
Clinical and Genetic Re-Evaluation of Inherited Retinal Degeneration Pedigrees following Initial Negative Findings on Panel-Based Next Generation Sequencing

 ,
,  ,
,
Abstract
1. Introduction
2. Methods
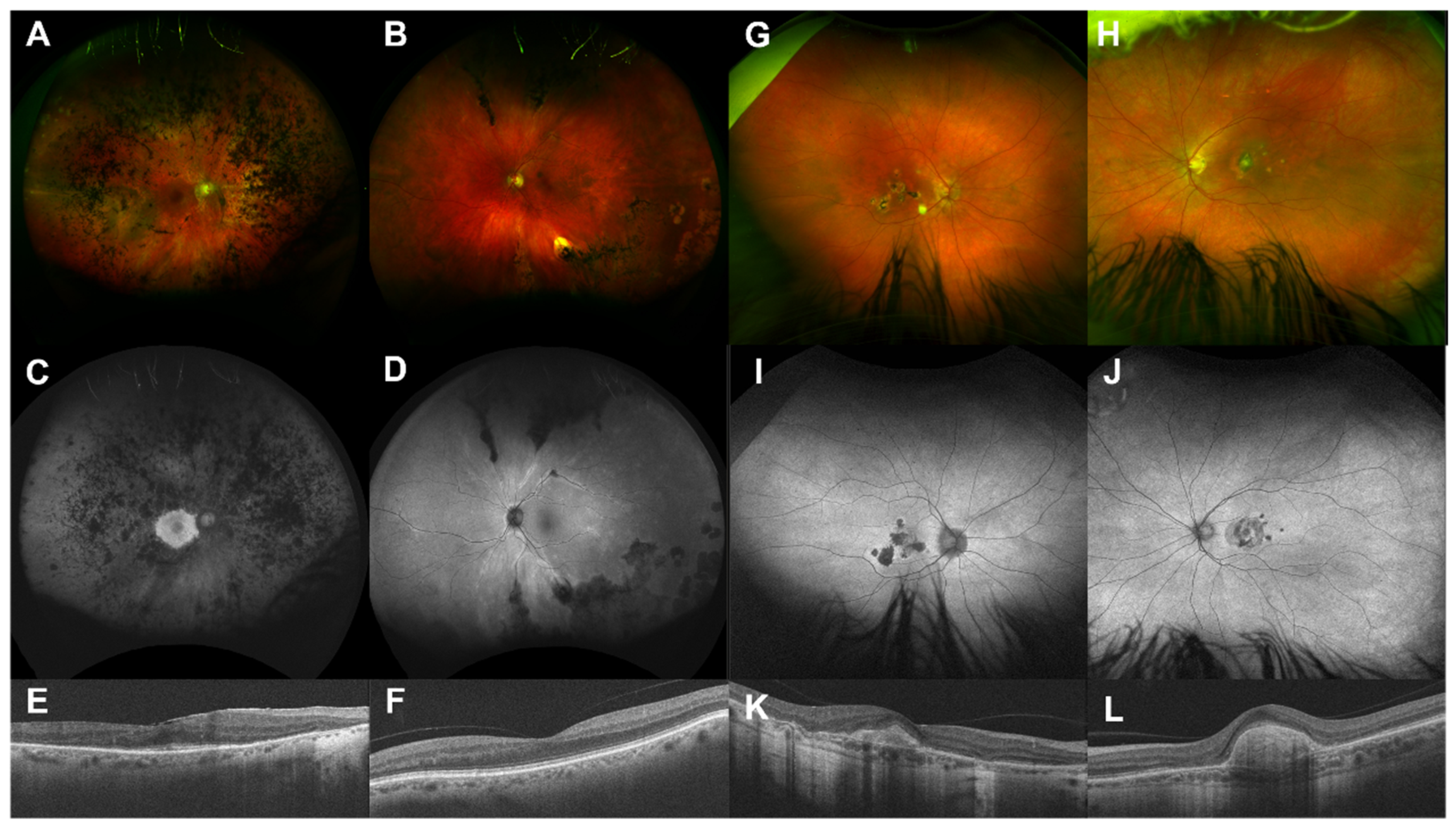
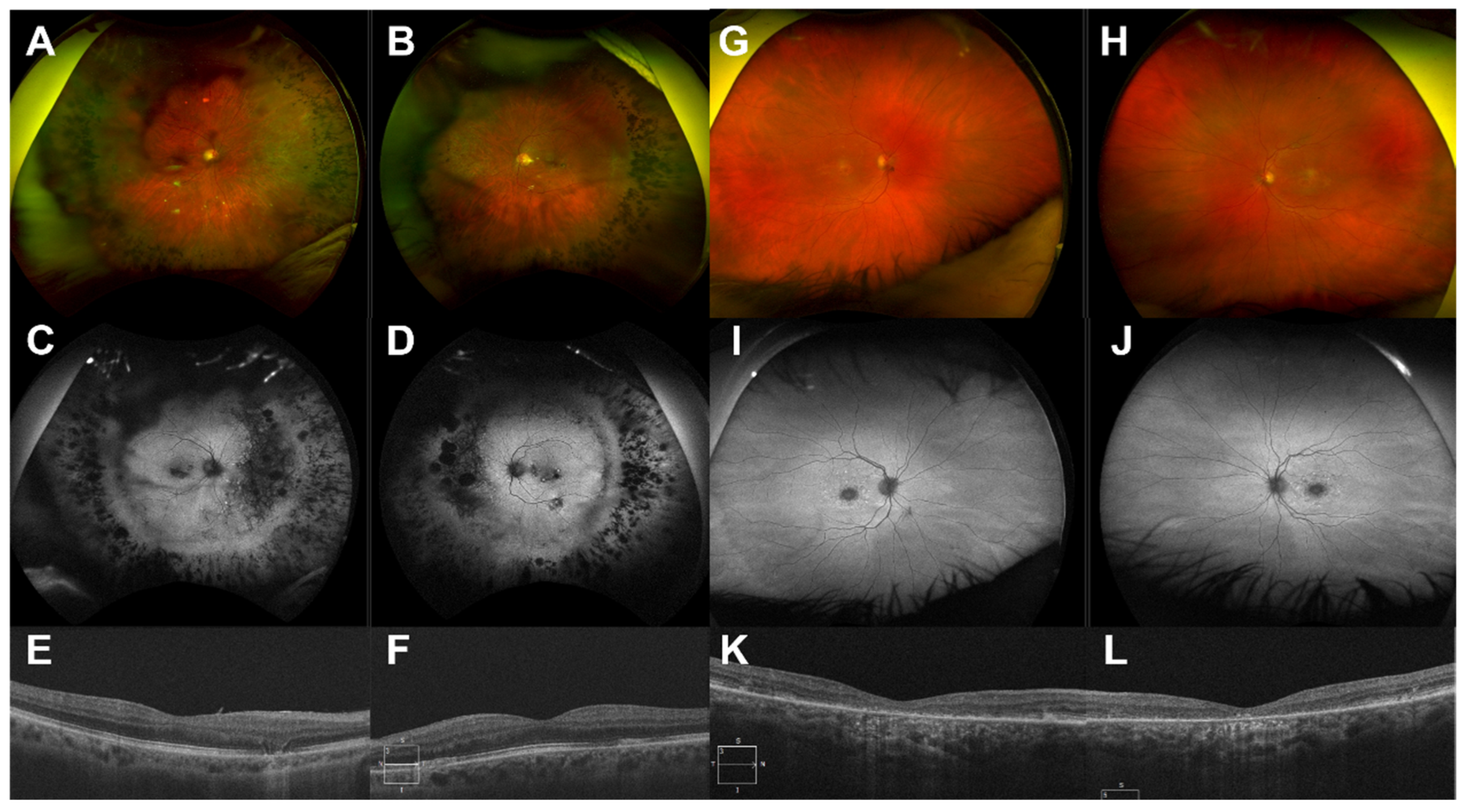
3. Results
4. Discussion
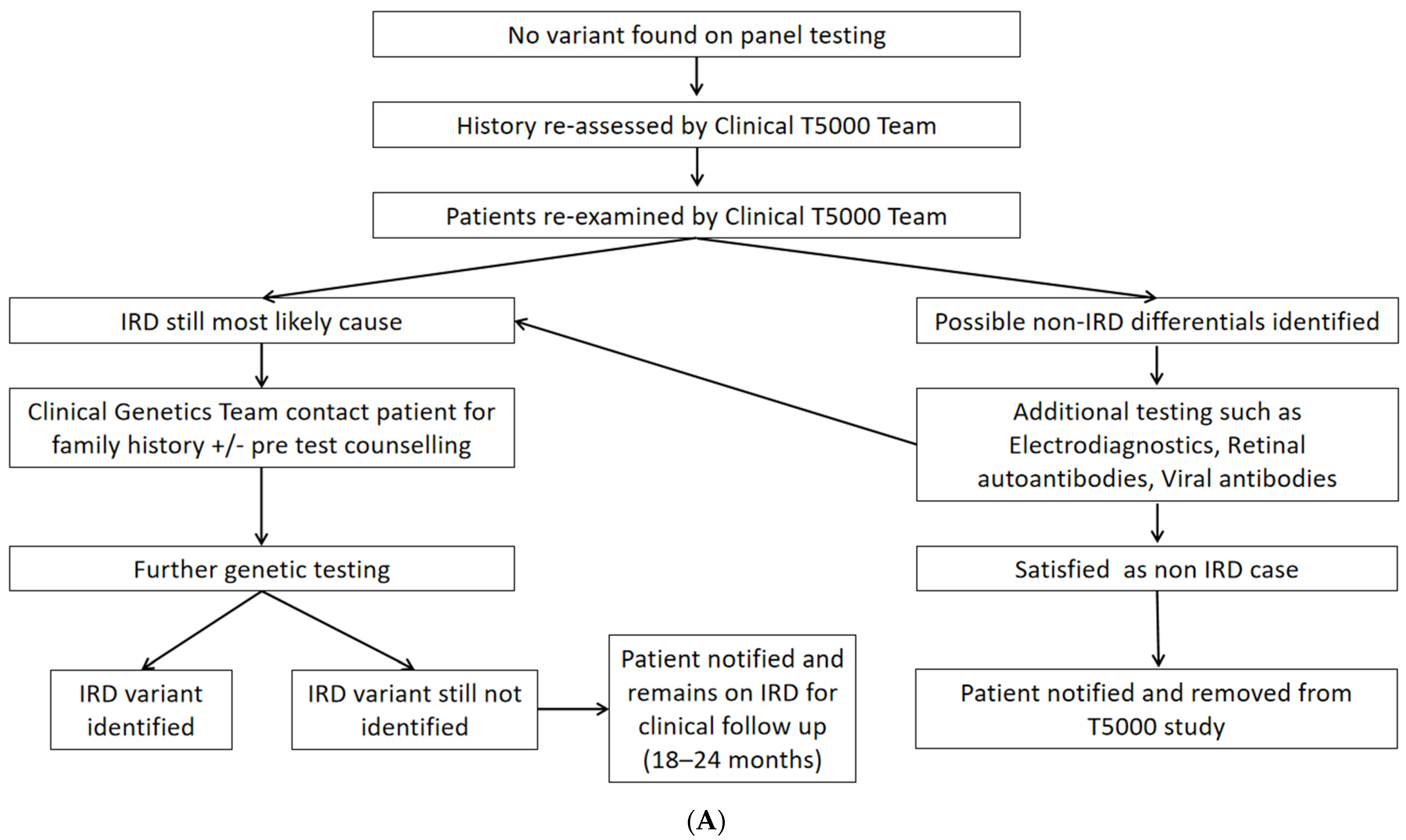
4.1. Clinical Reassessment
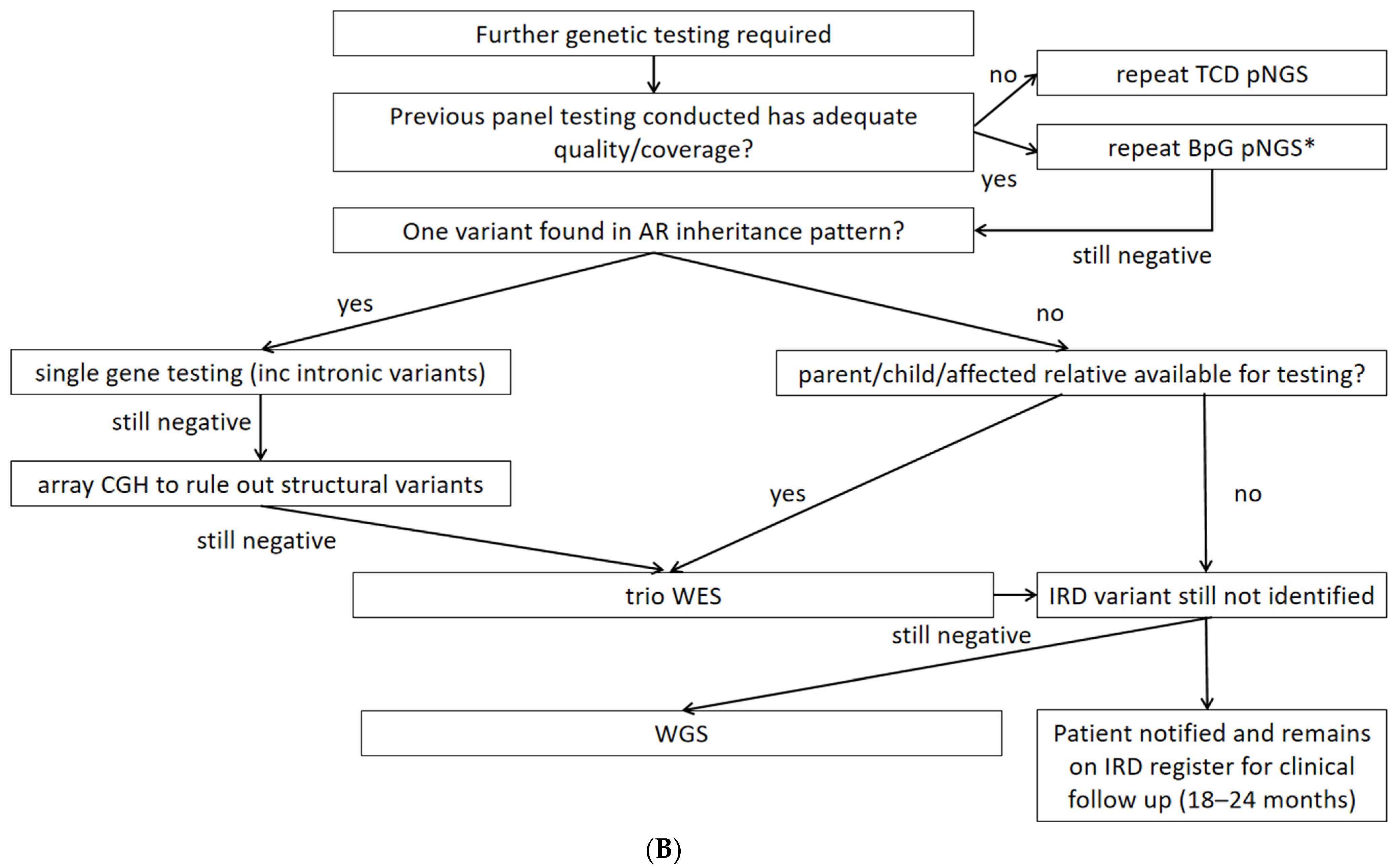
4.2. Second-Tier Genetic Testing Approaches and Costs
4.3. Examples of Resolution Problems and Their Solutions
4.4. Relevance to Gene Therapies
4.5. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Daiger, S.P. Summaries of Genes and Loci Causing Retinal Diseases (RetNet). Available online: https://sph.uth.edu/retnet/ (accessed on 5 October 2021).
- Heath Jeffery, R.C.; Mukhtar, S.A.; McAllister, I.L.; Morgan, W.H.; Mackey, D.A.; Chen, F.K. Inherited retinal diseases are the most common cause of blindness in the working-age population in Australia. Ophthalmic Genet. 2021, 42, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Liew, G.; Michaelides, M.; Bunce, C. A comparison of the causes of blindness certifications in England and Wales in working age adults (16–64 years), 1999–2000 with 2009–2010. BMJ Open 2014, 4, e004015. [Google Scholar] [CrossRef]
- Dockery, A.; Stephenson, K.; Keegan, D.; Wynne, N.; Silvestri, G.; Humphries, P.; Kenna, P.F.; Carrigan, M.; Farrar, G.J. Target 5000: Target capture sequencing for inherited retinal degenerations. Genes 2017, 8, 304. [Google Scholar] [CrossRef]
- Whelan, L.; Dockery, A.; Wynne, N.; Zhu, J.; Stephenson, K.; Silvestri, G.; Turner, J.; O’Byrne, J.J.; Carrigan, M.; Humphries, P.; et al. Findings from a genotyping study of over 1000 people with inherited retinal disorders in Ireland. Genes 2020, 11, 105. [Google Scholar] [CrossRef]
- Shah, M.; Shanks, M.; Packham, E.; Williams, J.; Haysmoore, J.; MacLaren, R.E.; Németh, A.H.; Clouston, P.; Downes, S.M. Next generation sequencing using phenotype-based panels for genetic testing in inherited retinal diseases. Ophthalmic Genet. 2020, 41, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Ellingford, J.M.; Barton, S.; Bhaskar, S.; Williams, S.G.; Sergouniotis, P.I.; O’Sullivan, J.; Lamb, J.A.; Perveen, R.; Hall, G.; Newman, W.G.; et al. Whole genome sequencing increases molecular diagnostic yield compared with current diagnostic testing for inherited retinal disease. Ophthalmology 2016, 123, 1143–1150. [Google Scholar] [CrossRef]
- Smedley, D.; Smith, K.R.; Martin, A.; Thomas, E.A.; McDonagh, E.M.; Cipriani, V.; Ellingford, J.M.; Arno, G.; Tucci, A.; Vandrovcova, J.; et al. 100,000 Genomes Pilot on Rare-Disease Diagnosis in Health Care—Preliminary Report. N. Engl. J. Med. 2021, 385, 1868–1880. [Google Scholar]
- Hart, M.R.; Biesecker, B.B.; Blout, C.L.; Christensen, K.D.; Amendola, L.M.; Bergstrom, K.L.; Biswas, S.; Bowling, K.M.; Brothers, K.B.; Conlin, L.K.; et al. Secondary findings from clinical genomic sequencing: Prevalence, patient perspectives, family history assessment, and health-care costs from a multisite study. Genet. Med. 2019, 21, 1100–1110. [Google Scholar] [CrossRef]
- Zhu, J.; Stephenson, K.A.; Farrar, G.J.; Turner, J.; O’Byrne, J.J.; Keegan, D. Management of significant secondary genetic findings in an ophthalmic genetics clinic. Eye 2021, 3, 1–3. [Google Scholar] [CrossRef]
- Stone, E.M.; Andorf, J.L.; Whitmore, S.S.; DeLuca, A.P.; Giacalone, J.C.; Streb, L.M.; Braun, T.A.; Mullins, R.F.; Scheetz, T.E.; Sheffield, V.C.; et al. Clinically focused molecular investigation of 1000 consecutive families with inherited retinal disease. Ophthalmology 2017, 124, 1314–1331. [Google Scholar] [CrossRef]
- Bush, R.A.; Zeng, Y.; Colosi, P.; Kjellstrom, S.; Hiriyanna, S.; Vijayasarathy, C.; Santos, M.; Li, J.; Wu, Z.; Sieving, P.A. Preclinical dose-escalation study of intravitreal AAV-RS1 gene therapy in a mouse model of X-linked retinoschisis: Dose-dependent expression and improved retinal structure and function. Hum. Gene Ther. 2016, 27, 376–389. [Google Scholar] [CrossRef]
- Cukras, C.; Wiley, H.E.; Jeffrey, B.G.; Sen, H.N.; Turriff, A.; Zeng, Y.; Vijayasarathy, C.; Marangoni, D.; Ziccardi, L.; Kjellstrom, S.; et al. Retinal AAV8-RS1 gene therapy for X-linked retinoschisis: Initial findings from a phase I/IIa trial by intravitreal delivery. Mol. Ther. 2018, 26, 2282–2294. [Google Scholar] [CrossRef] [PubMed]
- Cehajic-Kapetanovic, J.; Xue, K.; de la Camara, C.M.; Nanda, A.; Davies, A.; Wood, L.J.; Salvetti, A.P.; Fischer, M.D.; Aylward, J.W.; Barnard, A.R.; et al. Initial results from a first-in-human gene therapy trial on X-linked retinitis pigmentosa caused by mutations in RPGR. Nat. Med. 2020, 26, 354–359. [Google Scholar] [CrossRef]
- Méjécase, C.; Malka, S.; Guan, Z.; Slater, A.; Arno, G.; Moosajee, M. Practical guide to genetic screening for inherited eye diseases. Ther. Adv. Ophthalmol. 2020, 12, 2515841420954592. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, K.A.; Zhu, J.; Wynne, N.; Dockery, A.; Cairns, R.M.; Duignan, E.; Whelan, L.; Malone, C.P.; Dempsey, H.; Collins, K.; et al. Target 5000: A standardized all-Ireland pathway for the diagnosis and management of inherited retinal degenerations. Orph. J. Rare Dis. 2021, 16, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Genetics, B. Retinal Dystrophy Panel. Available online: https://blueprintgenetics.com/tests/panels/ophthalmology/retinal-dystrophy-panel/ (accessed on 8 November 2021).
- Koenig, R. Bardet-Biedl syndrome and Usher syndrome. Dev. Ophthalmol. 2003, 37, 126–140. [Google Scholar]
- Prokofyeva, E.; Troeger, E.; Wilke, R.; Zrenner, E. Early visual symptom patterns in inherited retinal dystrophies. Ophthalmologica 2011, 226, 151–156. [Google Scholar] [CrossRef]
- Sevgi, D.D.; Davoudi, S.; Comander, J.; Sobrin, L. Retinal pigmentary changes in chronic uveitis mimicking retinitis pigmentosa. Graefe’s Arch. Clin. Exp. Ophthalmol. 2017, 255, 1801–1810. [Google Scholar] [CrossRef]
- Haer-Wigman, L.; van Zelst-Stams, W.A.; Pfundt, R.; van den Born, L.I.; Klaver, C.C.; Verheij, J.B.; Hoyng, C.B.; Breuning, M.H.; Boon, C.J.; Kievit, A.J.; et al. Diagnostic exome sequencing in 266 Dutch patients with visual impairment. Eur. J. Hum. Genet. 2017, 25, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Van Nimwegen, K.J.; van Soest, R.A.; Veltman, J.A.; Nelen, M.R.; van der Wilt, G.J.; Vissers, L.E.; Grutters, J.P. Is the $1000 genome as near as we think? A cost analysis of next-generation sequencing. Clin. Chem. 2016, 62, 1458–1464. [Google Scholar] [CrossRef]
- Zampaglione, E.; Kinde, B.; Place, E.M.; Navarro-Gomez, D.; Maher, M.; Jamshidi, F.; Nassiri, S.; Mazzone, J.A.; Finn, C.; Schlegel, D.; et al. Copy-number variation contributes 9% of pathogenicity in the inherited retinal degenerations. Genet. Med. 2020, 22, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Van Cauwenbergh, C.; Van Schil, K.; Cannoodt, R.; Bauwens, M.; Van Laethem, T.; De Jaegere, S.; Steyaert, W.; Sante, T.; Menten, B.; Leroy, B.P.; et al. arrEYE: A customized platform for high-resolution copy number analysis of coding and noncoding regions of known and candidate retinal dystrophy genes and retinal noncoding RNAs. Genet. Med. 2017, 19, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Wetterstrand, K. DNA Sequencing Costs: Data from the NHGRI Genome Sequencing Program (GSP). Available online: www.genome.gov/sequencingcostsdata (accessed on 8 November 2021).
- Hims, M.M.; Diager, S.P.; Inglehearn, C.F. Retinitis pigmentosa: Genes, proteins and prospects. Dev. Ophthalmol. 2003, 37, 109–125. [Google Scholar]
- Ferrari, S.; Di Iorio, E.; Barbaro, V.; Ponzin, D.; SSorrentino, F.; Parmeggiani, F. Retinitis pigmentosa: Genes and disease mechanisms. Curr. Genom. 2011, 12, 238–249. [Google Scholar]
- Gerth, C.; Zawadzki, R.J.; Werner, J.S.; Héon, E. Retinal morphology in patients with BBS1 and BBS10 related Bardet–Biedl Syndrome evaluated by Fourier-domain optical coherence tomography. Vis. Res. 2008, 48, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Parmeggiani, F.; Barbaro, V.; De Nadai, K.; Lavezzo, E.; Toppo, S.; Chizzolini, M.; Palù, G.; Parolin, C.; Di Iorio, E. Identification of novel X-linked gain-of-function RPGR-ORF15 mutation in Italian family with retinitis pigmentosa and pathologic myopia. Sci. Rep. 2016, 6, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Dockery, A.; Whelan, L.; Humphries, P.; Farrar, G.J. Next-Generation Sequencing Applications for Inherited Retinal Diseases. Int. J. Mol. Sci. 2021, 22, 5684. [Google Scholar] [CrossRef] [PubMed]
- Cho, A.; de Carvalho, J.R.; Tanaka, A.J.; Jauregui, R.; Levi, S.R.; Bassuk, A.G.; Mahajan, V.B.; Tsang, S.H. Fundoscopy-directed genetic testing to re-evaluate negative whole exome sequencing results. Orphanet J. Rare Dis. 2020, 15, 1–11. [Google Scholar] [CrossRef]
- Ebenezer, N.D.; Michaelides, M.; Jenkins, S.A.; Audo, I.; Webster, A.R.; Cheetham, M.E.; Stockman, A.; Maher, E.R.; Ainsworth, J.R.; Yates, J.R.; et al. Identification of novel RPGR ORF15 mutations in X-linked progressive cone-rod dystrophy (XLCORD) families. Investig. Ophthalmol. Vis. Sci. 2005, 46, 1891–1898. [Google Scholar] [CrossRef]
- Farouni, R.; Djambazian, H.; Ferri, L.E.; Ragoussis, J.; Najafabadi, H.S. Model-based analysis of sample index hopping reveals its widespread artifacts in multiplexed single-cell RNA-sequencing. Nat. Commun. 2020, 11, 1–8. [Google Scholar] [CrossRef]
- Verbakel, S.K.; Fadaie, Z.; Klevering, B.J.; van Genderen, M.M.; Feenstra, I.; Cremers, F.P.; Hoyng, C.B.; Roosing, S. The identification of a RNA splice variant in TULP1 in two siblings with early-onset photoreceptor dystrophy. Mol. Genet. Genom. Med. 2019, 7, e660. [Google Scholar] [CrossRef]
- Khan, M.; Cornelis, S.S.; del Pozo-Valero, M.; Whelan, L.; Runhart, E.H.; Mishra, K.; Bults, F.; AlSwaiti, Y.; AlTalbishi, A.; De Baere, E.; et al. Resolving the dark matter of ABCA4 for 1054 Stargardt disease probands through integrated genomics and transcriptomics. Genet. Med. 2020, 22, 1235–1246. [Google Scholar] [CrossRef]
- Whelan, L.; Dockery, A.; Stephenson, K.A.J.; Zhu, J.; Kopcic, E.; Khan, M.; Corradi, Z.; Post, I.J.M.; Wynne, N.; Duignan, E.; et al. Enrichment of ABCA4 c.4539+2028C>T in Stargardt disease patients in Ireland—A detailed geno-type-phenotype analysis. Hum. Mutat. Current reference number humu-2021-0469. Hum. Mutat.
- Molina-Ramírez, L.P.; Kyle, C.; Ellingford, J.M.; Wright, R.; Taylor, A.; Bhaskar, S.S.; Campbell, C.; Jackson, H.; Fairclough, A.; Rousseau, A.; et al. Personalised virtual gene panels reduce interpretation workload and maintain diagnostic rates of proband-only clinical exome sequencing for rare disorders. J. Med. Genet. 2021. [Google Scholar] [CrossRef] [PubMed]
- Beryozkin, A.; Shevah, E.; Kimchi, A.; Mizrahi-Meissonnier, L.; Khateb, S.; Ratnapriya, R.; Lazar, C.H.; Blumenfeld, A.; Ben-Yosef, T.; Hemo, Y.; et al. Whole exome sequencing reveals mutations in known retinal disease genes in 33 out of 68 Israeli families with inherited retinopathies. Sci. Rep. 2015, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.J.; Lee, H.S.; Kim, K.; Choi, S.; Jang, I.; Cho, S.H.; Yoon, C.K.; Lee, E.K.; Yu, H.G. Whole-exome sequencing in 168 Korean patients with inherited retinal degeneration. BMC Med. Genom. 2021, 14, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Siemiatkowska, A.M.; Collin, R.W.; den Hollander, A.I.; Cremers, F.P. Genomic approaches for the discovery of genes mutated in inherited retinal degeneration. Cold Spring Harb. Perspect. Med. 2014, 4, a017137. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Qian, X.; Wang, J.; Wang, M.; Igelman, A.D.; Jones, K.D.; Li, Y.; Wang, K.; Goetz, K.E.; Birch, D.G.; Yang, P.; et al. Identification of Deep-Intronic Splice Mutations in a Large Cohort of Patients With Inherited Retinal Diseases. Front. Genet. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Zhang, W.; Wang, J.; Yang, K.; Han, Y.; Zhang, L. Review on the Application of Machine Learning Algorithms in the Sequence Data Mining of DNA. Front. Bioeng. Biotechnol. 2020, 8, 1032. [Google Scholar] [CrossRef] [PubMed]
- Hehir-Kwa, J.Y.; Claustres, M.; Hastings, R.J.; Van Ravenswaaij-Arts, C.; Christenhusz, G.; Genuardi, M.; Melegh, B.; Cambon-Thomsen, A.; Patsalis, P.; Vermeesch, J.; et al. Towards a European consensus for reporting incidental findings during clinical NGS testing. Eur. J. Hum. Genet. 2015, 23, 1601–1606. [Google Scholar] [CrossRef]
- Fadaie, Z.; Whelan, L.; Ben-Yosef, T.; Dockery, A.; Corradi, Z.; Gilissen, C.; Haer-Wigman, L.; Corominas, J.; Astuti, G.D.; de Rooij, L.; et al. Whole genome sequencing and in vitro splice assays reveal genetic causes for inherited retinal diseases. NPJ Genom. Med. 2021, 6, 1–11. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Mean Age, Years (SD) | Female (%) | n = (%) |
|---|---|---|---|
| Posterior Uveitis | 51 (16.39) | 57% | 7 (39%) |
| AMD | 76 (8.39) | 25% | 4 (22%) |
| Myopic/Pachychoroid Degeneration | 45.5 (3.54) | 50% | 2 (11%) |
| ION | 50 | 100% | 1 (6%) |
| Normal | 49 (12.92) | 75% | 4 (22%) |
| Pedigree | N = | Phenotype | Inheritance | Gene | Variant 1 | Variant 2 | Method | 1st-Tier Problem |
|---|---|---|---|---|---|---|---|---|
| 1–17 | 18 | Non-IRD | - | - | - | - | - | - |
| 18 | 1 | Refsum Disease | AR | PEX7 | c.875T > A, p.Leu292 * | c.40A > C, p.Thr14Pro | Single gene testing | Limited coverage |
| 19 | 2 | EOSRD | AR | CFAP410 | c.218G > C, p.Arg73Pro | c.218G > C, p.Arg73Pro | Repeat pNGS (R) | Misaligned reads, index hopping |
| 20 | 1 | sRP | AR | FLVCR1 | c.1022A > G, p.Tyr341Cys | c.1307 + 5G > T † | Repeat pNGS (R) | Additional phenotype information |
| 21 | 1 | STGD | AR | ABCA4 | c.752del, p.Phe251Serfs*11 † | c.5461 − 10T > C p.Thr1821Aspfs*6, Thr1821Valfs*13 | Single gene testing | Intronic variant |
| 22 | 1 | STGD | AR | ABCA4 | c.4363T > C, p.Cys1455Arg | c.4253 + 43G > A p.Ile1377Hisfs*3 | Single gene testing | Intronic variant |
| 23 | 1 | BBS | AR | BBS10 | c.2119_2120del, p.Val707 * | c.687del, p.Val230Phefs*7 | Repeat pNGS (R) | Poor coverage |
| 24 | 3 | nsRP | AD | RP1 | c.2321_2322insAlu | - | Repeat pNGS (A) | Complex structural variant |
| 25 | 1 | nsRP | AR | EYS | c.2620C > T, p.Gln874 * | c.(?_-538-1) _(2023+1_2024-1)del † | Single gene testing | Copy number variants |
| 26 | 3 | nsRP | XL | RPGR | c.2777_2778del, p.Glu926Glyfs*152 † | - | Repeat pNGS (A) | Low complexity ORF15 region |
| 27 | 2 | nsRP | XL | RPGR | c.2571_2572del, p.Glu859Glyfs*219 | - | Trio WES | Low complexity ORF15 region |
| 28 | 1 | sMD | AR | CNNM4 | c.1660G > T, p.Ala554Ser | Unresolved | Single gene testing | - |
| 29 | 1 | USH | AR | CDH23 | c.289-1G > A, p.Arg964Gln | Unresolved | Single gene testing | - |
| 30 | 1 | nsRP | AR | TRIM32 | c.691del, p.Ala231Glnfs*21 | Unresolved | Single gene testing | - |
| 31 | 1 | USH | AR | ADGRV1 | c.18025C > T, p.Arg6009 * | Unresolved | Single gene testing | - |
| 32 | 1 | BBS | AR | BBS1 | c.478C > T, p.Arg160Trp | Unresolved | Single gene testing | - |
| 33 | 3 | nsRP | AD * | - | Unresolved | - | Trio WES | - |
| 34 | 1 | nsRP | AR * | - | Unresolved | Unresolved | Trio WES | - |
| 35 | 9 | VRO | AD * | - | Unresolved | - | Trio WES | - |
| 36–52 | 16 | Still consistent with IRDs | - | - | Unresolved | Unresolved | - | Retest delayed due to SARS-CoV-2 pandemic |
| Test Type | Cost | N = Per Test Type | Total Cost | Resolve Rate, % (n=) |
|---|---|---|---|---|
| pNGS (resolved at research laboratory + accredited laboratory validation) | €600 (€250 + €350) | 441 | €240,450 * | 84.4% (372/441) |
| pNGS (negative research laboratory result + expanded accredited laboratory panel + validation at an accredited laboratory for other affected family members) | €1120 (€250 + €870 ± €350) | 5 + 3 variant confirmation | €6350 | 100% (5/5) |
| Single Gene Testing (negative research laboratory result + accredited single gene test + validation at an accredited laboratory for other affected family members) | €700 (€250 + €450 ± €350) | 9 | €6300 | 44% (4/9) |
| WES/trio WES (negative research laboratory result + accredited trio WES + validation at an accredited laboratory for other affected family members) | €2550 (€250 + €2300 ± €350) | 4 + 1 variant confirmation | €10,550 | 25% (1/4) ** |
| Gene | NCT Number | Technique | Phase | Status |
|---|---|---|---|---|
| Rod-Cone Dystrophies | ||||
| MERTK | NCT01482195 | AAV | 1/2 | Completed |
| PDE6B | NCT03328130 | AAV | 1/2 | Recruiting |
| RHO | NCT04123626 | AON | 1/2 | Recruiting |
| RPGR | NCT03252847 | AAV | 1/2 | Completed |
| NCT03116113 | AAV | 1/2 | Completed | |
| NCT03316560 | AAV | 1/2 | Recruiting | |
| RLBP1 | NCT03374657 | AAV | 1/2 | Recruiting |
| USH2A | NCT03780257 | AON | 1/2 | Not recruiting |
| MYO7A | NCT01505062 | LV | 1/2 | Terminated |
| Macular/Cone Dystrophies or Cone Dysfunction Syndromes | ||||
| RS1 | NCT02416622 | AAV | 1/2 | Terminated |
| NCT02317887 | AAV | 1/2 | Recruiting | |
| ABCA4 | NCT01367444 | LV | 1/2 | Terminated |
| CNGB3 | NCT03001310 | AAV | 1/2 | Completed |
| NCT02599922 | AAV | 1/2 | Recruiting | |
| CNGA3 | NCT03758404 | AAV | 1/2 | Completed |
| NCT02935517 | AAV | 1/2 | Recruiting | |
| NCT02610582 | AAV | 1/2 | Recruiting | |
| Leber Congenital Amaurosis | ||||
| RPE65 | NCT02781480 NCT02946879 | AAV | 1/2 | Recruiting |
| NCT00643747 | AAV | 1/2 | Completed | |
| NCT01496040 | AAV | 1/2 | Completed | |
| NCT00821340 | AAV | 1 | Completed | |
| NCT00749957 | AAV | 1/2 | Completed | |
| NCT00481546 | AAV | 1 | Completed | |
| GUCY2D | NCT03920007 | AAV | 1/2 | Recruiting |
| CEP290 | NCT03913143 | AON | 3 | Not recruiting |
| NCT03872479 | Gene editing | 1/2 | Recruiting | |
| Choroidal Dystrophies | ||||
| CHM | NCT02341807 | AAV | 1/2 | Completed |
| NCT02671539 | AAV | 2 | Completed | |
| NCT01461213 | AAV | 1/2 | Completed | |
| NCT02077361 | AAV | 1/2 | Not recruiting | |
| NCT02553135 | AAV | 1/2 | Completed | |
| NCT03507686 NCT03496012 | AAV | 2 3 | Completed Completed | |
| NCT02407678 | AAV | 2 | Completed | |
| NCT04483440 | AAV | 1 | Recruiting | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stephenson, K.A.J.; Zhu, J.; Dockery, A.; Whelan, L.; Burke, T.; Turner, J.; O’Byrne, J.J.; Farrar, G.J.; Keegan, D.J. Clinical and Genetic Re-Evaluation of Inherited Retinal Degeneration Pedigrees following Initial Negative Findings on Panel-Based Next Generation Sequencing. Int. J. Mol. Sci. 2022, 23, 995. https://doi.org/10.3390/ijms23020995
Stephenson KAJ, Zhu J, Dockery A, Whelan L, Burke T, Turner J, O’Byrne JJ, Farrar GJ, Keegan DJ. Clinical and Genetic Re-Evaluation of Inherited Retinal Degeneration Pedigrees following Initial Negative Findings on Panel-Based Next Generation Sequencing. International Journal of Molecular Sciences. 2022; 23(2):995. https://doi.org/10.3390/ijms23020995
Chicago/Turabian StyleStephenson, Kirk A. J., Julia Zhu, Adrian Dockery, Laura Whelan, Tomás Burke, Jacqueline Turner, James J. O’Byrne, G. Jane Farrar, and David J. Keegan. 2022. "Clinical and Genetic Re-Evaluation of Inherited Retinal Degeneration Pedigrees following Initial Negative Findings on Panel-Based Next Generation Sequencing" International Journal of Molecular Sciences 23, no. 2: 995. https://doi.org/10.3390/ijms23020995
APA StyleStephenson, K. A. J., Zhu, J., Dockery, A., Whelan, L., Burke, T., Turner, J., O’Byrne, J. J., Farrar, G. J., & Keegan, D. J. (2022). Clinical and Genetic Re-Evaluation of Inherited Retinal Degeneration Pedigrees following Initial Negative Findings on Panel-Based Next Generation Sequencing. International Journal of Molecular Sciences, 23(2), 995. https://doi.org/10.3390/ijms23020995