
The Landscape of Autophagy-Related (ATG) Genes and Functional Characterization of TaVAMP727 to Autophagy in Wheat

 ,
, 
Abstract
:1. Introduction
2. Results
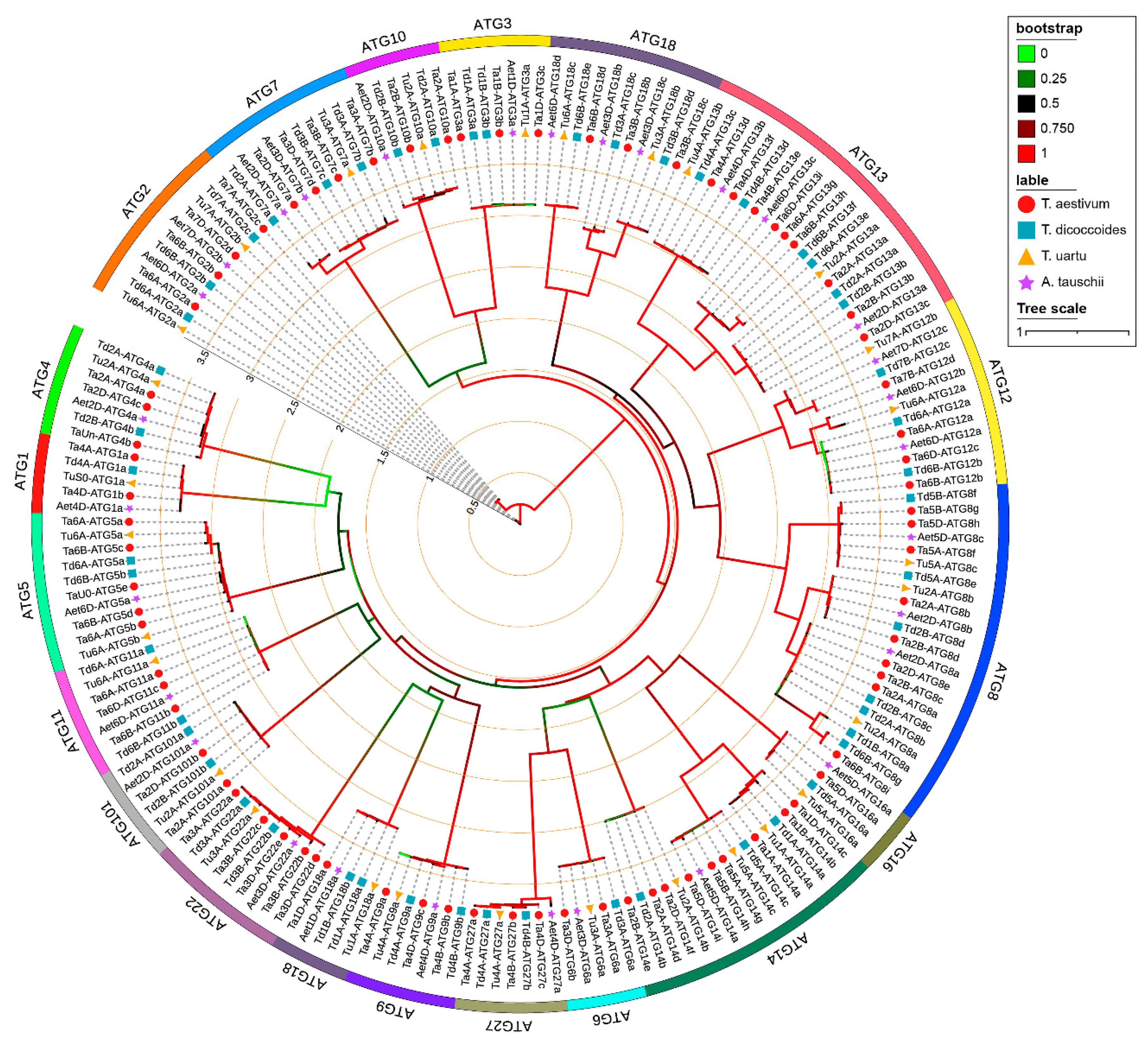
2.1. Identification of ATGs from Wheat and Its Diploid and Tetraploid Progenitors
2.2. Functional Annotation and Enrichment Analysis of Autophagy-Related Proteins
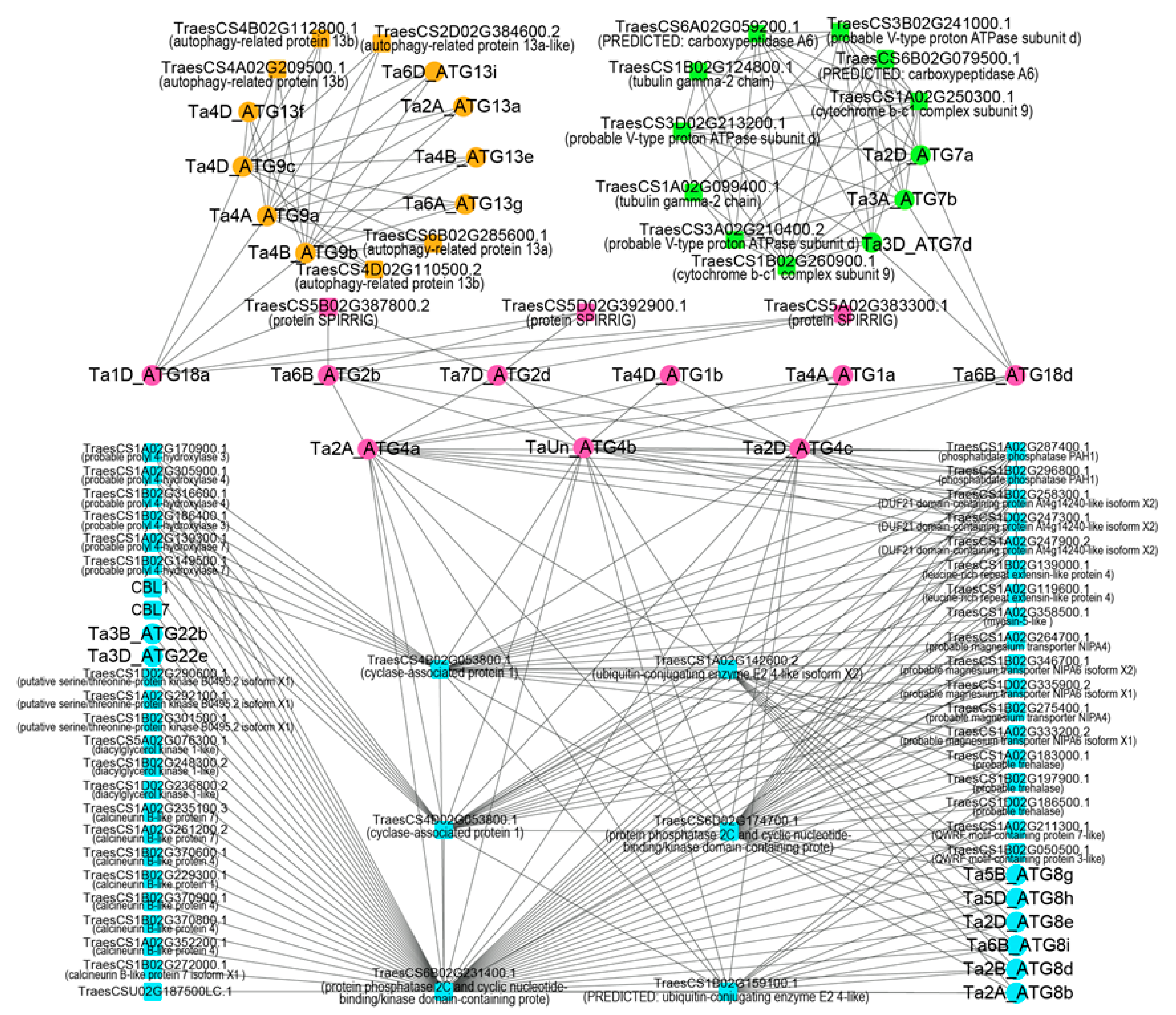
2.3. Protein-Protein Interaction Network of Autophagy-Related Proteins
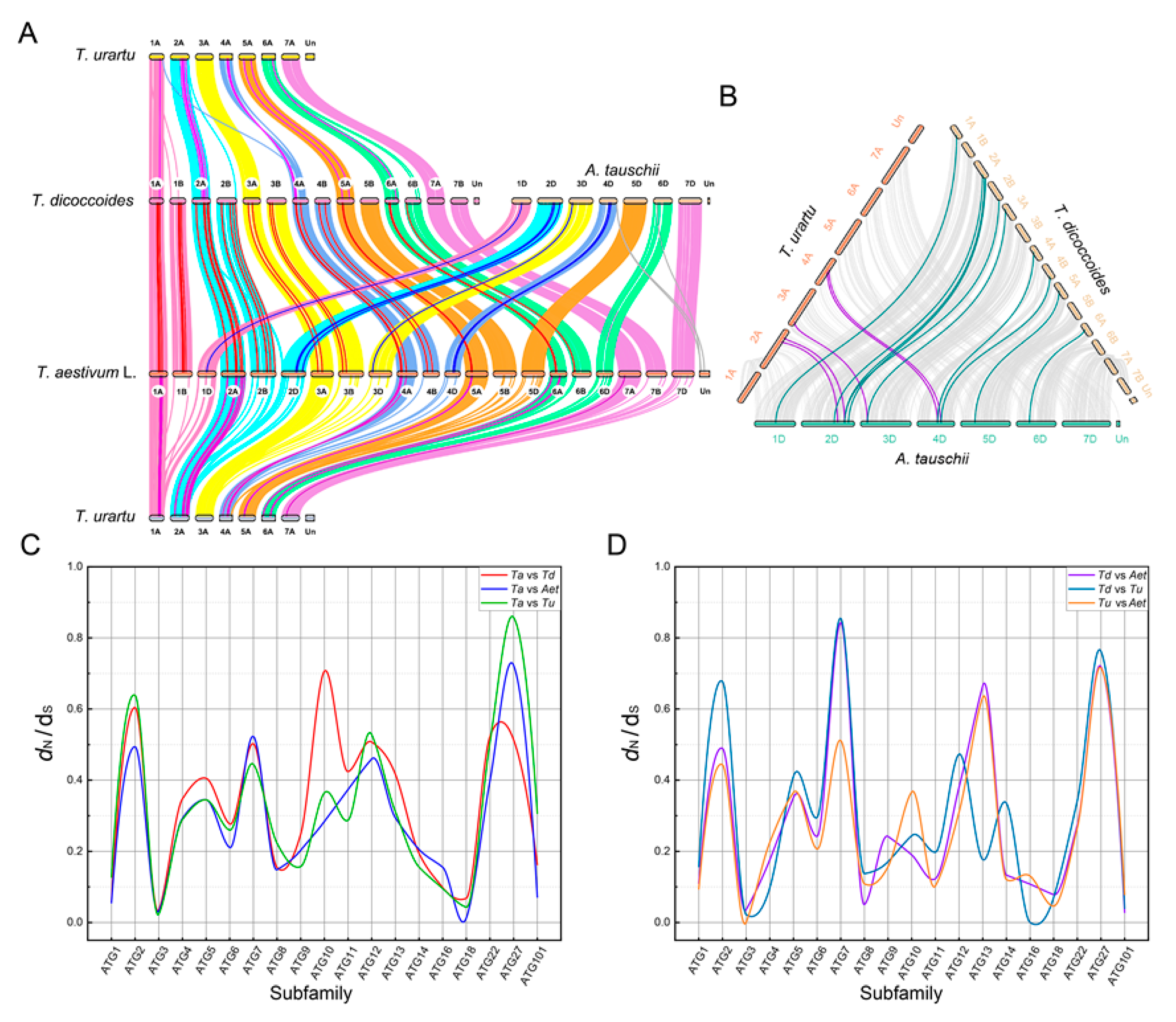
2.4. Synteny Events and Selection Pressure Analysis of ATGs in Wheat and Its Diploid and Tetraploid Progenitors
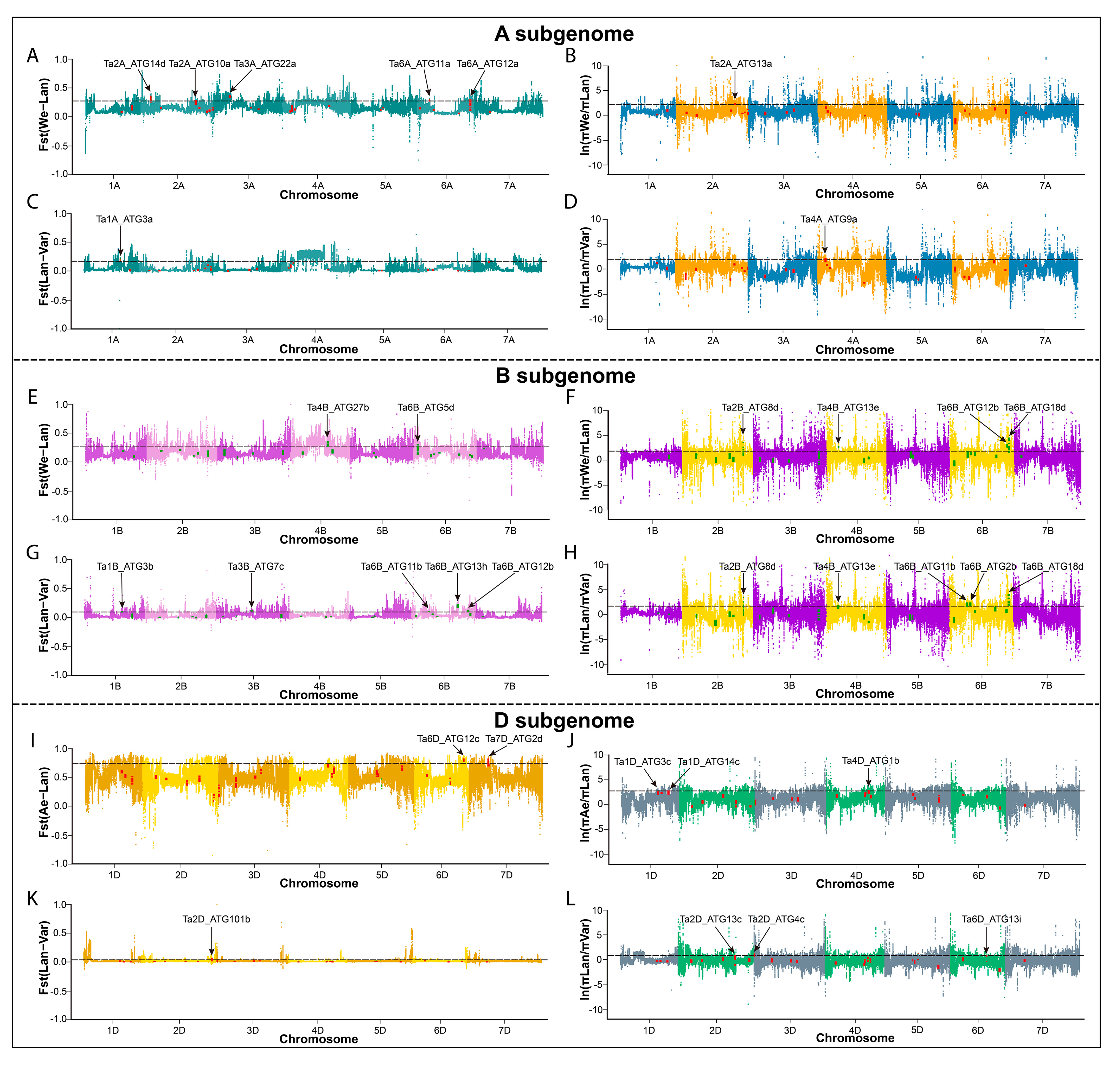
2.5. The Single Nucleotide Polymorphisms (SNPs) Analysis of TaATGs
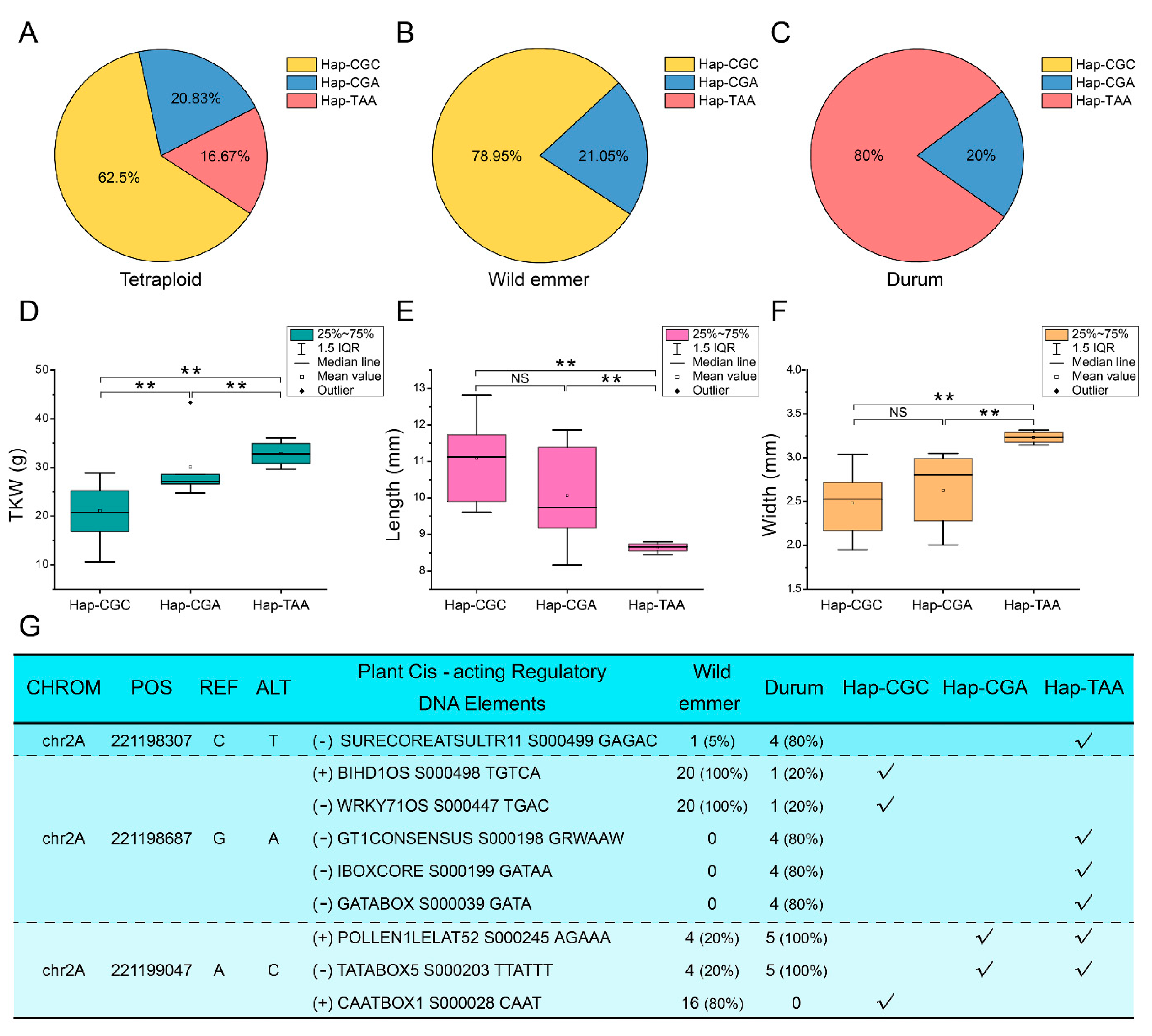
2.6. Correlation Analysis of the Variations at the Ta2A_ATG8a Locus with the Seed Size of Tetraploid Wheat
2.7. Effects of Overexpression TaVAMP727 in Arabidopsis
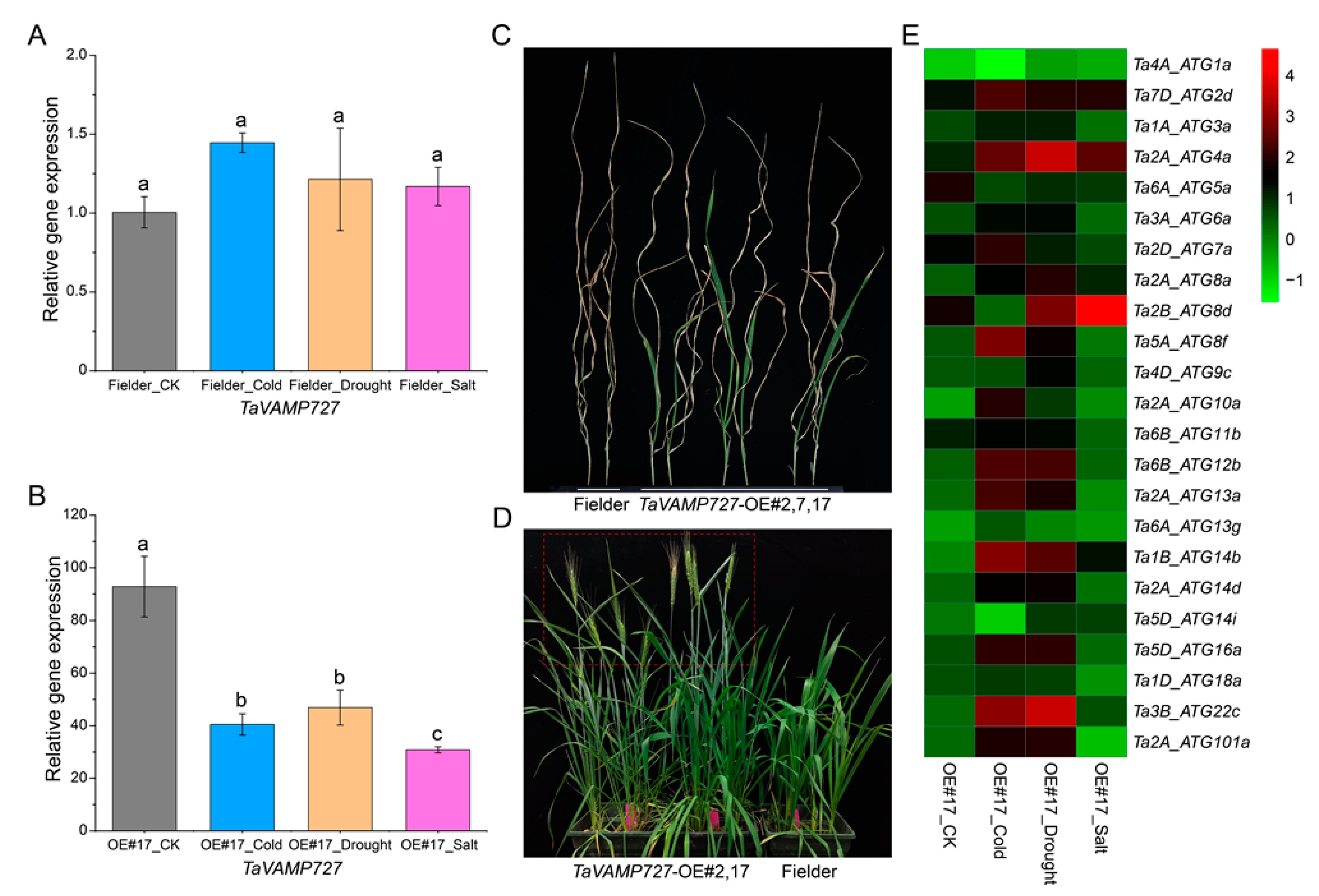
2.8. Effects of Overexpression TaVAMP727 in Wheat
3. Discussion
4. Materials and Methods
4.1. Identification of ATGs in Wheat and its Diploid and Tetraploid Progenitors
4.2. Bioinformatics Analysis
4.3. The Single Nucleotide Polymorphisms (SPNs) Analysis of TaATGs Based on the Resequence Data
4.4. Genetic Transformation of Arabidopsis and Wheat
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- van Doorn, W.G.; Papini, A. Ultrastructure of autophagy in plant cells: A review. Autophagy 2013, 9, 1922–1936. [Google Scholar] [CrossRef] [Green Version]
- Floyd, B.E.; Pu, Y.; Soto-Burgos, J.; Bassham, D.C. To Live or Die: Autophagy in Plants. In Plant Programmed Cell Death; Gunawardena, A.N., McCabe, P.F., Eds.; Springer International Publishing: Berlin, Germany, 2015; pp. 269–300. [Google Scholar]
- Suttangkakul, A.; Li, F.Q.; Chung, T.; Vierstra, R.D. The ATG1/ATG13 protein kinase complex is both a regulator and a target of autophagic recycling in Arabidopsis. Plant Cell 2011, 23, 3761–3779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, X.; Chung, K.P.; Cui, Y.; Lin, W.; Gao, C.; Kang, B.H.; Jiang, L. ATG9 regulates autophagosome progression from the endoplasmic reticulum in Arabidopsis. Proc. Natl. Acad. Sci. USA 2017, 114, E426–E435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marshall, R.S.; Vierstra, R.D. Autophagy: The master of bulk and selective recycling. Annu. Rev. Plant Biol. 2018, 69, 173–208. [Google Scholar] [CrossRef]
- Soto-Burgos, J.; Zhuang, X.; Jiang, L.; Bassham, D.C. Dynamics of autophagosome formation. Plant Physiol. 2018, 176, 219–229. [Google Scholar] [CrossRef] [Green Version]
- Soto-Burgos, J.; Bassham, D.C. SnRK1 activates autophagy via the TOR signaling pathway in Arabidopsis thaliana. PLoS ONE 2017, 12, e0182591. [Google Scholar] [CrossRef] [Green Version]
- Nikoletopoulou, V.; Markaki, M.; Palikaras, K.; Tavernarakis, N. Crosstalk between apoptosis, necrosis and autophagy. BBA-Mol. Cell Res. 2013, 1833, 3448–3459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.Y.; Li, L.S.; Hou, C.; Lai, Y.; Long, J.G.; Liu, J.K.; Zhong, Q.; Diao, J.J. SNARE-mediated membrane fusion in autophagy. Semin. Cell Dev. Biol. 2016, 60, 97–104. [Google Scholar] [CrossRef] [Green Version]
- Barz, S.; Kriegenburg, F.; Henning, A.; Bhattacharya, A.; Mancilla, H.; Sanchez-Martin, P.; Kraft, C. Atg1 kinase regulates autophagosome-vacuole fusion by controlling SNARE bundling. EMBO Rep. 2020, 21, e51869. [Google Scholar] [CrossRef]
- Yamamoto, H.; Kakuta, S.; Watanabe, T.M.; Kitamura, A.; Sekito, T.; Kondo-Kakuta, C.; Ichikawa, R.; Kinjo, M.; Ohsumi, Y. Atg9 vesicles are an important membrane source during early steps of autophagosome formation. J. Cell Biol. 2012, 198, 219–233. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.X.; Abudu, Y.P.; Kumar, S.; Bissa, B.; Choi, S.W.; Jia, J.Y.; Lazarou, M.; Eskelinen, E.L.; Johansen, T.; Deretic, V. Mammalian Atg8 proteins regulate lysosome and autolysosome biogenesis through SNAREs. EMBO J. 2019, 38, e101994. [Google Scholar] [CrossRef]
- Liu, R.; Zhi, X.Y.; Zhong, Q. ATG14 controls SNARE-mediated autophagosome fusion with a lysosome. Autophagy 2015, 11, 847–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takemoto, K.; Ebine, K.; Askani, J.C.; Kruger, F.; Gonzalez, Z.A.; Ito, E.; Goh, T.; Schumacher, K.; Nakano, A.; Ueda, T. Distinct sets of tethering complexes, SNARE complexes, and Rab GTPases mediate membrane fusion at the vacuole in Arabidopsis. Proc. Natl. Acad. Sci. USA 2018, 115, E2457–E2466. [Google Scholar] [CrossRef] [Green Version]
- Ebine, K.; Okatani, Y.; Uemura, T.; Goh, T.; Shoda, K.; Niihama, M.; Morita, M.T.; Spitzer, C.; Otegui, M.S.; Nakano, A.; et al. A SNARE complex unique to seed plants is required for protein storage vacuole biogenesis and seed development of Arabidopsis thaliana. Plant Cell 2008, 20, 3006–3021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, W.; Nie, X.; Cui, L.; Zhi, Y.; Zhang, T.; Du, X.; Song, W. Genome-wide sequence and expressional analysis of autophagy Gene family in bread wheat (Triticum aestivum L.). J. Plant Physiol. 2018, 229, 7–21. [Google Scholar] [CrossRef]
- Merkulova, E.A.; Guiboileau, A.; Naya, L.; Masclaux-Daubresse, C.; Yoshimoto, K. Assessment and optimization of autophagy monitoring methods in Arabidopsis roots indicate direct fusion of autophagosomes with vacuoles. Plant Cell Physiol. 2014, 55, 715–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenz, H.D.; Haller, E.; Melzer, E.; Kober, K.; Wurster, K.; Stahl, M.; Bassham, D.C.; Vierstra, R.D.; Parker, J.E.; Bautor, J.; et al. Autophagy differentially controls plant basal immunity to biotrophic and necrotrophic pathogens. Plant J. 2011, 66, 818–830. [Google Scholar] [CrossRef]
- Appels, R.; Eversole, K.; Feuillet, C.; Keller, B.; Rogers, J.; Stein, N.; Pozniak, C.J.; Stein, N.; Choulet, F.; Distelfeld, A.; et al. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 2018, 361, 6043. [Google Scholar]
- Berkman, P.J.; Visendi, P.; Lee, H.C.; Stiller, J.; Manoli, S.; Lorenc, M.T.; Lai, K.T.; Batley, J.; Fleury, D.; Simkova, H.; et al. Dispersion and domestication shaped the genome of bread wheat. Plant Biotechnol. J. 2013, 11, 564–571. [Google Scholar] [CrossRef] [Green Version]
- Avni, R.; Nave, M.; Barad, O.; Baruch, K.; Twardziok, S.O.; Gundlach, H.; Hale, I.; Mascher, M.; Spannagl, M.; Wiebe, K.; et al. Wild emmer genome architecture and diversity elucidate wheat evolution and domestication. Science 2017, 357, 93–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, M.C.; Gu, Y.Q.; Puiu, D.; Wang, H.; Twardziok, S.O.; Deal, K.R.; Huo, N.; Zhu, T.; Wang, L.; Wang, Y.; et al. Genome sequence of the progenitor of the wheat D genome Aegilops tauschii. Nature 2017, 551, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.Q.; Ma, B.; Shi, X.; Liu, H.; Dong, L.; Sun, H.; Cao, Y.; Gao, Q.; Zheng, S.; Li, Y.; et al. Genome sequence of the progenitor of wheat A subgenome Triticum urartu. Nature 2018, 557, 424–428. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Zhao, X.B.; Li, Y.W.; Xu, J.; Bi, A.Y.; Kang, L.P.; Xu, D.X.; Chen, H.F.; Wang, Y.; Wang, Y.G.; et al. Triticum population sequencing provides insights into wheat adaptation. Nat. Genet. 2020, 52, 1412–1422. [Google Scholar] [CrossRef]
- Cheng, H.; Liu, J.; Wen, J.; Nie, X.J.; Xu, L.H.; Chen, N.B.; Li, Z.X.; Wang, Q.L.; Zheng, Z.Q.; Li, M.; et al. Frequent intra- and inter-species introgression shapes the landscape of genetic variation in bread wheat. Genome Biol. 2019, 20, 136. [Google Scholar] [CrossRef] [Green Version]
- Pei, D.; Zhang, W.; Sun, H.; Wei, X.; Yue, J.; Wang, H. Identification of autophagy-related genes ATG4 and ATG8 from wheat (Triticum aestivum L.) and profiling of their expression patterns responding to biotic and abiotic stresses. Plant Cell Rep. 2014, 33, 1697–1710. [Google Scholar] [CrossRef] [PubMed]
- Yue, J.Y.; Sun, H.; Zhang, W.; Pei, D.; He, Y.; Wang, H.Z. Wheat homologs of yeast ATG6 function in autophagy and are implicated in powdery mildew immunity. BMC Plant Biol. 2015, 15, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Y.; Liu, W.; Hu, W.; Liu, G.; Wu, C.; Liu, W.; Zeng, H.; He, C.; Shi, H. Genome-wide analysis of autophagy-related genes in banana highlights MaATG8s in cell death and autophagy in immune response to Fusarium wilt. Plant Cell Rep. 2017, 36, 1237–1250. [Google Scholar] [CrossRef]
- Xia, K.; Liu, T.; Ouyang, J.; Wang, R.; Fan, T.; Zhang, M. Genome-wide identification, classification, and expression analysis of autophagy-associated gene homologues in rice (Oryza sativa L.). DNA Res. 2011, 18, 363–377. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Chen, M.; Wang, E.; Hu, L.; Hawkesford, M.J.; Zhong, L.; Chen, Z.; Xu, Z.; Li, L.; Zhou, Y.; et al. Genome-wide analysis of autophagy-associated genes in foxtail millet (Setaria italica L.) and characterization of the function of SiATG8a in conferring tolerance to nitrogen starvation in rice. BMC Genom. 2016, 17, 797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.M.; Zhao, P.; Wang, W.; Zou, J.; Cheng, T.H.; Peng, X.B.; Sun, M.X. A comprehensive, genome-wide analysis of autophagy-related genes identified in tobacco suggests a central role of autophagy in plant response to various environmental cues. DNA Res. 2015, 22, 245–257. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.H. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Liu, J.; Zhang, J.; Liu, S.; Du, J. Selective modes determine evolutionary rates, gene compactness and expression patterns in Brassica. Plant J. 2017, 91, 34–44. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Chen, H.; Lu, T. Molecular mechanisms of SNARE proteins in regulating autophagy. Yi Chuan 2014, 36, 547–551. [Google Scholar]
- Wendel, J.F. Genome evolution in polyploids. Plant Mol. Biol. 2000, 42, 225–249. [Google Scholar] [CrossRef]
- Altenhoff, A.M.; Studer, R.A.; Robinson-Rechavi, M.; Dessimoz, C. Resolving the ortholog conjecture: Orthologs tend to be weakly, but significantly, more similar in function than paralogs. PLoS Comput. Biol. 2012, 8, e1002514. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.; Guo, C.; Shan, H.; Kong, H. Divergence of duplicate genes in exon-intron structure. Proc. Natl. Acad. Sci. USA 2012, 109, 1187–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choulet, F.; Alberti, A.; Theil, S.; Glover, N.; Barbe, V.; Daron, J.; Pingault, L.; Sourdille, P.; Couloux, A.; Paux, E. Structural and functional partitioning of bread wheat chromosome 3B. Science 2014, 345, 1249721. [Google Scholar] [CrossRef]
- Huo, N.X.; Zhang, S.L.; Zhu, T.T.; Dong, L.L.; Wang, Y.; Mohr, T.; Hu, T.Z.; Liu, Z.Y.; Dvorak, J.; Luo, M.C.; et al. Gene duplication and evolution dynamics in the homeologous regions harboring multiple prolamin and resistance gene families in hexaploid wheat. Front. Plant Sci. 2018, 9, 673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Distefano, A.M.; Lopez, G.A.; Setzes, N.; Marchetti, F.; Cainzos, M.; Cascallares, M.; Zabaleta, E.; Pagnussat, G.C. Ferroptosis in plants: Triggers, proposed mechanisms and the role of iron in modulating cell death. J. Exp. Bot. 2020, 72, 2125–2135. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Ai, Y.; Sun, Q.; Ma, Y.; Cao, Y.; Wang, J.; Zhang, Z.; Wang, X. Membrane damage during ferroptosis is caused by oxidation of phospholipids catalyzed by the oxidoreductases POR and CYB5R1. Mol. Cell 2020, 81, 55–369. [Google Scholar] [CrossRef]
- Leary, A.Y.; Savage, Z.; Tumtas, Y.; Bozkurt, T.O. Contrasting and emerging roles of autophagy in plant immunity. Curr. Opin. Plant Biol. 2019, 52, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Ismayil, A.; Yang, M.; Liu, Y. Role of autophagy during plant-virus interactions. Semin. Cell Dev. Biol. 2020, 101, 36–40. [Google Scholar] [CrossRef]
- Kuzuoglu-Ozturk, D.; Yalcinkaya, O.C.; Akpinar, B.A.; Mitou, G.; Korkmaz, G.; Gozuacik, D.; Budak, H. Autophagy-related gene, TdAtg8, in wild emmer wheat plays a role in drought and osmotic stress response. Planta 2012, 236, 1081–1092. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.Y.; Bassham, D.C. Combating stress: The interplay between hormone signaling and autophagy in plants. J. Exp. Bot. 2020, 71, 1723–1733. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.; Park, E.; Dinesh-Kumar, S.P. Differential processing of Arabidopsis ubiquitin-like Atg8 autophagy proteins by Atg4 cysteine proteases. Proc. Natl. Acad. Sci. USA 2014, 111, 863–868. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Wandelmer, J.; Kriegenburg, F.; Rohringer, S.; Schuschnig, M.; Gómez-Sánchez, R.; Zens, B.; Abreu, S.; Hardenberg, R.; Hollenstein, D.; Gao, J. Atg4 proteolytic activity can be inhibited by Atg1 phosphorylation. Nat. Commun. 2017, 8, 295. [Google Scholar] [CrossRef]
- Steffens, A.; Brautigam, A.; Jakoby, M.; Hulskamp, M. The BEACH domain protein SPIRRIG is essential for Arabidopsis salt stress tolerance and functions as a regulator of transcript stabilization and localization. PLoS Biol. 2015, 13, e1002188. [Google Scholar] [CrossRef]
- Steffens, A.; Jakoby, M.; Hülskamp, M. Physical, functional and genetic interactions between the BEACH domain protein SPIRRIG and LIP5 and SKD1 and its role in endosomal trafficking to the vacuole in Arabidopsis. Front. Plant Sci. 2017, 8, 1969. [Google Scholar] [CrossRef] [Green Version]
- Memisoglu, G.; Eapen, V.V.; Yang, Y.; Klionsky, D.J.; Haber, J.E. PP2C phosphatases promote autophagy by dephosphorylation of the Atg1 complex. Proc. Natl. Acad. Sci. USA 2019, 116, 1613–1620. [Google Scholar] [CrossRef] [Green Version]
- He, Z.; Wu, J.; Sun, X.; Dai, M. The maize clade a PP2C phosphatases play critical roles in multiple abiotic stress responses. Int. J. Mol. Sci. 2019, 20, 3573. [Google Scholar] [CrossRef] [Green Version]
- Li, C.Y.; Li, X.W.; Xiao, J.; Liu, J.; Fan, X.; Fan, F.Y.; Lei, H.F. Genetic changes in the EPAS1 gene between Tibetan and Han ethnic groups and adaptation to the plateau hypoxic environment. Peerj 2019, 7, e794. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Soulay, F.; Saudemont, B.; Elmayan, T.; Marmagne, A.; Masclaux-Daubresse, C. Overexpression of ATG8 in Arabidopsis stimulates autophagic activity and increases nitrogen remobilization efficiency and grain filling. Plant Cell Physiol. 2018, 60, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Maruyama-Nakashita, A.; Nakamura, Y.; Watanabe-Takahashi, A.; Inoue, E.; Yamaya, T.; Takahashi, H. Identification of a novel cis-acting element conferring sulfur deficiency response in Arabidopsis roots. Plant J. 2010, 42, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.J.; Hawkesford, M.J.; McGrath, S.P. Sulphur assimilation and effects on yield and quality of wheat. J. Cereal Sci. 1999, 30, 1–17. [Google Scholar] [CrossRef]
- Vauclare, P.; Macherel, D.; Douce, R.; Bourguignon, J. The gene encoding T protein of the glycine decarboxylase complex involved in the mitochondrial step of the photorespiratory pathway in plants exhibits features of light-induced genes. Plant Mol. Biol. 1998, 37, 309–318. [Google Scholar] [CrossRef]
- Guan, M.; Moller, I.S.; Schjoerring, J.K. Two cytosolic glutamine synthetase isoforms play specific roles for seed germination and seed yield structure in Arabidopsis. J. Exp. Bot. 2015, 66, 203–212. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Liu, Y.; Zhu, X.F.; Jung, J.H.; Sun, Q.; Li, T.Y.; Chen, L.J.; Duan, Y.X.; Xuan, Y.H. SYP22 and VAMP727 regulate BRI1 plasma membrane targeting to control plant growth in Arabidopsis. New Phytol. 2019, 223, 1059–1065. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.F.; Liu, Y.; Gai, X.T.; Zhou, Y.; Xia, Z.Y.; Chen, L.J.; Duan, Y.X.; Xuan, Y.H. SNARE proteins SYP22 and VAMP727 negatively regulate plant defense. Plant Signal. Behav. 2019, 14, 1610300. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Yue, H.; Feng, K.; Deng, P.; Song, W.; Nie, X. Genome-wide identification, phylogeny and expressional profiles of mitogen activated protein kinase kinase kinase (MAPKKK) gene family in bread wheat (Triticum aestivum L.). BMC Genom. 2016, 17, 668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higo, K.; Ugawa, Y.; Iwamoto, M.; Korenaga, T. Plant cis-acting regulatory DNA elements (PLACE) database: 1999. Nucleic Acids Res. 1999, 27, 297–300. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernandez-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Krishnakumar, V.; Li, J.; Tiany; MichelMoser; Maria; Yim, W.C. jcvi: JCVI Utility Libraries; Zenodo, 2015; Available online: https://zenodo.org/record/31631#.YeEQeFkRU2z (accessed on 9 January 2022).
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Proc, G.P.D. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Xu, W.; Wang, Y.; Wang, X.; Hao, W.; Sun, M. Construction of the plant expression vectors carrying resistant genes to powdery mildew and adversities in wild species of Vvitis in China. Acta Bot. Boreali-Occident. Sin. 2005, 25, 851–857. [Google Scholar]
- Clough, S.J.; Bent, A.F. Floral dip: A simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 1998, 16, 735–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez, L.; Mauriat, M.; Guenin, S.; Pelloux, J.; Lefebvre, J.F.; Louvet, R.; Rusterucci, C.; Moritz, T.; Guerineau, F.; Bellini, C.; et al. The lack of a systematic validation of reference genes: A serious pitfall undervalued in reverse transcription-polymerase chain reaction (RT-PCR) analysis in plants. Plant Biotechnol. J. 2008, 6, 609–618. [Google Scholar] [CrossRef] [Green Version]
- Udvardi, M.K.; Czechowski, T.; Scheible, W.R. Eleven golden rules of quantitative RT-PCR. Plant Cell 2008, 20, 1736–1737. [Google Scholar] [CrossRef] [Green Version]
- Ishida, Y.; Tsunashima, M.; Hiei, Y.; Komari, T. Wheat (Triticum aestivum L.) transformation using immature embryos. Methods Mol. Biol. 2015, 1223, 189–198. [Google Scholar] [PubMed]
- Garrido, J.; Aguilar, M.; Prieto, P. Identification and validation of reference genes for RT-qPCR normalization in wheat meiosis. Sci. Rep. 2020, 10, 2726. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subfamily | T. aestivum L. | T. dicoccoides | T. urartu | A. tauschii | O. sativa L. | S. italica L. | N. tabacum L. | M. acuminata |
|---|---|---|---|---|---|---|---|---|
| ATG1 | 2 | 1 | 1 | 1 | 3 | 1 | 3 | 1 |
| ATG2 | 4 | 3 | 2 | 2 | 0 | 1 | 1 | 1 |
| ATG3 | 3 | 2 | 1 | 1 | 2 | 2 | 1 | 3 |
| ATG4 | 3 | 2 | 1 | 1 | 2 | 1 | 1 | 3 |
| ATG5 | 5 | 2 | 2 | 1 | 1 | 1 | 1 | 1 |
| ATG6 | 2 | 1 | 1 | 1 | 3 | 2 | 1 | 1 |
| ATG7 | 4 | 3 | 1 | 2 | 1 | 2 | 0 | 1 |
| ATG8 | 8 | 7 | 3 | 3 | 7 | 4 | 5 | 10 |
| ATG9 | 3 | 2 | 1 | 1 | 2 | 2 | 1 | 2 |
| ATG10 | 2 | 2 | 1 | 1 | 2 | 1 | 1 | 0 |
| ATG11 | 3 | 2 | 1 | 1 | 0 | 1 | 0 | 0 |
| ATG12 | 4 | 3 | 2 | 3 | 1 | 1 | 0 | 1 |
| ATG13 | 9 | 6 | 2 | 3 | 2 | 3 | 3 | 2 |
| ATG14 | 9 | 3 | 3 | 1 | 0 | 0 | 0 | 0 |
| ATG16 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 |
| ATG18 | 4 | 5 | 3 | 4 | 6 | 7 | 6 | 5 |
| ATG19 | 5 | 2 | 1 | 1 | 0 | 0 | 0 | 0 |
| ATG20 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| ATG27 | 3 | 2 | 1 | 1 | 0 | 0 | 0 | 0 |
| ATG101 | 2 | 2 | 1 | 1 | 0 | 0 | 0 | 0 |
| all | 76 | 51 | 29 | 30 | 33 | 30 | 25 | 32 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yue, W.; Zhang, H.; Sun, X.; Su, N.; Zhao, Q.; Yan, Z.; Weining, S.; Yue, H. The Landscape of Autophagy-Related (ATG) Genes and Functional Characterization of TaVAMP727 to Autophagy in Wheat. Int. J. Mol. Sci. 2022, 23, 891. https://doi.org/10.3390/ijms23020891
Yue W, Zhang H, Sun X, Su N, Zhao Q, Yan Z, Weining S, Yue H. The Landscape of Autophagy-Related (ATG) Genes and Functional Characterization of TaVAMP727 to Autophagy in Wheat. International Journal of Molecular Sciences. 2022; 23(2):891. https://doi.org/10.3390/ijms23020891
Chicago/Turabian StyleYue, Wenjie, Haobin Zhang, Xuming Sun, Ning Su, Qi Zhao, Zhaogui Yan, Song Weining, and Hong Yue. 2022. "The Landscape of Autophagy-Related (ATG) Genes and Functional Characterization of TaVAMP727 to Autophagy in Wheat" International Journal of Molecular Sciences 23, no. 2: 891. https://doi.org/10.3390/ijms23020891
APA StyleYue, W., Zhang, H., Sun, X., Su, N., Zhao, Q., Yan, Z., Weining, S., & Yue, H. (2022). The Landscape of Autophagy-Related (ATG) Genes and Functional Characterization of TaVAMP727 to Autophagy in Wheat. International Journal of Molecular Sciences, 23(2), 891. https://doi.org/10.3390/ijms23020891