
DFT Study of the Molecular and Electronic Structure of Metal-Free Tetrabenzoporphyrin and Its Metal Complexes with Zn, Cd, Al, Ga, In

 , and
, and
Abstract
1. Introduction
2. Results
2.1. Molecular Structure
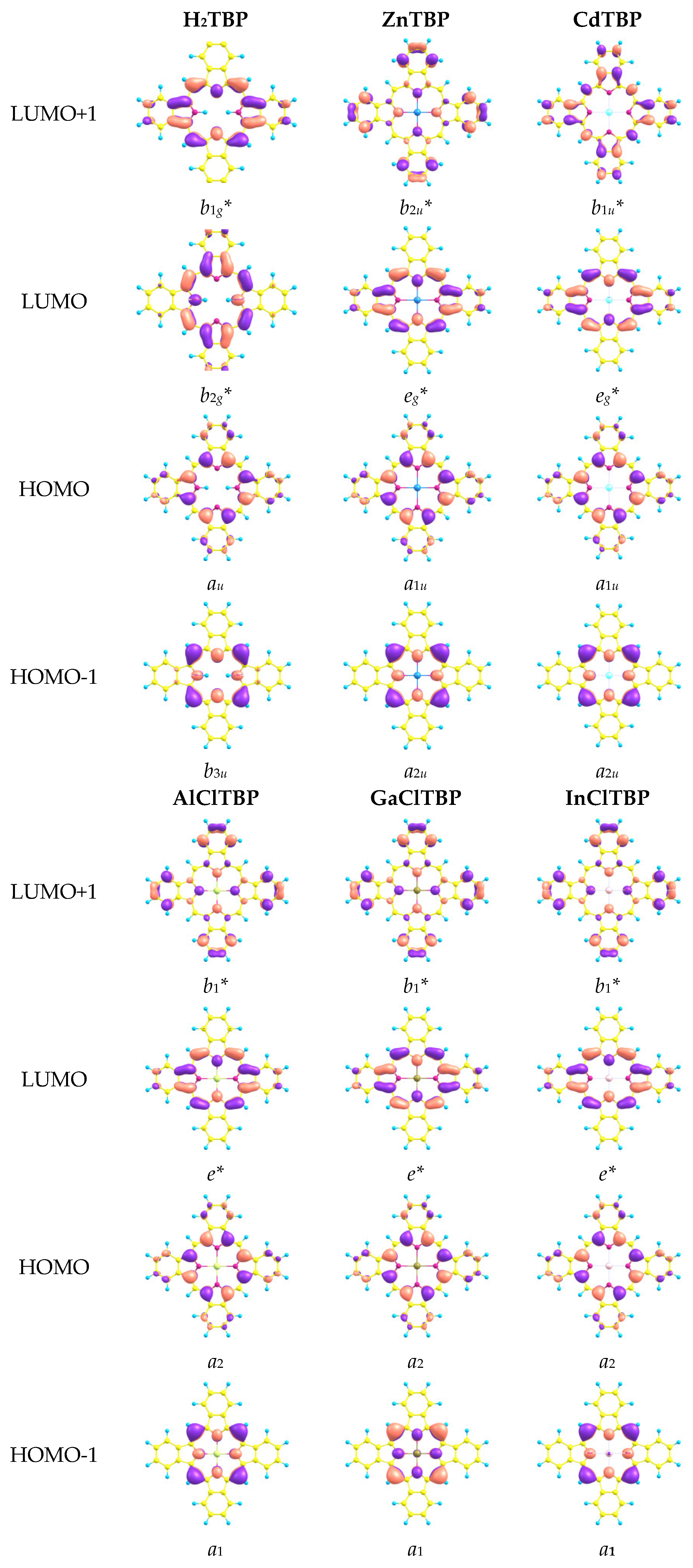
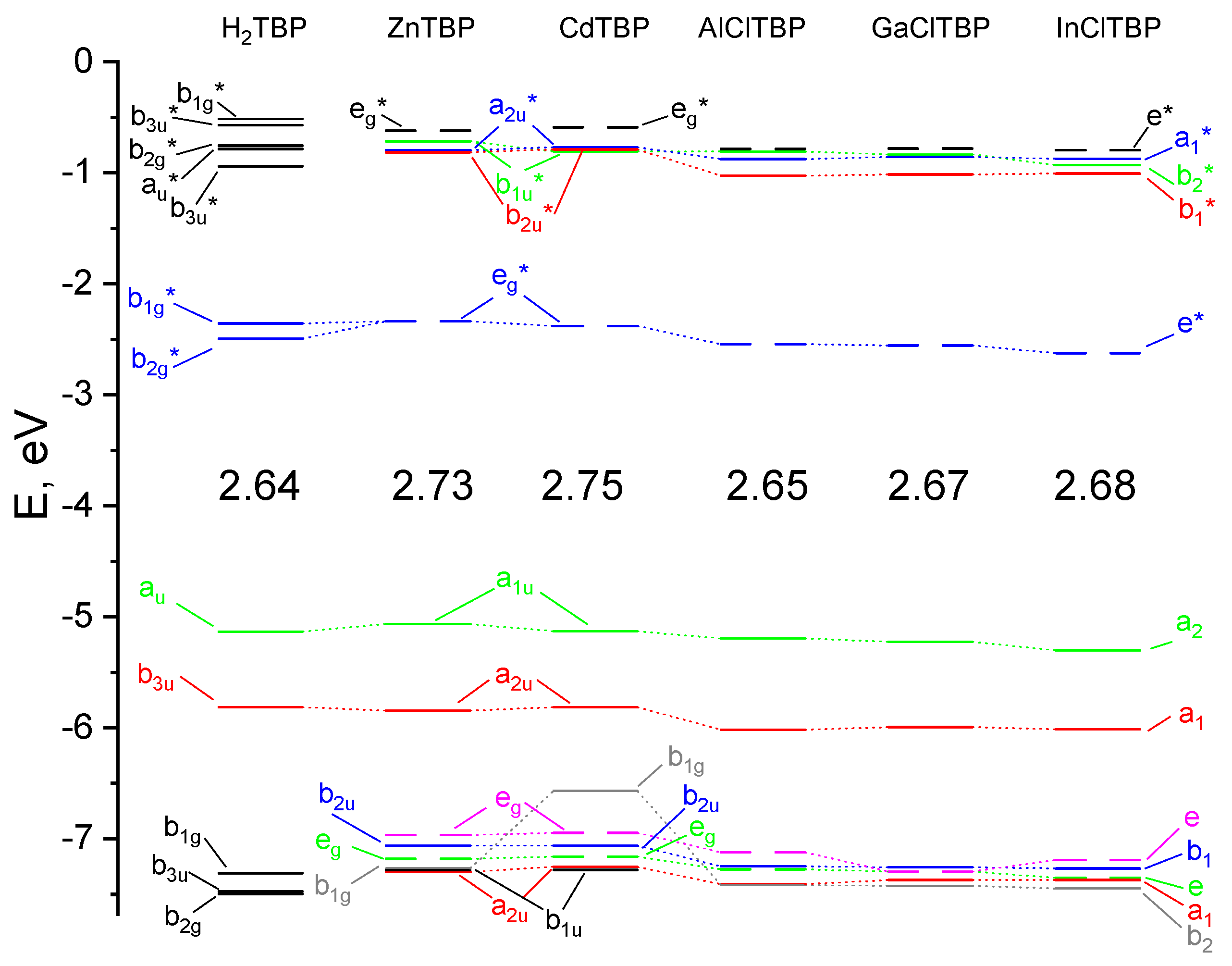
2.2. NBO-Analysis
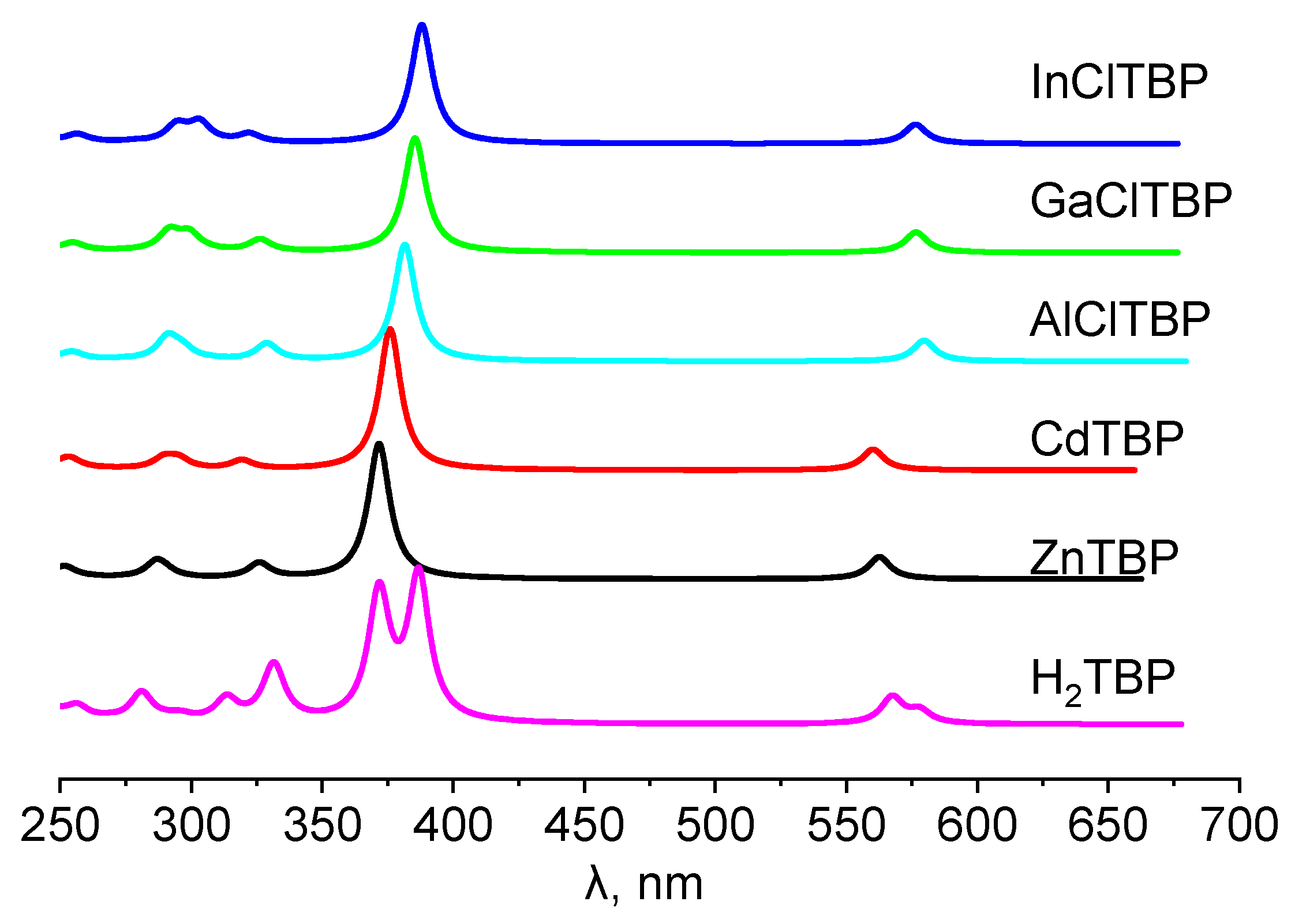
2.3. Electronic Spectra
3. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sorokin, A.B. Recent progress on exploring µ-oxo bridged binuclear porphyrinoid complexes in catalysis and material science. Coord. Chem. Rev. 2019, 389, 141–160. [Google Scholar] [CrossRef]
- Yu, Z.; Hagfeldt, A.; Sun, L. The application of transition metal complexes in hole-transporting layers for perovskite solar cells: Recent progress and future perspectives. Coord. Chem. Rev. 2020, 406, 213143. [Google Scholar] [CrossRef]
- Almeida-Marrero, V.; Van De Winckel, E.; Anaya-Plaza, E.; Torres, T.; De La Escosura, A. Porphyrinoid biohybrid materials as an emerging toolbox for biomedical light management. Chem. Soc. Rev. 2018, 47, 7369–7400. [Google Scholar] [CrossRef]
- Koifman, O.I.; Ageeva, T.A.; Beletskaya, I.P.; Averin, A.D.; Yakushev, A.A.; Tomilova, L.G.; Dubinina, T.V.; Tsivadze, A.Y.; Gorbunova, Y.G.; Martynov, A.G.; et al. Macroheterocyclic compounds-a key building block in new functional materials and molecular devices. Macroheterocycles 2020, 13, 311–467. [Google Scholar] [CrossRef]
- Longevial, J.; Clément, S.; Wytko, J.A.; Ruppert, R.; Weiss, J.; Richeter, S. Peripherally Metalated Porphyrins with Applications in Catalysis, Molecular Electronics and Biomedicine. Chem.-A Eur. J. 2018, 24, 15442–15460. [Google Scholar] [CrossRef]
- Zhou, Q.; Xu, S.; Yang, C.; Zhang, B.; Li, Z.; Deng, K. Modulation of peripheral substituents of cobalt thioporphyrazines and their photocatalytic activity. Appl. Catal. B Environ. 2016, 192, 108–115. [Google Scholar] [CrossRef]
- Borek, C.; Hanson, K.; Djurovich, P.I.; Thompson, M.E.; Aznavour, K.; Bau, R.; Sun, Y.; Forrest, S.R.; Brooks, J.; Michalski, L.; et al. Highly efficient, near-infrared electrophosphorescence from a Pt-metalloporphyrin complex. Angew. Chem.-Int. Ed. 2007, 46, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Srivatsan, A.; Missert, J.R.; Upadhyay, S.K.; Pandey, R.K. Porphyrin-based photosensitizers and the corresponding multifunctional nanoplatforms for cancer-imaging and phototherapy. J. Porphyr. Phthalocyanines 2015, 19, 109–134. [Google Scholar] [CrossRef]
- Abrahamse, H.; Hamblin, M.R. New photosensitizers for photodynamic therapy. Biochem. J. 2016, 473, 347–364. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, S.A.; Wilson, D.F. Porphyrin dendrimers as biological oxygen sensors. In Designing Dendrimers; John Wiley & Sons: New York, NY, USA, 2012; pp. 465–503. ISBN 978-0-470-43355-3. [Google Scholar]
- Quaranta, M.; Borisov, S.M.; Klimant, I. Indicators for optical oxygen sensors. Bioanal. Rev. 2012, 4, 115–157. [Google Scholar] [CrossRef] [PubMed]
- Papkovsky, D.B.; Dmitriev, R.I. Biological detection by optical oxygen sensing. Chem. Soc. Rev. 2013, 42, 8700–8732. [Google Scholar] [CrossRef] [PubMed]
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.W.; Gollnick, S.O.; Hahn, S.M.; Hamblin, M.R.; Juzeniene, A.; Kessel, D.; et al. Photodynamic therapy of cancer: An update. CA Cancer J. Clin. 2011, 61, 250–281. [Google Scholar] [CrossRef] [PubMed]
- Kelkar, S.S.; Reineke, T.M. Theranostics: Combining imaging and therapy. Bioconjug. Chem. 2011, 22, 1879–1903. [Google Scholar] [CrossRef]
- Juweid, M.E.; Mottaghy, F.M. Current and future aspects of nuclear molecular therapies: A model of theranostics. Methods 2011, 55, 193–195. [Google Scholar] [CrossRef]
- Scherer, R.L.; McIntyre, J.O.; Matrisian, L.M. Imaging matrix metalloproteinases in cancer. Cancer Metastasis Rev. 2008, 27, 679–690. [Google Scholar] [CrossRef]
- Danhier, F.; Ansorena, E.; Silva, J.M.; Coco, R.; Le Breton, A.; Préat, V. PLGA-based nanoparticles: An overview of biomedical applications. J. Control. Release 2012, 161, 505–522. [Google Scholar] [CrossRef] [PubMed]
- Van De Wiele, C.; Oltenfreiter, R. Imaging probes targeting matrix metalloproteinases. Cancer Biother. Radiopharm. 2006, 21, 409–417. [Google Scholar] [CrossRef]
- O’Connor, A.E.; Gallagher, W.M.; Byrne, A.T. Porphyrin and nonporphyrin photosensitizers in oncology: Preclinical and clinical advances in photodynamic therapy. Photochem. Photobiol. 2009, 85, 1053–1074. [Google Scholar] [CrossRef]
- Bonnett, R. Photosensitizers of the porphyrin and phthalocyanine series for photodynamic therapy. Chem. Soc. Rev. 1995, 24, 19–33. [Google Scholar] [CrossRef]
- Balaz, M.; Collins, H.A.; Dahlstedt, E.; Anderson, H.L. Synthesis of hydrophilic conjugated porphyrin dimers for one-photon and two-photon photodynamic therapy at NIR wavelengths. Org. Biomol. Chem. 2009, 7, 874–888. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.; Sutty, S.; Klenkler, R.; Aziz, H. Renewed interest in metal phthalocyanine donors for small molecule organic solar cells. Sol. Energy Mater. Sol. Cells 2014, 124, 217–226. [Google Scholar] [CrossRef]
- Travkin, V.V.; Stuzhin, P.A.; Okhapkin, A.I.; Korolyov, S.A.; Pakhomov, G.L. Organic tandem Schottky junction cells with high open circuit voltage. Synth. Met. 2016, 212, 51–54. [Google Scholar] [CrossRef]
- Kadish, K.M.; Smith, K.M.; Guilard, R. The Porphyrin Handbook: Inorganic, Organometallic and Coordination Chemistry; Elsevier: Amsterdam, The Netherlands, 2003; Volume 3, p. 425. [Google Scholar]
- Milgrom, L.R. The Colours of Life: An Introduction to the Chemistry of Porphyrins and Related Compounds; Oxford University Press: New York, NY, USA, 1997. [Google Scholar]
- Otlyotov, A.A.; Ryzhov, I.V.; Kuzmin, I.A.; Zhabanov, Y.A.; Mikhailov, M.S.; Stuzhin, P.A. Peculiarities of electronic structure and chemical bonding in iron and cobalt metal complexes of porphyrazine and tetra(1,2,5-thiadiazole)porphyrazine. J. Porphyr. Phthalocyanines 2020, 24, 1146–1154. [Google Scholar] [CrossRef]
- Otlyotov, A.A.; Ryzhov, I.V.; Kuzmin, I.A.; Zhabanov, Y.A.; Mikhailov, M.S.; Stuzhin, P.A. Dft study of molecular and electronic structure of ca(Ii) and zn(ii) complexes with porphyrazine and tetrakis(1,2,5-thiadiazole)porphyrazine. Int. J. Mol. Sci. 2020, 21, 2923. [Google Scholar] [CrossRef]
- Zhabanov, Y.A.; Ryzhov, I.V.; Kuzmin, I.A.; Eroshin, A.V.; Stuzhin, P.A. DFT Study of Molecular and Electronic Structure of Y, La and Lu Complexes with Porphyrazine and Tetrakis(1,2,5-thiadiazole)porphyrazine. Molecules 2020, 26, 113. [Google Scholar] [CrossRef] [PubMed]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef]
- Zhabanov, Y.A.; Tverdova, N.V.; Giricheva, N.I.; Girichev, G.V.; Stuzhin, P.A. DFT Study of molecular and electronic structure of magnesium (II) tetra(1,2,5-chalcogenadiazolo) porphyrazines, [TXDPzMg] (X = O, S, Se, Te). J. Porphyr. Phthalocyanines 2017, 21, 439–452. [Google Scholar] [CrossRef]
- Lebedeva (Yablokova), I.A.; Ivanova, S.S.; Novakova, V.; Zhabanov, Y.A.; Stuzhin, P.A. Perfluorinated porphyrazines. 3. Synthesis, spectral-luminescence and electrochemical properties of perfluorinated octaphenylporphyrazinatozinc(II). J. Fluor. Chem. 2018, 214, 86–93. [Google Scholar] [CrossRef]
- Stuzhin, P.A.; Skvortsov, I.A.; Zhabanov, Y.A.; Somov, N.V.; Razgonyaev, O.V.; Nikitin, I.A.; Koifman, O.I. Subphthalocyanine azaanalogues—Boron(III) subporphyrazines with fused pyrazine fragments. Dye. Pigment. 2019, 162, 888–897. [Google Scholar] [CrossRef]
- Otlyotov, A.A.; Zhabanov, Y.A.; Pogonin, A.E.; Kuznetsova, A.S.; Islyaikin, M.K.; Girichev, G.V. Gas-phase structures of hemiporphyrazine and dicarbahemiporphyrazine: Key role of interactions inside coordination cavity. J. Mol. Struct. 2019, 1184, 576–582. [Google Scholar] [CrossRef]
- Hamdoush, M.; Nikitin, K.; Skvortsov, I.; Somov, N.; Zhabanov, Y.; Stuzhin, P.A. Influence of heteroatom substitution in benzene rings on structural features and spectral properties of subphthalocyanine dyes. Dye. Pigment. 2019, 170, 107584. [Google Scholar] [CrossRef]
- Eroshin, A.V.; Otlyotov, A.A.; Zhabanov, Y.A.; Veretennikov, V.V.; Islyaikin, M.K. Complexes of ca(Ii), ni(ii) and zn(ii) with hemi-and dicarbahemiporphyrazines: Molecular structure and features of metal-ligand bonding. Macroheterocycles 2021, 14, 119–129. [Google Scholar] [CrossRef]
- Zhabanov, Y.A.; Eroshin, A.V.; Stuzhin, P.A.; Ryzhov, I.V.; Kuzmin, I.A.; Finogenov, D.N. Molecular structure, thermodynamic and spectral characteristics of metal-free and nickel complex of tetrakis(1,2,5-thiadiazolo)porphyrazine. Molecules 2021, 26, 2945. [Google Scholar] [CrossRef]
- Bursch, M.; Hansen, A.; Pracht, P.; Kohn, J.T.; Grimme, S. Theoretical study on conformational energies of transition metal complexes. Phys. Chem. Chem. Phys. 2021, 23, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Maurer, L.R.; Bursch, M.; Grimme, S.; Hansen, A. Assessing Density Functional Theory for Chemically Relevant Open-Shell Transition Metal Reactions. J. Chem. Theory Comput. 2021, 17, 6134–6151. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Sect. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Sliznev, V.V.; Pogonin, A.E.; Ischenko, A.A.; Girichev, G.V. Vibrational spectra of cobalt(II), nickel(II), copper(II), zinc(II) Etioporphyrins-II, MN4C32H36. Macroheterocycles 2014, 7, 60–72. [Google Scholar] [CrossRef][Green Version]
- Luc, N.Q.; Dang, V.S.; Tran, Q.T.; Pham, V.T.; Mai, A.T. Density Function Theory calculation, and phthalonitrile process for a synthesis of single crystal zinc phthalocyanine. Mater. Sci. Semicond. Process. 2020, 113, 105025. [Google Scholar] [CrossRef]
- Ruan, C.Y.; Mastryukov, V.; Fink, M. Electron diffraction studies of metal phthalocyanines, MPc, where M = Sn, Mg, and Zn (reinvestigation). J. Chem. Phys. 1999, 111, 3035–3041. [Google Scholar] [CrossRef]
- Allred, A.L.; Rochow, E.G. A scale of electronegativity based on electrostatic force. J. Inorg. Nucl. Chem. 1958, 5, 264–268. [Google Scholar] [CrossRef]
- Mann, J.B.; Meek, T.L.; Allen, L.C. Configuration energies of the main group elements. J. Am. Chem. Soc. 2000, 122, 2780–2783. [Google Scholar] [CrossRef]
- Gouterman, M. Spectra of porphyrins. J. Mol. Spectrosc. 1961, 6, 138–163. [Google Scholar] [CrossRef]
- Gouterman, M.; Wagnière, G.H.; Snyder, L.C. Spectra of porphyrins. Part II. Four orbital model. J. Mol. Spectrosc. 1963, 11, 108–127. [Google Scholar] [CrossRef]
- Ehrenberg, B.; Johnson, F.M. Spectroscopic studies of tetrabenzoporphyrins: MgTBP, ZnTBP and H2TBP. Spectrochim. Acta Part A Mol. Spectrosc. 1990, 46, 1521–1532. [Google Scholar] [CrossRef]
- VanCott, T.C.; Koralewski, M.; Metcalf, D.H.; Schatz, P.N.; Williamson, B.E. Magnetooptical spectroscopy of zinc tetrabenzoporphyrin in an argon matrix. J. Phys. Chem. 1993, 97, 7417–7426. [Google Scholar] [CrossRef]
- Koehorst, R.B.M.; Kleibeuker, J.F.; Schaafsma, T.J.; De Bie, D.A.; Geurtsen, B.; Henrie, R.N.; Van Der Plas, H.C. Preparation and spectroscopic properties of pure tetrabenzoporphyrins. J. Chem. Soc. Perkin Trans. 1981, 7, 1005–1009. [Google Scholar] [CrossRef]
- Stillman, M.; Mack, J.; Kobayashi, N. Theoretical aspects of the spectroscopy of porphyrins and phthalocyanines. J. Porphyr. Phthalocyanines 2002, 6, 296–300. [Google Scholar] [CrossRef]
- Nemykin, V.N.; Hadt, R.G.; Belosludov, R.V.; Mizuseki, H.; Kawazoe, Y. Influence of molecular geometry, exchange-correlation functional, and solvent effects in the modeling of vertical excitation energies in phthalocyanines using time-dependent density functional theory (TDDFT) and polarized continuum model TDDFT methods: Can modern computational chemistry methods explain experimental controversies? J. Phys. Chem. A 2007, 111, 12901–12913. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Rev A.1; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Schuchardt, K.L.; Didier, B.T.; Elsethagen, T.; Sun, L.; Gurumoorthi, V.; Chase, J.; Li, J.; Windus, T.L. Basis set exchange: A community database for computational sciences. J. Chem. Inf. Model. 2007, 47, 1045–1052. [Google Scholar] [CrossRef]
- Feller, D. The role of databases in support of computational chemistry calculations. J. Comput. Chem. 1996, 17, 1571–1586. [Google Scholar] [CrossRef]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibson, T.D.; Windus, T.L. New Basis Set Exchange: An Open, Up-to-Date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814–4820. [Google Scholar] [CrossRef] [PubMed]
- Andrae, D.; Häußermann, U.; Dolg, M.; Stoll, H.; Preuß, H. Energy-adjusted ab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Metz, B.; Stoll, H.; Dolg, M. Small-core multiconfiguration-Dirac-Hartree-Fock-adjusted pseudopotentials for post-d main group elements: Application to PbH and PbO. J. Chem. Phys. 2000, 113, 2563–2569. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A.; Vreven, J.T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03, Revision B.03; Gaussian Inc.: Wallingford, CT, USA, 2003. [Google Scholar]
- Granovsky, A.A. Firefly Version 8. Available online: http://classic.chem.msu.su/gran/firefly/index.html (accessed on 15 April 2021).
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Zhurko, G.A.; Zhurko, D.A. ChemCraft Version 1.6 (Build 312). Available online: http://www.chemcraftprog.com/index.html (accessed on 1 December 2021).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| H2TBP | ZnTBP | CdTBP | AlClTBP | GaClTBP | InClTBP | |
|---|---|---|---|---|---|---|
| Symmetry | D2h | D4h | D4h | C4v | C4v | C4v |
| Distances | ||||||
| M-N | 1.012 (2.340) 2 | 2.063 | 2.152 | 2.044 | 2.075 | 2.184 |
| M-Cl | - | - | - | 2.154 | 2.196 | 2.360 |
| N…Nopp | 4.268 (4.106) | 4.125 | 4.304 | 4.006 | 4.060 | 4.195 |
| N…Nadj | 2.961 | 2.917 | 3.043 | 2.833 | 2.871 | 2.966 |
| N-Cα | 1.362 (1.353) | 1.363 | 1.355 | 1.370 | 1.366 | 1.361 |
| Cα-Cβ | 1.439 (1.457) | 1.446 | 1.453 | 1.440 | 1.441 | 1.446 |
| Cβ-Cβ | 1.407 (1.399) | 1.401 | 1.409 | 1.395 | 1.397 | 1.404 |
| Cβ-Cγ | 1.394 (1.389) | 1.393 | 1.391 | 1.394 | 1.393 | 1.392 |
| Cγ-Cδ | 1.379 (1.385) | 1.381 | 1.383 | 1.380 | 1.380 | 1.381 |
| Cδ-Cδ | 1.404 (1.398) | 1.402 | 1.399 | 1.403 | 1.402 | 1.401 |
| Cα-Cm | 1.379 (1.390) | 1.383 | 1.398 | 1.373 | 1.376 | 1.387 |
| Cα-Cm-Cα | 128.0 | 127.4 | 130.3 | 125.3 | 126.0 | 128.3 |
| r(M-X) 3 | - | 0 | 0 | 0.407 | 0.453 | 0.678 |
| Bond angles | ||||||
| N-Cα-Cm | 126.2 (125.9) | 125.6 | 125.6 | 125.6 | 125.8 | 125.8 |
| N-Cα-Cβ | 106.3 (110.7) | 109.5 | 107.6 | 110.6 | 109.9 | 108.6 |
| A 4 | 180.0 | 180.0 | 180.0 | 176.3 | 176.4 | 171.6 |
| ZnTBP | CdTBP | AlClTBP | GaClTBP | InClTBP | |
|---|---|---|---|---|---|
| E(HOMO), eV | −5.06 | −5.13 | −5.19 | −5.22 | −5.30 |
| E(LUMO), eV | −2.34 | −2.38 | −2.54 | −2.55 | −2.62 |
| ∆E, eV | 2.72 | 2.75 | 2.65 | 2.67 | 2.68 |
| q(M) NPA, e | 1.304 | 1.333 | 1.776 | 1.673 | 1.718 |
| q(N) NPA, e | −0.573 | −0.570 | −0.607 | −0.584 | −0.579 |
| q(Cl) NPA, e | −0.574 | −0.545 | −0.563 | ||
| configuration | 4s0.353d9.974p0.37 | 5s0.415d9.955p0.31 | 3s0.423p0.76 | 4s0.554p0.76 | 5s0.545p0.73 |
| ∑ E(d-a), kcal mol−1 | 312.2 | 303.8 | 451.7 | 495.6 | 471.9 |
| Q(M–N) | 0.283 | 0.274 | 0.320 | 0.335 | 0.330 |
| r(M–N), Å | 2.063 | 2.152 | 2.044 | 2.075 | 2.184 |
| State | Composition (%) | λ, nm | f | exp λ, nm |
|---|---|---|---|---|
| H2TBP | ||||
| 11B1u | (31) (69) | 578 | 0.11 | 663.5 (Py) [48] |
| 11B2u | (19) (81) | 568 | 0.23 | |
| 21B1u | (8) (59) (28) | 387 | 1.24 | 431.8 (Py) [48] |
| 21B2u | (74) (18) | 372 | 1.10 | 416.1 (Py) [48] |
| 31B1u | (88) (6) | 332 | 0.35 | |
| 31B2u | (93) | 331 | 0.15 | |
| ZnTBP | ||||
| 11 Eu | (21) (79) | 563 | 0.19 | 613(Ar matrix) [49] 628.5 (Py) [48] |
| 21 Eu | (73) (20) | 372 | 1.17 | 433.3(Py) [48] |
| 31 Eu | (94) | 326 | 0.13 | |
| CdTBP | ||||
| 11Eu | (23) (77) | 560 | 0.18 | 628 (Py) [50] |
| 21 Eu | (71) (22) | 376 | 1.21 | 434 (Py) [50] |
| 31Eu | (18) (78) | 320 | 0.08 | |
| AlClTBP | ||||
| 11E | (20) (80) | 580 | 0.18 | |
| 21E | (73) (19) | 382 | 1.01 | |
| 31E | (93) | 329 | 0.14 | |
| GaClTBP | ||||
| 11E | (21) (79) | 577 | 0.17 | |
| 21E | (73) (20) | 385 | 0.99 | |
| 31E | (5) (93) | 326 | 0.10 | |
| InClTBP | ||||
| 11E | (22) (77) | 577 | 0.17 | |
| 21E | (72) (22) | 388 | 1.03 | |
| 31E | (23) (73) | 322 | 0.07 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eroshin, A.V.; Otlyotov, A.A.; Kuzmin, I.A.; Stuzhin, P.A.; Zhabanov, Y.A. DFT Study of the Molecular and Electronic Structure of Metal-Free Tetrabenzoporphyrin and Its Metal Complexes with Zn, Cd, Al, Ga, In. Int. J. Mol. Sci. 2022, 23, 939. https://doi.org/10.3390/ijms23020939
Eroshin AV, Otlyotov AA, Kuzmin IA, Stuzhin PA, Zhabanov YA. DFT Study of the Molecular and Electronic Structure of Metal-Free Tetrabenzoporphyrin and Its Metal Complexes with Zn, Cd, Al, Ga, In. International Journal of Molecular Sciences. 2022; 23(2):939. https://doi.org/10.3390/ijms23020939
Chicago/Turabian StyleEroshin, Alexey V., Arseniy A. Otlyotov, Ilya A. Kuzmin, Pavel A. Stuzhin, and Yuriy A. Zhabanov. 2022. "DFT Study of the Molecular and Electronic Structure of Metal-Free Tetrabenzoporphyrin and Its Metal Complexes with Zn, Cd, Al, Ga, In" International Journal of Molecular Sciences 23, no. 2: 939. https://doi.org/10.3390/ijms23020939
APA StyleEroshin, A. V., Otlyotov, A. A., Kuzmin, I. A., Stuzhin, P. A., & Zhabanov, Y. A. (2022). DFT Study of the Molecular and Electronic Structure of Metal-Free Tetrabenzoporphyrin and Its Metal Complexes with Zn, Cd, Al, Ga, In. International Journal of Molecular Sciences, 23(2), 939. https://doi.org/10.3390/ijms23020939