
Antibodies against Angiotensin II Type 1 and Endothelin 1 Type A Receptors in Cardiovascular Pathologies



{kind=link}
Abstract
1. Introduction
2. Angiotensin II (AngII)
3. Endothelin (ET) 1
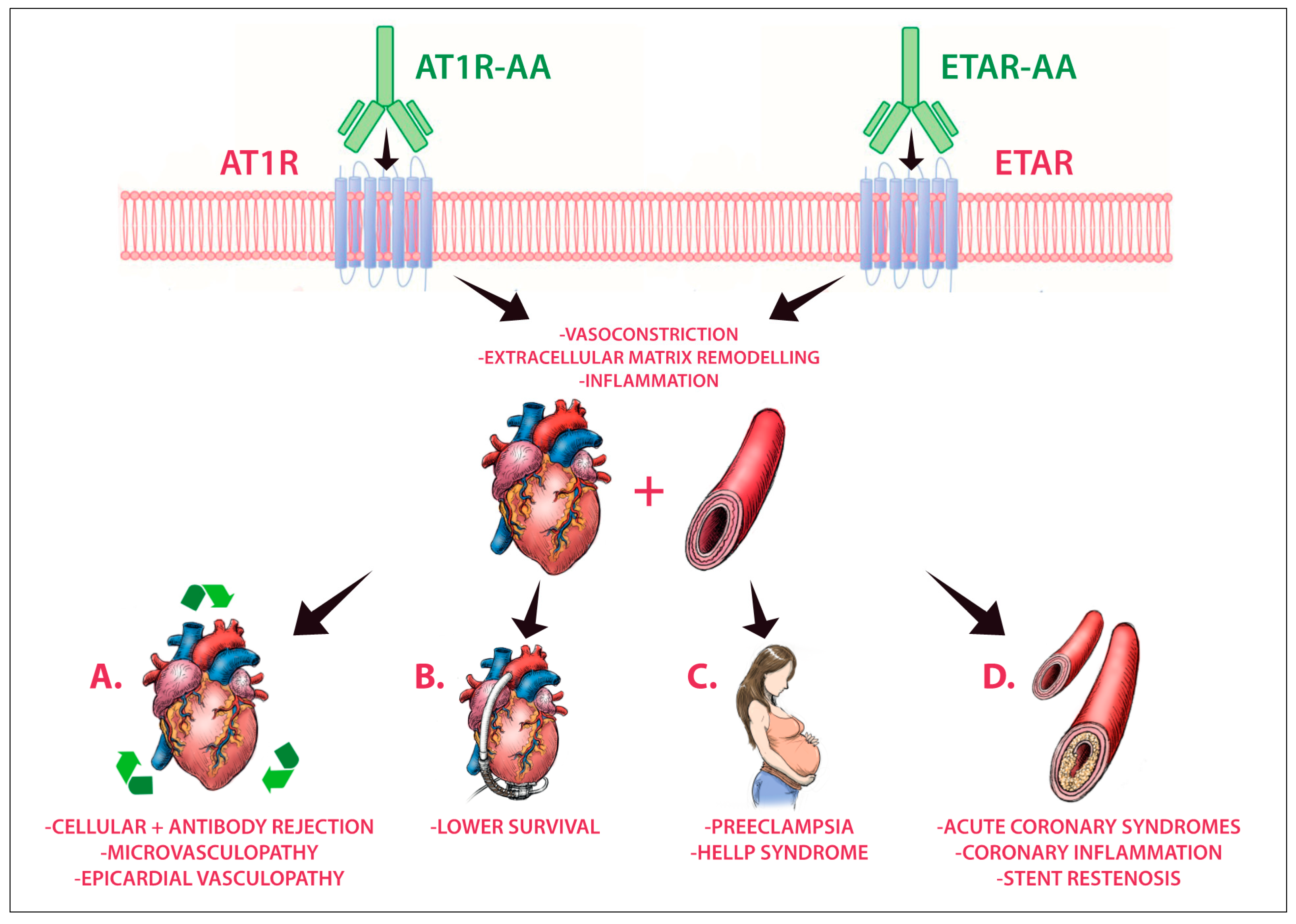
4. AT1R-AAs and ETAR-AAs
5. Towards Cardiovascular Pathologies
5.1. Preeclampsia
5.2. Heart Transplantation and Mechanical Circulatory Support (MCS)
5.3. Acute Coronary Syndromes
6. The Beginning of a New Era?
Author Contributions
Funding
Conflicts of Interest
References
- Riemekasten, G.; Philippe, A.; Näther, M.; Slowinski, T.; Müller, D.N.; Heidecke, H.; Matucci-Cerinic, M.; Czirják, L.; Lukitsch, I.; Becker, M.; et al. Involvement of functional autoantibodies against vascular receptors in systemic sclerosis. Ann. Rheum. Dis. 2011, 70, 530–536. [Google Scholar] [CrossRef]
- de Gasparo, M.; Catt, K.J.; Inagami, T.; Wright, J.W.; Unger, T. International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol. Rev. 2000, 52, 415–472. [Google Scholar]
- Schneider, M.P.; Boesen, E.I.; Pollock, D.M. Contrasting actions of endothelin ETA and ETB receptors in cardiovascular disease. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 731–759. [Google Scholar] [CrossRef] [PubMed]
- Becker, M.O.; Kill, A.; Kutsche, M.; Guenther, J.; Rose, A.; Tabeling, C.; Witzenrath, M.; Kühl, A.A.; Heidecke, H.; Ghofrani, H.A.; et al. vascular receptor autoantibodies in pulmonary arterial hypertension associated with systemic sclerosis. Am. J. Respir. Crit. Care Med. 2014, 190, 808–817. [Google Scholar] [CrossRef] [PubMed]
- Dragun, D.; Bräsen, J.H.; Schönemann, C.; Fritsche, L.; Budde, K.; Neumayer, H.-H.; Luft, F.C.; Wallukat, G. Patients with steroid refractory acute vascular rejection develop agonistic antibodies targeting angiotensin II type 1 receptor. Transplant. Proc. 2003, 35, 2104–2105. [Google Scholar] [CrossRef]
- Dragun, D.; Müller, D.N.; Bräsen, J.H.; Fritsche, L.; Nieminen-Kelhä, M.; Dechend, R.; Kintscher, U.; Rudolph, B.; Hoebeke, J.; Eckert, D.; et al. Angiotensin II type 1-receptor activating antibodies in renal-allograft rejection. N. Engl. J. Med. 2005, 352, 558–569. [Google Scholar] [CrossRef]
- Banasik, M.; Boratyńska, M.; Kościelska-Kasprzak, K.; Kamińska, D.; Zmonarski, S.; Mazanowska, O.; Krajewska, M.; Bartoszek, D.; Żabińska, M.; Myszka, M.; et al. Non-HLA antibodies: Angiotensin II type 1 receptor (Anti-AT1R) and endothelin-1 type A receptor (Anti-ETAR) are associated with renal allograft injury and graft loss. Transplant. Proc. 2014, 46, 2618–2621. [Google Scholar] [CrossRef]
- Catar, R.A.; Wischnewski, O.; Chen, L.; Heidecke, H.; Rutz, C.; Schülein, R.; Dragun, D.; Philippe, A.; Kusch, A. Non-HLA antibodies targeting angiotensin II type 1 receptors and endothelin-1 type A receptors impair endothelial repair via a Β2-arrestin link to the MTOR pathway. Kidney Int. 2021. [Google Scholar] [CrossRef]
- Cozzi, E.; Calabrese, F.; Schiavon, M.; Feltracco, P.; Seveso, M.; Carollo, C.; Loy, M.; Cardillo, M.; Rea, F. Immediate and catastrophic antibody-mediated rejection in a lung transplant recipient with anti-angiotensin II receptor type 1 and anti-endothelin-1 receptor type A antibodies. Am. J. Transplant 2017, 17, 557–564. [Google Scholar] [CrossRef]
- Ohe, H.; Uchida, Y.; Yoshizawa, A.; Hirao, H.; Taniguchi, M.; Maruya, E.; Yurugi, K.; Hishida, R.; Maekawa, T.; Uemoto, S.; et al. Association of anti-human leukocyte antigen and anti-angiotensin II type 1 receptor antibodies with liver allograft fibrosis after immunosuppression withdrawal. Transplantation 2014, 98, 1105–1111. [Google Scholar] [CrossRef]
- OʼLeary, J.G.; Demetris, A.J.; Philippe, A.; Freeman, R.; Cai, J.; Heidecke, H.; Smith, C.; Hart, B.; Jennings, L.W.; Catar, R.; et al. Non-HLA antibodies impact on C4d staining, stellate cell activation and fibrosis in liver allografts. Transplantation 2017, 101, 2399–2409. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, J.G.; Philippe, A.; Freeman, R.; Heidecke, H.; Jennings, L.W.; Catar, R.; Klintmalm, G.B.; Dragun, D. Non-HLA autoantibodies at 1 year negatively affect 5-year native renal function in liver transplant recipients. Transplant. Proc. 2021, 53, 1019–1024. [Google Scholar] [CrossRef]
- Miedema, J.; Schreurs, M.; van der Sar–van der Brugge, S.; Paats, M.; Baart, S.; Bakker, M.; Hoek, R.; Dik, W.A.; Endeman, H.; Van Der Velden, V.; et al. Antibodies against angiotensin ii receptor type 1 and endothelin a receptor are associated with an unfavorable COVID19 disease course. Front. Immunol. 2021, 12, 684142. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in COVID-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Budding, K.; van de Graaf, E.A.; Hoefnagel, T.; Kwakkel-van Erp, J.M.; van Kessel, D.A.; Dragun, D.; Hack, C.E.; Otten, H.G. Anti-ETAR and anti-AT1R autoantibodies are elevated in patients with endstage cystic fibrosis. J. Cyst. Fibros. 2015, 14, 42–45. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, J.L.; Sigmund, C.D. Minireview: Overview of the renin-angiotensin system—An endocrine and paracrine system. Endocrinology 2003, 144, 2179–2183. [Google Scholar] [CrossRef]
- Bonnardeaux, A.; Davies, E.; Jeunemaitre, X.; Féry, I.; Charru, A.; Clauser, E.; Tiret, L.; Cambien, F.; Corvol, P.; Soubrier, F. Angiotensin II type 1 receptor gene polymorphisms in human essential hypertension. Hypertension 1994, 24, 63–69. [Google Scholar] [CrossRef]
- Wierzbicki, A.S.; Lambert-Hammill, M.; Lumb, P.J.; Crook, M.A. Renin-angiotensin system polymorphisms and coronary events in familial hypercholesterolemia. Hypertension 2000, 36, 808–812. [Google Scholar] [CrossRef][Green Version]
- Berge, K.E.; Bakken, A.; Bøhn, M.; Erikssen, J.; Berg, K. A DNA polymorphism at the angiotensin ii type 1 receptor (AT1R) locus and myocardial infarction. Clin. Genet. 1997, 52, 71–76. [Google Scholar] [CrossRef]
- Dandona, P.; Dhindsa, S.; Ghanim, H.; Chaudhuri, A. Angiotensin II and inflammation: The effect of angiotensin-converting enzyme inhibition and angiotensin ii receptor blockade. J. Hum. Hypertens. 2007, 21, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.K.; Griendling, K.K. Angiotensin II cell signaling: Physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Cell Physiol. 2007, 292, C82–C97. [Google Scholar] [CrossRef]
- Griendling, K.K.; Delafontaine, P.; Rittenhouse, S.E.; Gimbrone, M.A.; Alexander, R.W. Correlation of Receptor sequestration with sustained diacylglycerol accumulation in angiotensin ii-stimulated cultured vascular smooth muscle cells. J. Biol. Chem. 1987, 262, 14555–14562. [Google Scholar] [CrossRef]
- Lassègue, B.; Alexander, R.W.; Nickenig, G.; Clark, M.; Murphy, T.J.; Griendling, K.K. Angiotensin II down-regulates the vascular smooth muscle AT1 receptor by transcriptional and post-transcriptional mechanisms: Evidence for homologous and heterologous regulation. Mol. Pharmacol. 1995, 48, 601–609. [Google Scholar] [PubMed]
- Touyz, R.M.; He, G.; Deng, L.-Y.; Schiffrin, E.L. Role of extracellular signal-regulated kinases in angiotensin ii-stimulated contraction of smooth muscle cells from human resistance arteries. Circulation 1999, 99, 392–399. [Google Scholar] [CrossRef]
- Griendling, K.K.; Lassègue, B.; Alexander, R.W. Angiotensin receptors and their therapeutic implications. Annu. Rev. Pharmacol. Toxicol. 1996, 36, 281–306. [Google Scholar] [CrossRef]
- Bedecs, K.; Elbaz, N.; Sutren, M.; Masson, M.; Susini, C.; Strosberg, A.D.; Nahmias, C. Angiotensin II type 2 receptors mediate inhibition of mitogen-activated protein kinase cascade and functional activation of SHP-1 tyrosine phosphatase. Biochem. J. 1997, 325, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Munzenmaier, D.H.; Greene, A.S. Opposing actions of angiotensin II on microvascular growth and arterial blood pressure. Hypertension 1996, 27, 760–765. [Google Scholar] [CrossRef]
- Bumpus, F.M.; Catt, K.J.; Chiu, A.T.; DeGasparo, M.; Goodfriend, T.; Husain, A.; Peach, M.J.; Taylor, D.G.; Timmermans, P.B. Nomenclature for angiotensin receptors. A report of the nomenclature committee of the council for high blood pressure research. Hypertension 1991, 17, 720–721. [Google Scholar] [CrossRef]
- Wang, Y.; Del Borgo, M.; Lee, H.W.; Baraldi, D.; Hirmiz, B.; Gaspari, T.A.; Denton, K.M.; Aguilar, M.-I.; Samuel, C.S.; Widdop, R.E. Anti-fibrotic potential of AT2 receptor agonists. Front. Pharmacol. 2017, 8, 564. [Google Scholar] [CrossRef] [PubMed]
- Horinouchi, T.; Terada, K.; Higashi, T.; Miwa, S. Endothelin receptor signaling: New insight into its regulatory mechanisms. J. Pharmacol. Sci. 2013, 123, 85–101. [Google Scholar] [CrossRef]
- Rubin, L.J. Endothelin receptor antagonists for the treatment of pulmonary artery hypertension. Life Sci. 2012, 91, 517–521. [Google Scholar] [CrossRef]
- Frumkin, L.R. The pharmacological treatment of pulmonary arterial hypertension. Pharmacol. Rev. 2012, 64, 583–620. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, J.; Goresky, C.A.; Fournier, A. Pulmonary clearance of circulating endothelin-1 in dogs in vivo: Exclusive role of ETB receptors. J. Appl. Physiol. 1996, 81, 1510–1515. [Google Scholar] [CrossRef] [PubMed]
- Warner, T.D.; Mitchell, J.A.; de Nucci, G.; Vane, J.R. Endothelin-1 and rndothelin-3 release EDRF from isolated perfused arterial vessels of the rat and rabbit. J. Cardiovasc. Pharmacol. 1989, 13, S85–S88. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, M.; Kurihara, H.; Kimura, S.; Tomobe, Y.; Kobayashi, M.; Mitsui, Y.; Yazaki, Y.; Goto, K.; Masaki, T. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 1988, 332, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Stewart, D.J.; Kubac, G.; Costello, K.B.; Cernacek, P. Increased plasma endothelin-1 in the early hours of acute myocardial infarction. J. Am. Coll. Cardiol. 1991, 18, 38–43. [Google Scholar] [CrossRef]
- Galiuto, L.; DeMaria, A.N.; del Balzo, U.; May-Newman, K.; Flaim, S.F.; Wolf, P.L.; Kirchengast, M.; Iliceto, S. Ischemia-reperfusion injury at the microvascular level: Treatment by endothelin A–selective antagonist and evaluation by myocardial contrast echocardiography. Circulation 2000, 102, 3111–3116. [Google Scholar] [CrossRef]
- Kirchengast, M.; Hergenröder, S.; Schult, S.; Münter, K.; Rübsamen, K. Endothelin-1 and unstable angina: Effect of either endothelin ETA or ETB receptor antagonism in a locally injured canine coronary artery. Eur. J. Pharmacol. 1998, 341, 187–190. [Google Scholar] [CrossRef]
- Stewart, D.J.; Levy, R.D.; Cernacek, P.; Langleben, D. Increased plasma endothelin-1 in pulmonary hypertension: Marker or mediator of disease? Ann. Intern. Med. 1991, 114, 464–469. [Google Scholar] [CrossRef]
- Cody, R.J.; Haas, G.J.; Binkley, P.F.; Capers, Q.; Kelley, R. Plasma endothelin correlates with the extent of pulmonary hypertension in patients with chronic congestive heart failure. Circulation 1992, 85, 504–509. [Google Scholar] [CrossRef]
- Philogene, M.C.; Johnson, T.; Vaught, A.J.; Zakaria, S.; Fedarko, N. Antibodies against angiotensin II type 1 and endothelin A receptors: Relevance and pathogenicity. Hum. Immunol. 2019, 80, 561–567. [Google Scholar] [CrossRef]
- Zhang, Q.; Reed, E.F. The importance of non-HLA antibodies in transplantation. Nat. Rev. Nephrol. 2016, 12, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Banasik, M.; Boratyńska, M.; Kościelska-Kasprzak, K.; Krajewska, M.; Mazanowska, O.; Kamińska, D.; Bartoszek, D.; Zabińska, M.; Myszka, M.; Nowakowska, B.; et al. The impact of non-HLA antibodies directed against endothelin-1 type A receptors (ETAR) on early renal transplant outcomes. Transpl. Immunol. 2014, 30, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Sheng, L.; Huang, P.; Li, J.; Zhang, C.-H.; Yang, J.; Liao, Y.-H.; Li, L.-D. Agonistic autoantibodies against the angiotensin AT1 receptor increase in unstable angina patients after stent implantation. Coron. Artery Dis. 2014, 25, 691–697. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, Z.; Chen, Y.; Li, S.; Lv, Y.; Zhou, W.; Liao, M.; Zhu, F.; Zhou, Z.; Cheng, X.; et al. Autoantibodies targeting AT1 receptor from patients with acute coronary syndrome upregulate proinflammatory cytokines expression in endothelial cells involving NF- κ B pathway. J. Immunol. Res. 2014, 2014, 342693. [Google Scholar] [CrossRef] [PubMed]
- Bjerre, A.; Tangeraas, T.; Heidecke, H.; Dragun, D.; Dechend, R.; Staff, A.C. Angiotensin II Type 1 Receptor Antibodies in Childhood Kidney Transplantation. Pediatr. Transplant. 2016, 20, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Lukitsch, I.; Kehr, J.; Chaykovska, L.; Wallukat, G.; Nieminen-Kelhä, M.; Batuman, V.; Dragun, D.; Gollasch, M. Renal ischemia and transplantation predispose to vascular constriction mediated by angiotensin II type 1 receptor-activating antibodies. Transplant. J. 2012, 94, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Dechend, R.; Dragun, D.; Herse, F.; Riemekasten, G.; Schulze-Forster, K.; Müller, D.N.; Heidecke, H. Activating Autoantibodies against the AT1- Receptor in Vascular Disease. Transplantationsmedizin 2012, 24, 20–26. [Google Scholar]
- Zhang, S.; Zheng, R.; Yang, L.; Zhang, X.; Zuo, L.; Yang, X.; Bai, K.; Song, L.; Tian, J.; Yang, J.; et al. Angiotensin type 1 receptor autoantibody from preeclamptic patients induces human fetoplacental vasoconstriction. J. Cell. Physiol. 2013, 228, 142–148. [Google Scholar] [CrossRef]
- Buttrup Larsen, S.; Wallukat, G.; Schimke, I.; Sandager, A.; Tvilum Christensen, T.; Uldbjerg, N.; Tørring, N. Functional autoantibodies against endothelin-1 receptor type A and angiotensin II receptor type 1 in patients with preeclampsia. Pregnancy Hypertens. 2018, 14, 189–194. [Google Scholar] [CrossRef]
- Gareau, A.J.; Wiebe, C.; Pochinco, D.; Gibson, I.W.; Ho, J.; Rush, D.N.; Nickerson, P.W. Pre-transplant AT 1 R antibodies correlate with early allograft rejection. Transpl. Immunol. 2018, 46, 29–35. [Google Scholar] [CrossRef]
- Darrah, E.; Kim, A.; Zhang, X.; Boronina, T.; Cole, R.N.; Fava, A.; Giles, J.T.; Bingham, C.O., III; Chalmers, M.J.; Griffin, P.R.; et al. Proteolysis by granzyme B enhances presentation of autoantigenic peptidylarginine deiminase 4 epitopes in rheumatoid arthritis. J. Proteome Res. 2017, 16, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Mirocha, J.; Aintablian, T.; Dimbil, S.; Moriguchi, J.; Arabia, F.; Kobashigawa, J.A.; Reinsmoen, N. Revealing a New mode of sensitization induced by mechanical circulatory support devices: Impact of anti-AT1R antibodies. Clin. Transpl. 2018, 32, e13178. [Google Scholar] [CrossRef]
- Abadir, P.M.; Jain, A.; Powell, L.J.; Xue, Q.-L.; Tian, J.; Hamilton, R.G.; Bennett, D.A.; Finucane, T.; Walston, J.D.; Fedarko, N.S. Discovery and validation of agonistic angiotensin receptor autoantibodies as biomarkers of adverse outcomes. Circulation 2017, 135, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.L.; Herlitz, H.; Schulze, W.; Wallukat, G.; Micke, P.; Eftekhari, P.; Sjögren, K.G.; Hjalmarson, A.; Müller-Esterl, W.; Hoebeke, J. Autoantibodies against the angiotensin receptor (AT1) in patients with hypertension. J. Hypertens. 2000, 18, 945–953. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.-H.; Wei, Y.-M.; Wang, M.; Wang, Z.-H.; Yuan, H.-T.; Cheng, L.-X. Autoantibodies against AT1-receptor and alpha1-adrenergic receptor in patients with hypertension. Hypertens. Res. 2002, 25, 641–646. [Google Scholar] [CrossRef]
- Zhu, F.; Sun, Y.; Wang, M.; Ma, S.; Chen, X.; Cao, A.; Chen, F.; Qiu, Y.; Liao, Y. Correlation between HLA-DRB1, HLA-DQB1 polymorphism and autoantibodies against angiotensin AT1 receptors in chinese patients with essential hypertension. Clin. Cardiol. 2011, 34, 302–308. [Google Scholar] [CrossRef]
- Rossitto, G.; Regolisti, G.; Rossi, E.; Negro, A.; Nicoli, D.; Casali, B.; Toniato, A.; Caroccia, B.; Seccia, T.M.; Walther, T.; et al. Elevation of angiotensin-II type-1-receptor autoantibodies titer in primary aldosteronism as a result of aldosterone-producing adenoma. Hypertension 2013, 61, 526–533. [Google Scholar] [CrossRef]
- Meyer, L.S.; Gong, S.; Reincke, M.; Williams, T.A. Angiotensin II type 1 receptor autoantibodies in primary aldosteronism. Horm. Metab. Res. 2020, 52, 379–385. [Google Scholar] [CrossRef]
- Piazza, M.; Seccia, T.M.; Caroccia, B.; Rossitto, G.; Scarpa, R.; Persichitti, P.; Basso, D.; Rossi, G.P. AT1AA (Angiotensin II type-1 receptor autoantibodies): Cause or consequence of human primary aldosteronism? Hypertension 2019, 74, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.M.; Pearson, G.D.; Cutler, J.A.; Lindheimer, M.D. Summary of the NHLBI working group on research on hypertension during pregnancy. Hypertens. Pregnancy 2003, 22, 109–127. [Google Scholar] [CrossRef]
- Sibai, B.; Dekker, G.; Kupferminc, M. Pre-Eclampsia. Lancet 2005, 365, 785–799. [Google Scholar] [CrossRef]
- Duley, L. The global impact of pre-eclampsia and eclampsia. Semin. Perinatol. 2009, 33, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Gant, N.F.; Daley, G.L.; Chand, S.; Whalley, P.J.; MacDonald, P.C. A Study of angiotensin II pressor response throughout primigravid pregnancy. J. Clin. Invest. 1973, 52, 2682–2689. [Google Scholar] [CrossRef]
- Baker, P.N.; Kilby, M.D.; Broughton Pipkin, F. The effect of angiotensin II on platelet intracellular free calcium concentration in human pregnancy. J. Hypertens. 1992, 10, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Wallukat, G.; Homuth, V.; Fischer, T.; Lindschau, C.; Horstkamp, B.; Jüpner, A.; Baur, E.; Nissen, E.; Vetter, K.; Neichel, D.; et al. Patients with Preeclampsia develop agonistic autoantibodies against the angiotensin AT1 receptor. J. Clin. Invest. 1999, 103, 945–952. [Google Scholar] [CrossRef]
- Velloso, E.P.; Pimentel, R.L.; Braga, J.F.; Cabral, A.C.V.; Reis, Z.S.N.; Bader, M.; Santos, R.A.S.; Wallukat, G. Identification of a novel agonist-like autoantibody in preeclamptic patients. Am. J. Hypertens. 2015, 29, 405–412. [Google Scholar] [CrossRef]
- Dechend, R.; Homuth, V.; Wallukat, G.; Kreuzer, J.; Park, J.K.; Theuer, J.; Juepner, A.; Gulba, D.C.; Mackman, N.; Haller, H.; et al. AT1 receptor agonistic antibodies from preeclamptic patients cause vascular cells to express tissue factor. Circulation 2000, 101, 2382–2387. [Google Scholar] [CrossRef]
- Dechend, R.; Viedt, C.; Müller, D.N.; Ugele, B.; Brandes, R.P.; Wallukat, G.; Park, J.-K.; Janke, J.; Barta, P.; Theuer, J.; et al. AT1 receptor agonistic antibodies from preeclamptic patients stimulate NADPH oxidase. Circulation 2003, 107, 1632–1639. [Google Scholar] [CrossRef]
- Xia, Y. Maternal autoantibodies from preeclamptic patients activate angiotensin receptors on human trophoblast cells. J. Soc. Gynecol. Investig. 2003, 10, 82–93. [Google Scholar] [CrossRef]
- Zhou, C.C.; Zhang, Y.; Irani, R.A.; Zhang, H.; Mi, T.; Popek, E.J.; Hicks, M.J.; Ramin, S.M.; Kellems, R.E.; Xia, Y. Angiotensin receptor agonistic autoantibodies induce pre-eclampsia in pregnant mice. Nat. Med. 2008, 14, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Kellems, R.E. Angiotensin receptor agonistic autoantibodies and hypertension: Preeclampsia and beyond. Circ. Res. 2013, 113, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Tanaka, H. The pathogenesis of coronary arteriosclerosis (‘‘chronic Rejection’’) in transplanted hearts. Clin. Transplant 1994, 8, 313–318. [Google Scholar] [PubMed]
- Hiemann, N.E.; Meyer, R.; Wellnhofer, E.; Schoenemann, C.; Heidecke, H.; Lachmann, N.; Hetzer, R.; Dragun, D. Non-HLA antibodies targeting vascular receptors enhance alloimmune response and microvasculopathy after heart transplantation. Transplant. J. 2012, 94, 919–924. [Google Scholar] [CrossRef] [PubMed]
- Urban, M.; Slavcev, A.; Gazdic, T.; Ivak, P.; Besik, J.; Netuka, I. The impact of angiotensin II type 1 receptor antibodies on post-heart transplantation outcome in heart mate II bridged recipients. Interact. Cardiovasc. Thorac. Surg. 2016, 22, 292–297. [Google Scholar] [CrossRef]
- Peters, S.; Trümmel, M.; Meyners, W.; Koehler, B.; Westermann, K. Valsartan versus ACE inhibition after bare metal stent implantation–Results of the VALVACE trial. Int. J. Cardiol. 2005, 98, 331–335. [Google Scholar] [CrossRef]
- Peters, S.; Götting, B.; Trümmel, M.; Rust, H.; Brattström, A. Valsartan for prevention of restenosis after stenting of type B2/C lesions: The VAL-PREST trial. J. Invasive Cardiol. 2001, 13, 93–97. [Google Scholar]
- Pearl, M.H.; Chen, L.; ElChaki, R.; Elashoff, D.; Gjertson, D.W.; Rossetti, M.; Weng, P.L.; Zhang, Q.; Reed, E.F.; Chambers, E.T. Endothelin type A receptor antibodies are associated with angiotensin II type 1 receptor antibodies, vascular inflammation, and decline in renal function in pediatric kidney transplantation. Kidney Int. Rep. 2020, 5, 1925–1936. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Civieri, G.; Iop, L.; Tona, F. Antibodies against Angiotensin II Type 1 and Endothelin 1 Type A Receptors in Cardiovascular Pathologies. Int. J. Mol. Sci. 2022, 23, 927. https://doi.org/10.3390/ijms23020927
Civieri G, Iop L, Tona F. Antibodies against Angiotensin II Type 1 and Endothelin 1 Type A Receptors in Cardiovascular Pathologies. International Journal of Molecular Sciences. 2022; 23(2):927. https://doi.org/10.3390/ijms23020927
Chicago/Turabian StyleCivieri, Giovanni, Laura Iop, and Francesco Tona. 2022. "Antibodies against Angiotensin II Type 1 and Endothelin 1 Type A Receptors in Cardiovascular Pathologies" International Journal of Molecular Sciences 23, no. 2: 927. https://doi.org/10.3390/ijms23020927
APA StyleCivieri, G., Iop, L., & Tona, F. (2022). Antibodies against Angiotensin II Type 1 and Endothelin 1 Type A Receptors in Cardiovascular Pathologies. International Journal of Molecular Sciences, 23(2), 927. https://doi.org/10.3390/ijms23020927