
Lipocalin 2 Influences Bone and Muscle Phenotype in the MDX Mouse Model of Duchenne Muscular Dystrophy




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
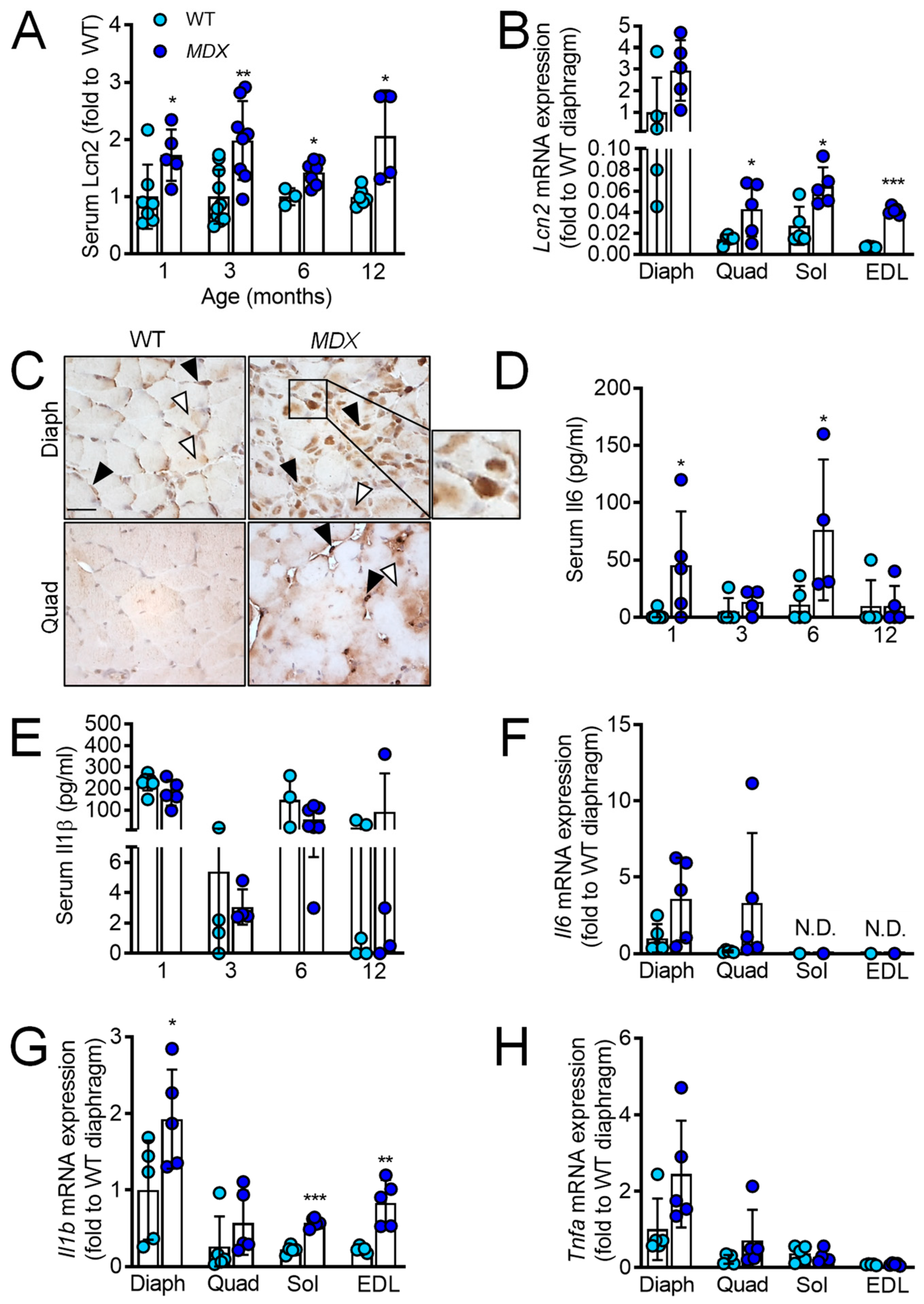
2.1. Lcn2 Levels Are Increased in MDX Mice and Correlate with Il1b and Tnfa Levels
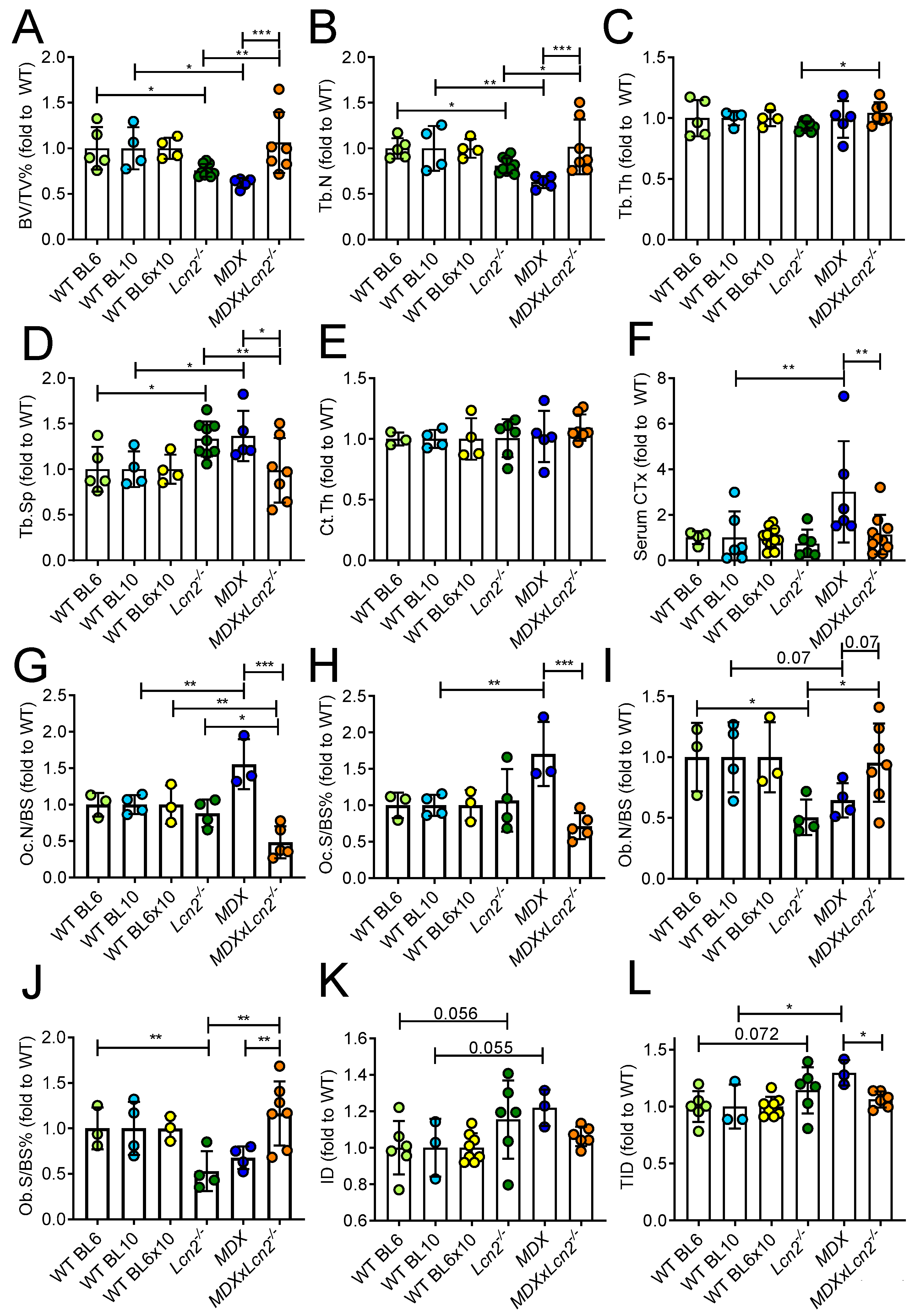
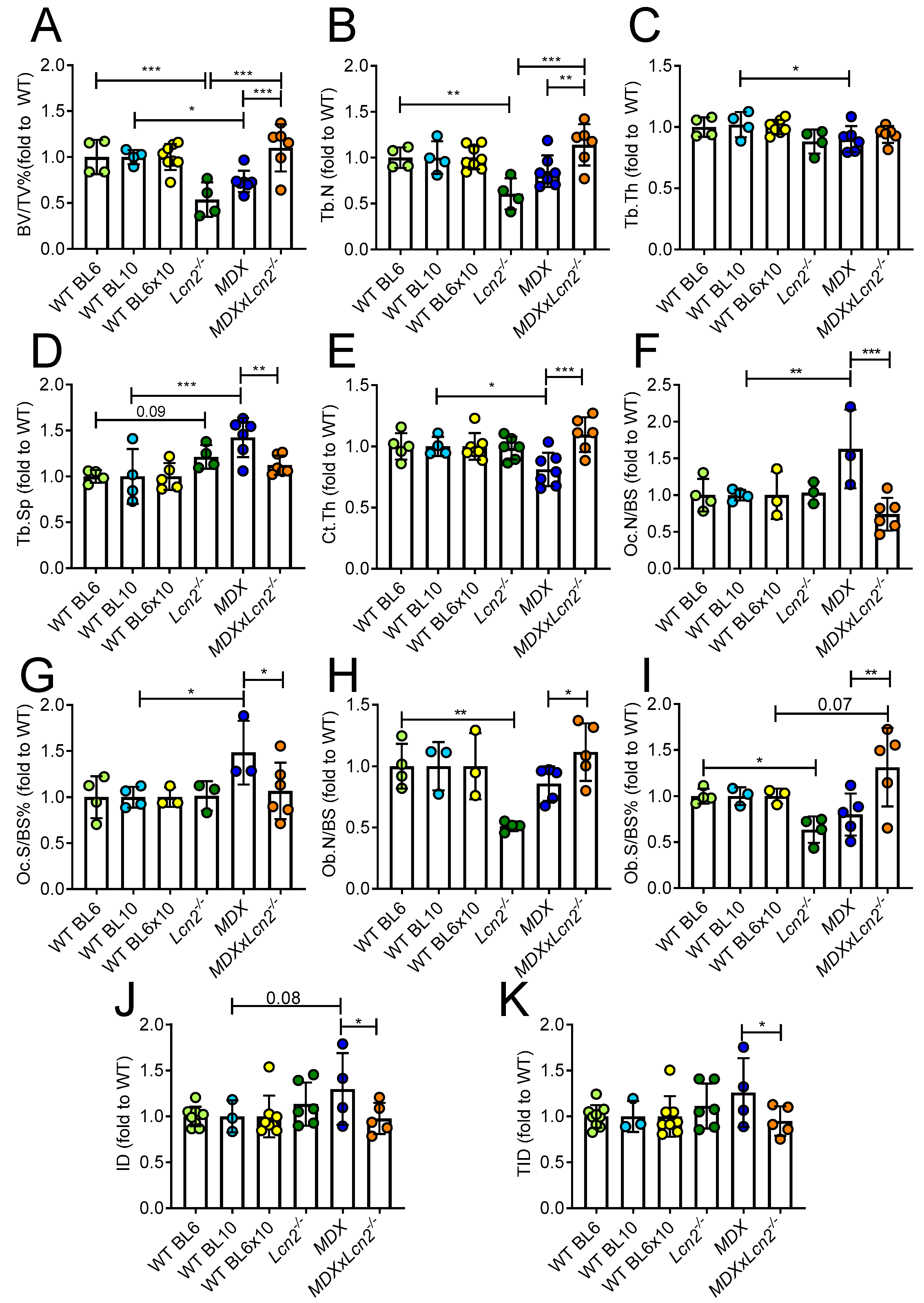
2.2. Genetic Ablation of Lcn2 Improves the Bone Phenotype of MDX Mice
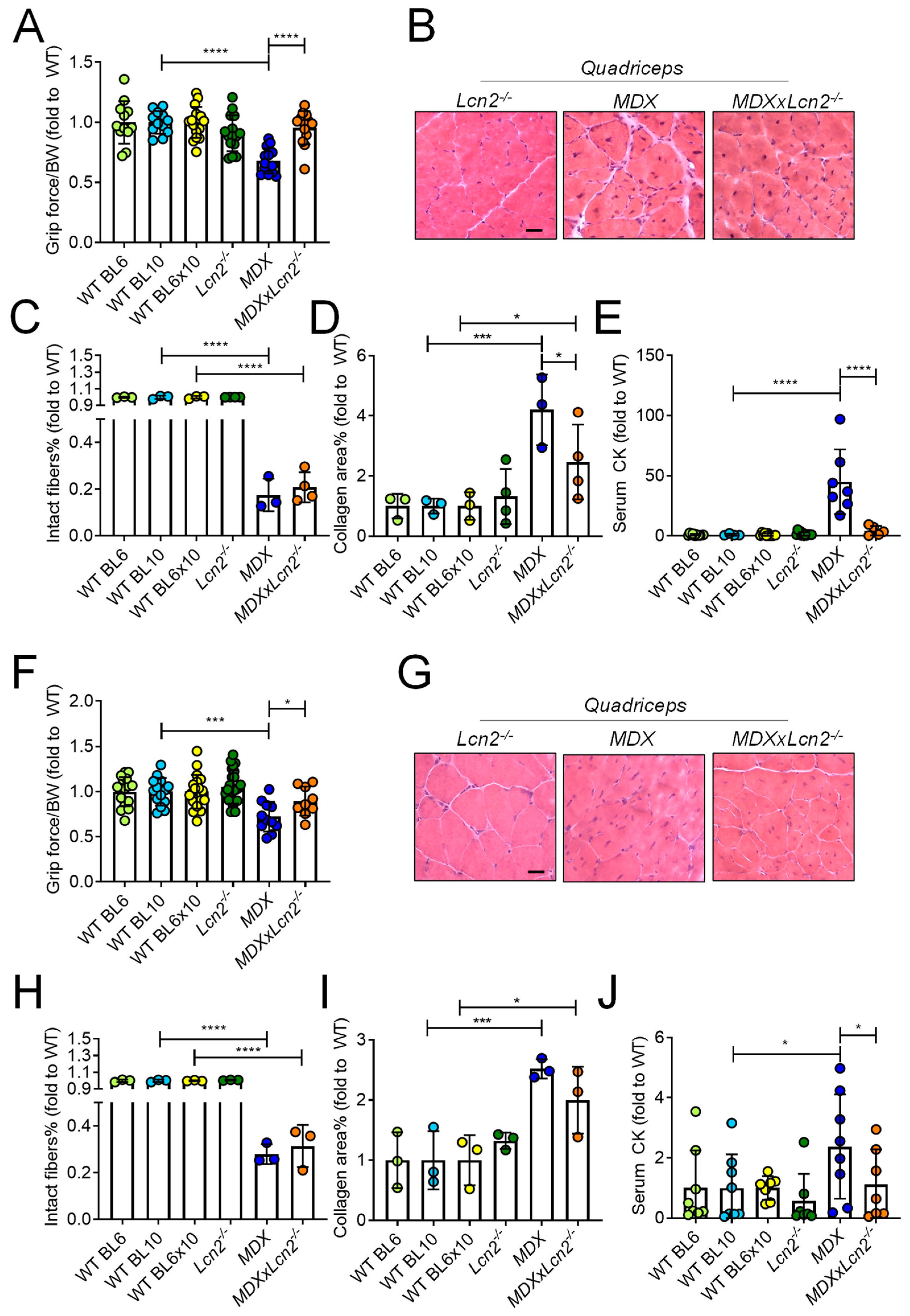
2.3. Genetic Ablation of Lcn2 Improves the Muscle Phenotype of MDX Mice
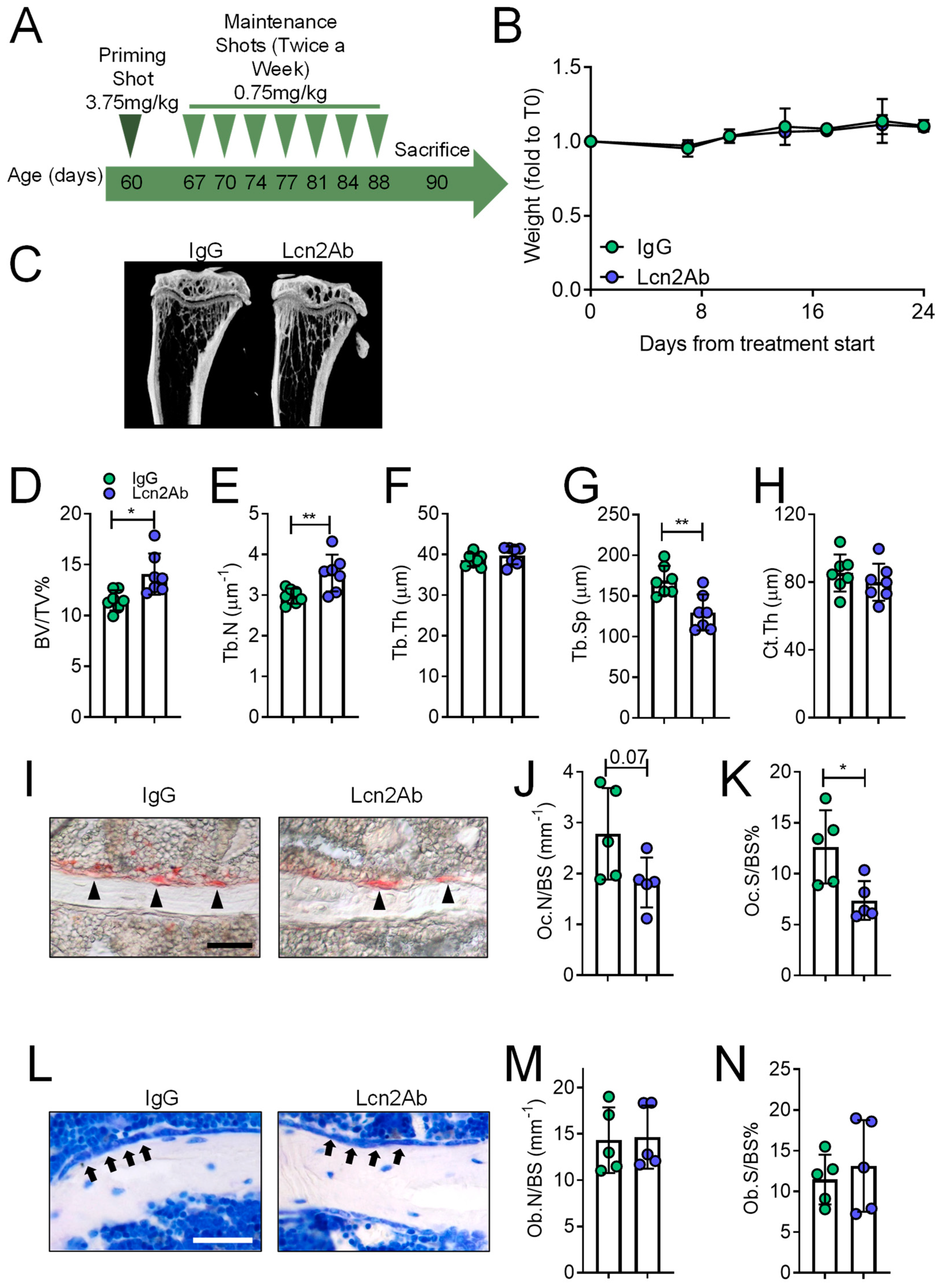
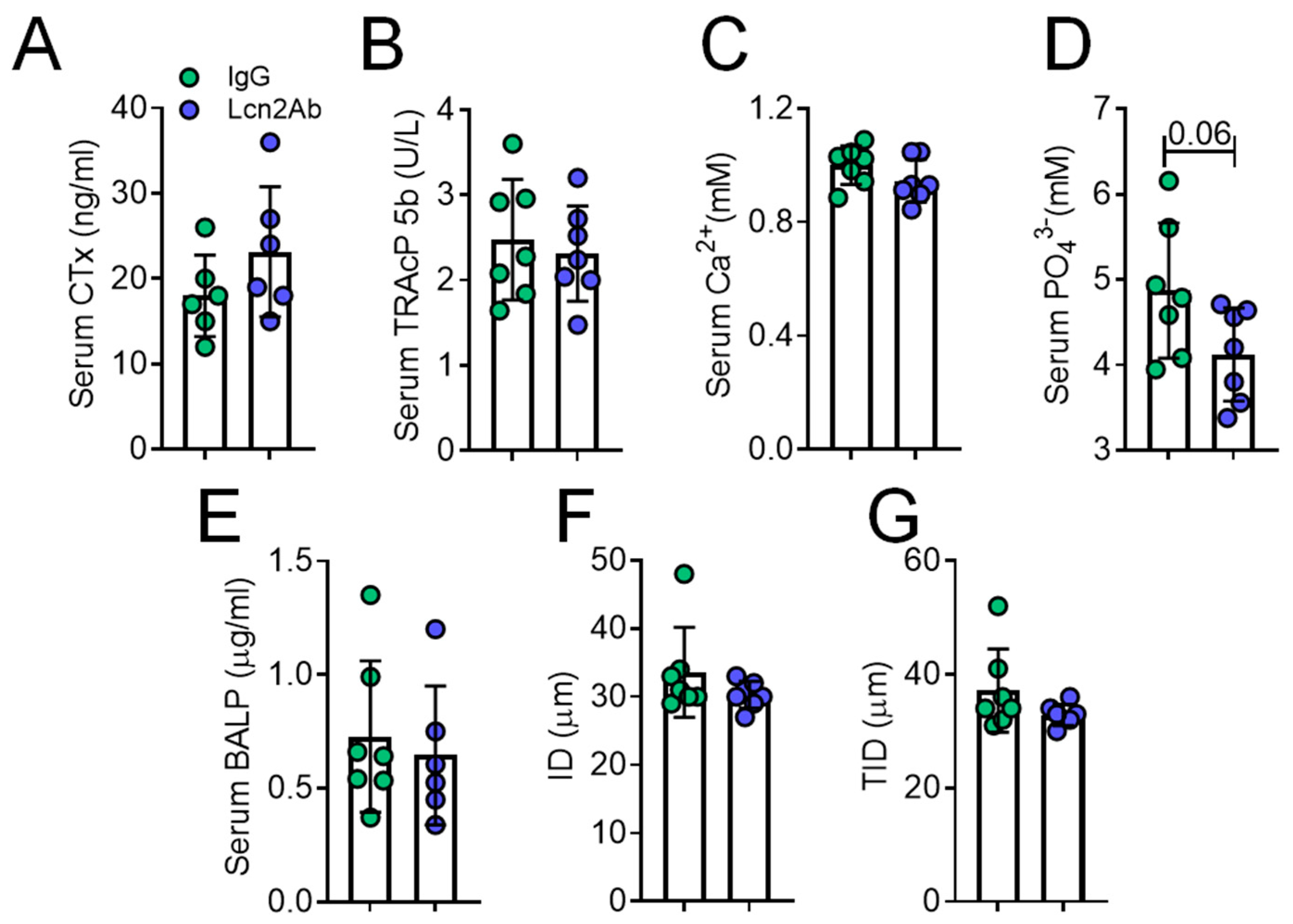
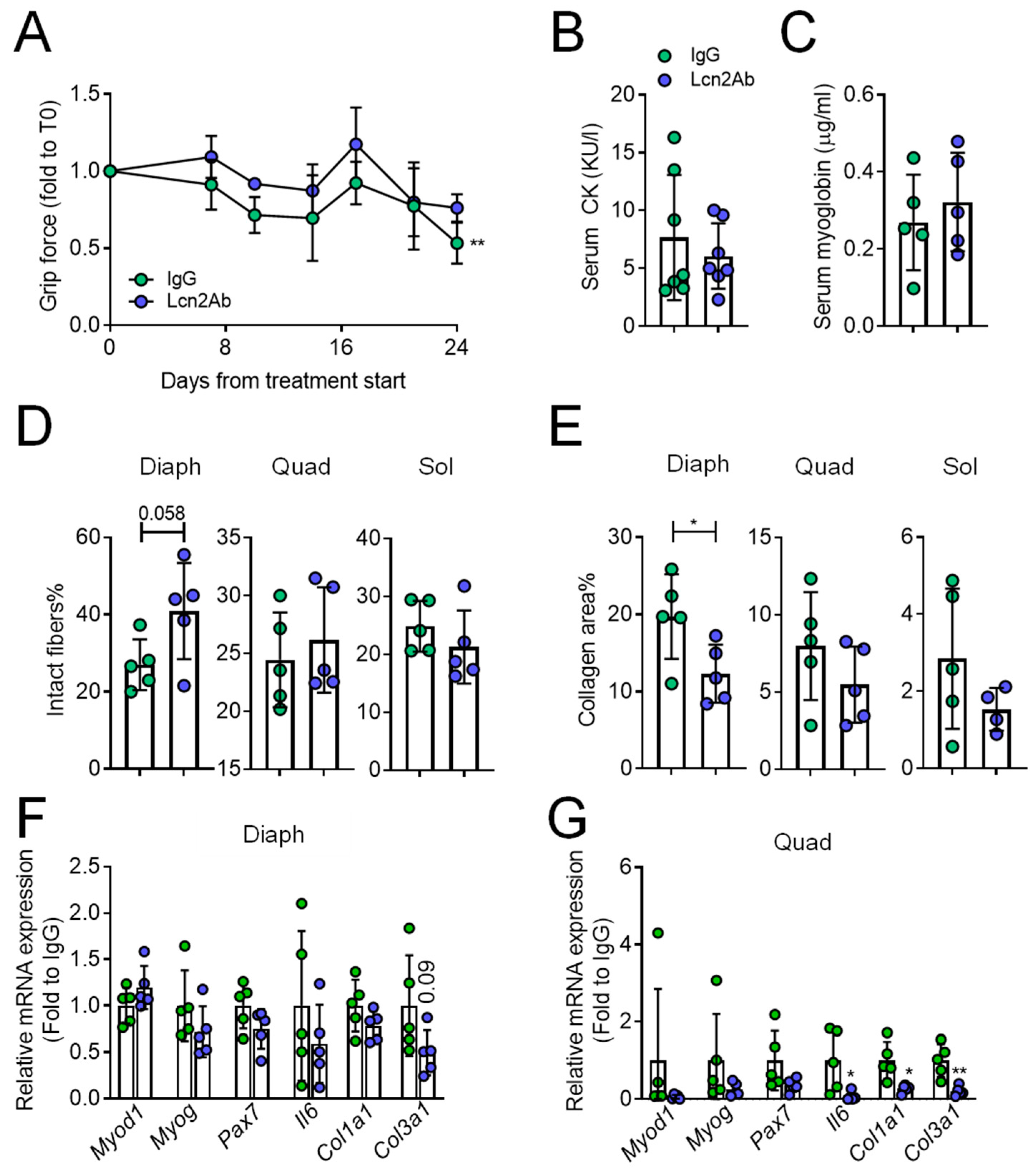
2.4. Lcn2 Blockade via a Monoclonal Antibody Improves Bone and Muscle Phenotype of MDX Mice
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Study Approval
4.3. Animals
4.4. Forelimb Grip Strenght Tests
4.5. Anti-Lcn2 Antibody Treatment
4.6. Comparative Real-Time RT-PCR
4.7. Histology and Histopathology
4.8. Bone Histomorphometry
4.9. Micro Computed Tomography (µCT) Analysis
4.10. Biodent® Mechanical Testing
4.11. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Lögdberg, L.; Wester, L. Immunocalins: A lipocalin subfamily that modulates immune and inflammatory responses. Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 2000, 1482, 284–297. [Google Scholar] [CrossRef]
- Mishra, J.; Qing, M.A.; Prada, A.; Mitsnefes, M.; Zahedi, K.; Yang, J.; Barasch, J.; Devarajan, P. Identification of neutrophil gelatinase-associated lipocalin as a novel early urinary biomarker for ischemic renal injury. J. Am. Soc. Nephrol. 2003, 14, 2534–2543. [Google Scholar] [CrossRef] [Green Version]
- Costa, D.; Lazzarini, E.; Canciani, B.; Giuliani, A.; Spanò, R.; Marozzi, K.; Manescu, A.; Cancedda, R.; Tavella, S. Altered bone development and turnover in transgenic mice over-expressing Lipocalin-2 in bone. J. Cell. Physiol. 2013, 228, 2210–2221. [Google Scholar] [CrossRef]
- Rucci, N.; Capulli, M.; Piperni, S.G.; Cappariello, A.; Lau, P.; Frings-Meuthen, P.; Heer, M.; Teti, A. Lipocalin 2: A new mechanoresponding gene regulating bone homeostasis. J. Bone Miner. Res. 2015, 30, 357–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosialou, I.; Shikhel, S.; Liu, J.M.; Maurizi, A.; Luo, N.; He, Z.; Huang, Y.; Zong, H.; Friedman, R.A.; Barasch, J.; et al. MC4R-dependent suppression of appetite by bone-derived lipocalin 2. Nature 2017, 543, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Capulli, M.; Ponzetti, M.; Maurizi, A.; Gemini-Piperni, S.; Berger, T.; Mak, T.W.; Teti, A.; Rucci, N. A Complex Role for Lipocalin 2 in Bone Metabolism: Global Ablation in Mice Induces Osteopenia Caused by an Altered Energy Metabolism. J. Bone Miner. Res. 2018, 33, 1141–1153. [Google Scholar] [CrossRef]
- Gambara, G.; Salanova, M.; Ciciliot, S.; Furlan, S.; Gutsmann, M.; Schiffl, G.; Ungethuem, U.; Volpe, P.; Gunga, H.C.; Blottner, D. Microgravity-induced transcriptome adaptation in mouse paraspinal longissimus dorsi muscle highlights insulin resistance-linked genes. Front. Physiol. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebalka, I.A.; Monaco, C.M.F.; Varah, N.E.; Berger, T.; D’souza, D.M.; Zhou, S.; Mak, T.W.; Hawke, T.J. Loss of the adipokine lipocalin-2 impairs satellite cell activation and skeletal muscle regeneration. Am. J. Physiol. Cell Physiol. 2018, 315, C714–C721. [Google Scholar] [CrossRef]
- Ponzetti, M.; Aielli, F.; Ucci, A.; Cappariello, A.; Lombardi, G.; Teti, A.; Rucci, N. Lipocalin 2 increases after high-intensity exercise in humans and influences muscle gene expression and differentiation in mice. J. Cell. Physiol. 2021, jcp.30501. [Google Scholar] [CrossRef] [PubMed]
- Marques, F.Z.; Prestes, P.R.; Byars, S.G.; Ritchie, S.C.; Würtz, P.; Patel, S.K.; Booth, S.A.; Rana, I.; Minoda, Y.; Berzins, S.P.; et al. Experimental and Human Evidence for Lipocalin-2 (Neutrophil Gelatinase-Associated Lipocalin [NGAL]) in the Development of Cardiac Hypertrophy and heart failure. J. Am. Heart Assoc. 2017, 6, e005971. [Google Scholar] [CrossRef]
- Sung, H.K.; Chan, Y.K.; Han, M.; Jahng, J.W.S.; Song, E.; Danielson, E.; Berger, T.; Mak, T.W.; Sweeney, G. Lipocalin-2 (NGAL) Attenuates Autophagy to Exacerbate Cardiac Apoptosis Induced by Myocardial Ischemia. J. Cell. Physiol. 2017, 232, 2125–2134. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Ahn, J.H.; Chang, S.Y.; Eguchi, M.; Ogier, A.; Han, S.J.; Park, Y.S.; Shim, C.Y.; Jang, Y.S.; Yang, B.; et al. Lipocalin-2 induces cardiomyocyte apoptosis by increasing intracellular iron accumulation. J. Biol. Chem. 2012, 287, 4808–4817. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Hanawa, H.; Ota, Y.; Hasegawa, G.; Hao, K.; Asami, F.; Watanabe, R.; Yoshida, T.; Toba, K.; Yoshida, K.; et al. Lipocalin-2/neutrophil gelatinase-B associated lipocalin is strongly induced in hearts of rats with autoimmune myocarditis and in human myocarditis. Circ. J. 2010, 74, 523–530. [Google Scholar] [CrossRef] [Green Version]
- Moser, H. Duchenne muscular dystrophy: Pathogenetic aspects and genetic prevention. Hum. Genet. 1984, 66, 17–40. [Google Scholar] [CrossRef]
- Petrof, B.J.; Shrager, J.B.; Stedman, H.H.; Kelly, A.M.; Sweeney, H.L. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. USA 1993, 90, 3710–3714. [Google Scholar] [CrossRef] [Green Version]
- De Paepe, B.; De Bleecker, J.L. Cytokines and chemokines as regulators of skeletal muscle inflammation: Presenting the case of Duchenne muscular dystrophy. Mediat. Inflamm. 2013, 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Apkon, S.D.; Blackwell, A.; Brumbaugh, D.; Case, L.E.; Clemens, P.R.; Hadjiyannakis, S.; Pandya, S.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018, 17, 251–267. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, M.L.; Mazzanti, A.; Galbiati, E.; Saraifoger, S.; Dubini, A.; Cornelio, F.; Morandi, L. Bone mineral density and bone metabolism in Duchenne muscular dystrophy. Osteoporos. Int. 2003, 14, 761–767. [Google Scholar] [CrossRef]
- Takata, S.; Yasui, N. Disuse osteoporosis. J. Med. Investig. 2001, 48, 147–156. [Google Scholar]
- Alexandre, C.; Vico, L. Pathophysiology of bone loss in disuse osteoporosis. Jt. Bone Spine. 2011, 78, 572–576. [Google Scholar] [CrossRef]
- Ginaldi, L.; Di Benedetto, M.C.; De Martinis, M. Osteoporosis, inflammation and ageing. Immun. Ageing 2005, 2. [Google Scholar] [CrossRef] [Green Version]
- Buckner, J.L.; Bowden, S.A.; Mahan, J.D. Optimizing bone health in Duchenne muscular dystrophy. Int. J. Endocrinol. 2015, 2015:928385. [Google Scholar] [CrossRef]
- Rufo, A.; Del Fattore, A.; Capulli, M.; Carvello, F.; De Pasquale, L.; Ferrari, S.; Pierroz, D.; Morandi, L.; De Simone, M.; Rucci, N.; et al. Mechanisms inducing low bone density in Duchenne muscular dystrophy in mice and humans. J. Bone Miner. Res. 2011, 26, 1891–1903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Alman, B.A.; Apkon, S.D.; Blackwell, A.; Case, L.E.; Cripe, L.; Hadjiyannakis, S.; Olson, A.K.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: Respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol. 2018, 17, 347–361. [Google Scholar] [CrossRef] [Green Version]
- De Almeida Hermes, T.; Mâncio, R.D.; Macedo, A.B.; Mizobuti, D.S.; Da Rocha, G.L.; Alves Cagnon, V.H.; Minatel, E. Tempol treatment shows phenotype improvement in mdx mice. PLoS ONE 2019, 14. [Google Scholar] [CrossRef]
- Yin, H.; Moulton, H.M.; Seow, Y.; Boyd, C.; Boutilier, J.; Iverson, P.; Wood, M.J.A. Cell-penetrating peptide-conjugated antisense oligonucleotides restore systemic muscle and cardiac dystrophin expression and function. Hum. Mol. Genet. 2008, 17, 3909–3918. [Google Scholar] [CrossRef] [Green Version]
- Capulli, M.; Rufo, A.; Teti, A.; Rucci, N. Global transcriptome analysis in mouse calvarial osteoblasts highlights sets of genes regulated by modeled microgravity and identifies A “mechanoresponsive osteoblast gene signature”. J. Cell. Biochem. 2009, 107, 240–252. [Google Scholar] [CrossRef]
- Abella, V.; Scotece, M.; Conde, J.; Gómez, R.; Lois, A.; Pino, J.; Gómez-Reino, J.J.; Lago, F.; Mobasheri, A.; Gualillo, O. The potential of lipocalin-2/NGAL as biomarker for inflammatory and metabolic diseases. Biomarkers 2015, 20, 565–571. [Google Scholar] [CrossRef] [Green Version]
- Xiao, X.; Yeoh, B.S.; Vijay-Kumar, M. Lipocalin 2: An Emerging Player in Iron Homeostasis and Inflammation. Annu. Rev. Nutr. 2017, 37, 103–130. [Google Scholar] [CrossRef]
- Moschen, A.R.; Adolph, T.E.; Gerner, R.R.; Wieser, V.; Tilg, H. Lipocalin-2: A Master Mediator of Intestinal and Metabolic Inflammation. Trends Endocrinol. Metab. 2017, 28, 388–397. [Google Scholar] [CrossRef]
- Rosenberg, A.S.; Puig, M.; Nagaraju, K.; Hoffman, E.P.; Villalta, S.A.; Rao, V.A.; Wakefield, L.M.; Woodcock, J. Immune-mediated pathology in Duchenne muscular dystrophy. Sci. Transl. Med. 2015, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conde, J.; Lazzaro, V.; Scotece, M.; Abella, V.; Villar, R.; López, V.; Gonzalez-Gay, M.; Pino, J.; Gómez, R.; Mera, A.; et al. Corticoids synergize with IL-1 in the induction of LCN2. Osteoarthr. Cartil. 2017, 25, 1172–1178. [Google Scholar] [CrossRef] [Green Version]
- Berger, T.; Togawa, A.; Duncan, G.S.; Elia, A.J.; You-Ten, A.; Wakeham, A.; Fong, H.E.H.; Cheung, C.C.; Mak, T.W. Lipocalin 2-deficient mice exhibit increased sensitivity to Escherichia coli infection but not to ischemia-reperfusion injury. Proc. Natl. Acad. Sci. USA 2006, 103, 1834–1839. [Google Scholar] [CrossRef] [Green Version]
- Cheng, L.; Xing, H.; Mao, X.; Li, L.; Li, X.; Li, Q. Lipocalin-2 promotes M1 macrophages polarization in a mouse cardiac ischaemia-reperfusion injury model. Scand. J. Immunol. 2015, 81, 31–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelosi, L.; Berardinelli, M.G.; De Pasquale, L.; Nicoletti, C.; D’Amico, A.; Carvello, F.; Moneta, G.M.; Catizone, A.; Bertini, E.; De Benedetti, F.; et al. Functional and Morphological Improvement of Dystrophic Muscle by Interleukin 6 Receptor Blockade. EBioMedicine 2015, 2, 285–293. [Google Scholar] [CrossRef] [Green Version]
- Gutpel, K.M.; Hrinivich, W.T.; Hoffman, L.M. Skeletal muscle fibrosis in the mdx/utrn+/-mouse validates its suitability as a murine model of duchenne muscular dystrophy. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Dempster, D.W.; Compston, J.E.; Drezner, M.K.; Glorieux, F.H.; Kanis, J.A.; Malluche, H.; Meunier, P.J.; Ott, S.M.; Recker, R.R.; Parfitt, A.M. Standardized nomenclature, symbols, and units for bone histomorphometry: A 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J. Bone Miner. Res. 2013, 28, 2–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldkamp, L.A.; Davis, L.C.; Kress, J.W. Practical cone-beam algorithm. J. Opt. Soc. Am. A 1984, 1, 612. [Google Scholar] [CrossRef] [Green Version]
- Lorensen, W.E.; Cline, H.E. Marching Cubes: A High Resolution 3D Surface Construction Algorithm. Comput. Graph. 1987, 21, 163–169. [Google Scholar] [CrossRef]
- Vinet, L.; Zhedanov, A. A “missing” family of classical orthogonal polynomials. J. Phys. A Math. Theor. 2011, 44, 085201. [Google Scholar] [CrossRef]
- Bouxsein, M.L.; Boyd, S.K.; Christiansen, B.A.; Guldberg, R.E.; Jepsen, K.J.; Müller, R. Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J. Bone Miner. Res. 2010, 25, 1468–1486. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ponzetti, M.; Ucci, A.; Maurizi, A.; Giacchi, L.; Teti, A.; Rucci, N. Lipocalin 2 Influences Bone and Muscle Phenotype in the MDX Mouse Model of Duchenne Muscular Dystrophy. Int. J. Mol. Sci. 2022, 23, 958. https://doi.org/10.3390/ijms23020958
Ponzetti M, Ucci A, Maurizi A, Giacchi L, Teti A, Rucci N. Lipocalin 2 Influences Bone and Muscle Phenotype in the MDX Mouse Model of Duchenne Muscular Dystrophy. International Journal of Molecular Sciences. 2022; 23(2):958. https://doi.org/10.3390/ijms23020958
Chicago/Turabian StylePonzetti, Marco, Argia Ucci, Antonio Maurizi, Luca Giacchi, Anna Teti, and Nadia Rucci. 2022. "Lipocalin 2 Influences Bone and Muscle Phenotype in the MDX Mouse Model of Duchenne Muscular Dystrophy" International Journal of Molecular Sciences 23, no. 2: 958. https://doi.org/10.3390/ijms23020958
APA StylePonzetti, M., Ucci, A., Maurizi, A., Giacchi, L., Teti, A., & Rucci, N. (2022). Lipocalin 2 Influences Bone and Muscle Phenotype in the MDX Mouse Model of Duchenne Muscular Dystrophy. International Journal of Molecular Sciences, 23(2), 958. https://doi.org/10.3390/ijms23020958