google Scholar] [CrossRef]
- Wei, R.; Cao, J.; Yao, S. Matrine Promotes Liver Cancer Cell Apoptosis by Inhibiting Mitophagy and Pink1/Parkin Pathways. Cell Stress Chaperones 2018, 23, 1295–1309. [Google Scholar]
- Kang, X.; Wang, H.; Li, Y.; Xiao, Y.; Zhao, L.; Zhang, T.; Zhou, S.; Zhou, X.; Li, Y.; Shou, Z.; et al. Alantolactone Induces Apoptosis through Ros-Mediated Akt Pathway and Inhibition of Pink1-Mediated Mitophagy in Human Hepg2 Cells. Artif. Cells NanoMed. Biotechnol. 2019, 47, 1961–1970. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. Swiss-Model: Homology Modelling of Protein Structures and Complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Ferro, F.; Servais, S.; Besson, P.; Roger, S.; Dumas, J.F.; Brisson, L. Autophagy and Mitophagy in Cancer Metabolic Remodelling. Semin. Cell Dev. Biol. 2020, 98, 129–138. [Google Scholar] [CrossRef]
- Karvela, M.; Baquero, P.; Kuntz, E.M.; Mukhopadhyay, A.; Mitchell, R.; Allan, E.K.; Chan, E.; Kranc, K.R.; Calabretta, B.; Salomoni, P.; et al. Atg7 Regulates Energy Metabolism, Differentiation and Survival of Philadelphia-Chromosome-Positive Cells. Autophagy 2016, 12, 936–948. [Google Scholar] [CrossRef]
- Wei, H.; Wei, S.; Gan, B.; Peng, X.; Zou, W.; Guan, J.L. Suppression of Autophagy by Fip200 Deletion Inhibits Mammary Tumorigenesis. Genes Dev. 2011, 25, 1510–1527. [Google Scholar] [CrossRef]
- Zhang, F.; Liang, J.; Feng, D.; Liu, S.; Wu, J.; Tang, Y.; Liu, Z.; Lu, Y.; Wang, X.; Wei, X. Integrated Analysis of Energy Metabolism Signature-Identified Distinct Subtypes of Bladder Urothelial Carcinoma. Front. Cell Dev. Biol. 2022, 10, 814735. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Xu, D.; Zhang, T.; Hu, W.; Feng, Z. Gain-of-Function Mutant P53 in Cancer Progression and Therapy. J. Mol. Cell Biol. 2020, 12, 674–687. [Google Scholar] [CrossRef]
- Hjelmeland, A.; Zhang, J. Metabolic, Autophagic, and Mitophagic Activities in Cancer Initiation and Progression. BioMed. J. 2016, 39, 98–106. [Google Scholar] [CrossRef][Green Version]
- Liu, K.; Lee, J.; Kim, J.Y.; Wang, L.; Tian, Y.; Chan, S.T.; Cho, C.; Machida, K.; Chen, D.; Ou, J.-H.J. Mitophagy Controls the Activities of Tumor Suppressor P53 to Regulate Hepatic Cancer Stem Cells. Mol. Cell 2017, 68, 281–292.e5. [Google Scholar] [CrossRef]
- Chen, Y.Y.; Wang, W.H.; Che, L.; Lan, Y.; Zhang, L.Y.; Zhan, D.L.; Huang, Z.-Y.; Lin, Z.-N.; Lin, Y.-C. Bnip3l-Dependent Mitophagy Promotes Hbx-Induced Cancer Stemness of Hepatocellular Carcinoma Cells Via Glycolysis Metabolism Reprogramming. Cancers 2020, 12, 655. [Google Scholar] [CrossRef]
- Ziegler, P.K.; Bollrath, J.; Pallangyo, C.K.; Matsutani, T.; Canli, O.; De Oliveira, T.; Diamanti, M.A.; Müller, N.; Gamrekelashvili, J.; Putoczki, T.; et al. Mitophagy in Intestinal Epithelial Cells Triggers Adaptive Immunity During Tumorigenesis. Cell 2018, 174, 88–101.e16. [Google Scholar] [CrossRef]
- Claude-Taupin, A.; Jia, J.; Bhujabal, Z.; Garfa-Traore, M.; Kumar, S.; da Silva, G.P.D.; Javed, R.; Gu, Y.; Allers, L.; Peters, R.; et al. Atg9a Protects the Plasma Membrane from Programmed and Incidental Permeabilization. Nat. Cell Biol. 2021, 23, 846–858. [Google Scholar] [CrossRef] [PubMed]
- Guardia, C.M.; Tan, X.F.; Lian, T.; Rana, M.S.; Zhou, W.; Christenson, E.T.; Lowry, A.J.; Faraldo-Gómez, J.D.; Bonifacino, J.S.; Jiang, J.; et al. Structure of Human Atg9a, the Only Transmembrane Protein of the Core Autophagy Machinery. Cell Rep. 2020, 31, 107837. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Yamamoto, H.; Kinch, L.N.; Garza, C.M.; Takahashi, S.; Otomo, C.; Grishin, N.V.; Forli, S.; Mizushima, N.; Otomo, T. Structure, Lipid Scrambling Activity and Role in Autophagosome Formation of Atg9a. Nat. Struct. Mol. Biol. 2020, 27, 1194–1201. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Lin, N.; Dong, D.; Ma, J.; Su, J.; Sun, L. Pgam5: A Crucial Role in Mitochondrial Dynamics and Programmed Cell Death. Eur. J. Cell Biol. 2021, 100, 151144. [Google Scholar] [CrossRef]
- Yu, B.; Ma, J.; Li, J.; Wang, D.; Wang, Z.; Wang, S. Mitochondrial Phosphatase Pgam5 Modulates Cellular Senescence by Regulating Mitochondrial Dynamics. Nat. Commun. 2020, 11, 2549. [Google Scholar] [CrossRef]
- Liang, M.Z.; Ke, T.L.; Chen, L. Mitochondrial Protein Pgam5 Emerges as a New Regulator in Neurological Diseases. Front. Mol. NeuroSci. 2021, 14, 730604. [Google Scholar] [CrossRef]
- Zhang, S.L.; Tang, H.B.; Hu, J.T.; Zang, Z.L.; Ding, X.; Li, S.; Yang, H. Pgam5-Cypd Pathway Is Involved in Bromocriptine-Induced Rip3/Mlkl-Dependent Necroptosis of Prolactinoma Cells. BioMed. Pharm. 2019, 111, 638–648. [Google Scholar] [CrossRef]
- Lamark, T.; Svenning, S.; Johansen, T. Regulation of Selective Autophagy: The P62/Sqstm1 Paradigm. Essays Biochem. 2017, 61, 609–624. [Google Scholar]
- Drake, L.E.; Springer, M.Z.; Poole, L.P.; Kim, C.J.; Macleod, K.F. Expanding Perspectives on the Significance of Mitophagy in Cancer. Semin. Cancer Biol. 2017, 47, 110–124. [Google Scholar] [CrossRef]
- Moscat, J.; Karin, M.; Diaz-Meco, M.T. P62 in Cancer: Signaling Adaptor Beyond Autophagy. Cell 2016, 167, 606–609. [Google Scholar]
- Nguyen, T.D.; Shaid, S.; Vakhrusheva, O.; Koschade, S.E.; Klann, K.; Tholken, M.; Brandts, C.H. Loss of the Selective Autophagy Receptor P62 Impairs Murine Myeloid Leukemia Progression and Mitophagy. Blood 2019, 133, 168–179. [Google Scholar] [CrossRef]
- Schaaf, M.B.; Keulers, T.G.; Vooijs, M.A.; Rouschop, K.M. Lc3/Gabarap Family Proteins: Autophagy-(Un)Related Functions. FASEB J. 2016, 30, 3961–3978. [Google Scholar] [CrossRef]
- Berthier, A.; Seguin, S.; Sasco, A.J.; Bobin, J.Y.; De Laroche, G.; Datchary, J.; Saez, S.; Rodriguez-Lafrasse, C.; Tolle, F.; Fraichard, A.; et al. High Expression of Gabarapl1 Is Associated with a Better Outcome for Patients with Lymph Node-Positive Breast Cancer. Br. J. Cancer 2010, 102, 1024–1031. [Google Scholar] [CrossRef]
- Liu, C.; Xia, Y.; Jiang, W.; Liu, Y.; Yu, L. Low Expression of Gabarapl1 Is Associated with a Poor Outcome for Patients with Hepatocellular Carcinoma. Oncol. Rep. 2014, 31, 2043–2048. [Google Scholar] [CrossRef]
- Xu, W.; Zhao, D.X.; Huang, X.W.; Zhang, M.; Yin, M.Y.; Liu, L.; Wu, H.Y.; Weng, Z.; Xu, C.F. The prognostic value and clinical significance of mitophagy-related genes in hepatocellular carcinoma. Front. Genet. 2022, 13, 917584. [Google Scholar] [CrossRef]
- Chen, H.; Wang, J.H.; Zeng, R.J.; Luo, Y.J.; Guo, K.H.; Wu, H.H.; Yang, Q.; Jiang, R.; Sha, W.H.; Zhuo, Z.W. Development and Validation of a Novel Mitophagy-Related Gene Prognostic Signature for Hepatocellular Carcinoma Based on Immunoscore Classification of Tumor. J. Oncol. 2021, 2021, 5070099. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Z.; Sun, J.J.; Qian, Y.B. Identification of HCC Subtypes With Different Prognosis and Metabolic Patterns Based on Mitophagy. Front. Cell Dev. Biol. 2021, 9, 799507. [Google Scholar] [CrossRef]
- Liang, S.W.; Zhu, C.X.; Suo, C.X.; Wei, H.R.; Yu, Y.X.; Gu, X.M.; Chen, L.; Yuan, M.Q.; Shen, S.Q.; Li, S.T.; et al. Mitochondrion-Localized SND1 Promotes Mitophagy and Liver Cancer Progression Through PGAM5. Front. Oncol. 2022, 12, 857968. [Google Scholar] [CrossRef]
- Teo, M.Y.; Rosenberg, J.E. Nivolumab for the Treatment of Urothelial Cancers. Expert Rev. Anticancer. 2018, 18, 215–221. [Google Scholar] [CrossRef]
- Ding, M.M.; Li, Y.A.; Lu, Z.; Hou, G. Identification of Potential Immune Checkpoint Inhibitor Targets in Gliomas via Bioinformatic Analyses. BioMed. Res. Int. 2022, 2022, 1734847. [Google Scholar] [CrossRef]
- Liu, Z.; Zhao, Q.; Zuo, Z.X.; Yuan, S.Q.; Yu, K.; Zhang, Q.; Zhang, X.; Shen, H.; Ju, H.-Q.; Cheng, H.; et al. Systematic Analysis of the Aberrances and Functional Implications of Ferroptosis in Cancer. iScience 2020, 23, 101302. [Google Scholar] [CrossRef]
- Li, Y.; Xiao, J.; Bai, J.; Tian, Y.; Qu, Y.; Chen, X.; Wang, Q.; Li, X.; Zhang, Y.; Xu, J. Molecular Characterization and Clinical Relevance of M(6)a Regulators across 33 Cancer Types. Mol. Cancer 2019, 18, 137. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, C.; Wu, Z.; Wang, L.; Yang, Q.; Huang, J.; Huang, J. A Mitophagy-Related Gene Signature for Subtype Identification and Prognosis Prediction of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2022, 23, 12123. https://doi.org/10.3390/ijms232012123
Liu C, Wu Z, Wang L, Yang Q, Huang J, Huang J. A Mitophagy-Related Gene Signature for Subtype Identification and Prognosis Prediction of Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2022; 23(20):12123. https://doi.org/10.3390/ijms232012123
Chicago/Turabian StyleLiu, Chang, Zhen Wu, Liping Wang, Qian Yang, Ji Huang, and Jichang Huang. 2022. "A Mitophagy-Related Gene Signature for Subtype Identification and Prognosis Prediction of Hepatocellular Carcinoma" International Journal of Molecular Sciences 23, no. 20: 12123. https://doi.org/10.3390/ijms232012123
APA StyleLiu, C., Wu, Z., Wang, L., Yang, Q., Huang, J., & Huang, J. (2022). A Mitophagy-Related Gene Signature for Subtype Identification and Prognosis Prediction of Hepatocellular Carcinoma. International Journal of Molecular Sciences, 23(20), 12123. https://doi.org/10.3390/ijms232012123