
Skeletal Muscle Microvascular Dysfunction in Obesity-Related Insulin Resistance: Pathophysiological Mechanisms and Therapeutic Perspectives


Abstract
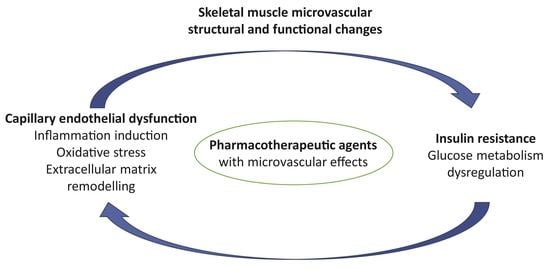
{kind=link}
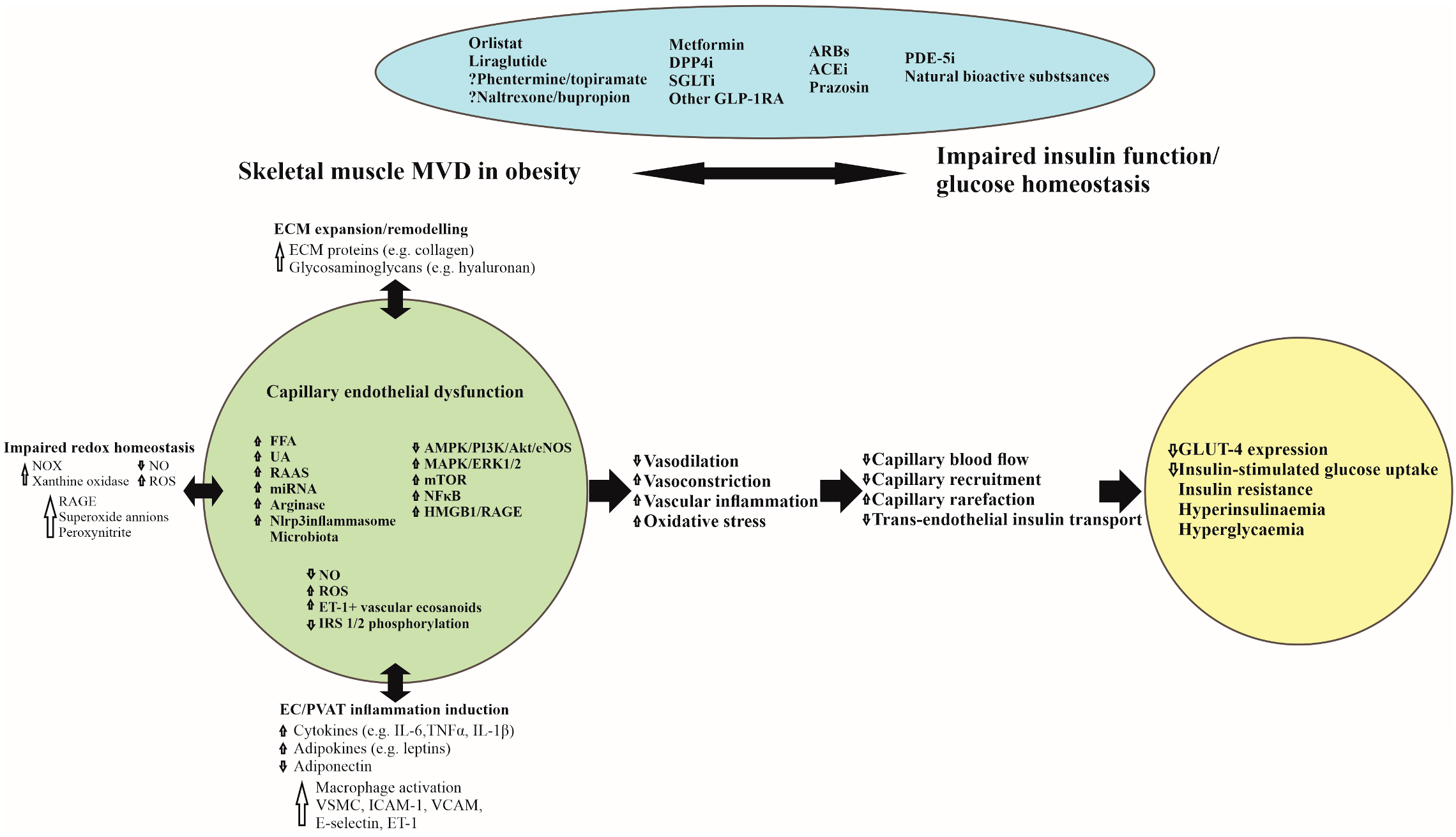{kind=link}
1. Introduction
2. Vascular and Metabolic Physiology of Skeletal Muscle Microcirculation
2.1. Anatomical Background
2.2. The Microvascular Endothelium Is the Key Regulator of Vascular Homeostasis
2.3. Role of Insulin in the Regulation of Microvascular Tone
2.4. Functional Capillary Recruitment
2.5. Assessment of Skeletal Muscle Microvascular Structure and Function
3. Skeletal Muscle Microvascular Dysfunction in Obesity
3.1. Skeletal Muscle Microvascular Functional and Structural Dynamics in Obesity
3.2. Endothelial Dysfunction Is the Key Driver of Microvascular Dysfunction in Obesity
3.3. Endothelial and Perivascular Adipose Tissue Inflammatory Mediators
3.4. Dysregulation of Redox Homeostasis
3.5. The Role of Extracellular Matrix Remodelling
4. Crossroads of Microvascular Pathophysiology and Pharmacology: Clinical Perspectives in Obesity Treatment
4.1. Current Paradigm
4.2. Targeting Microvascular Inflammatory Phenotype and Endothelial Dysfunction as a Therapeutic Strategy for Insulin Dysfunction in Obesity
4.2.1. Current Anti-Obesity Drugs
4.2.2. Anti-Hyperglycaemic and Other Agents
4.2.3. Experimental Phytochemicals and Dietary Interventions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AMPK | 5’-Adenosine Monophosphate-activated Protein Kinase |
| Akt | Protein Kinase B |
| AGEs | Advanced Glycation End Products |
| Ang | Angiotensin II |
| ARBs | Angiotensin Receptor Blockers |
| ARE | Antioxidant response element |
| BMI | Body Mass Index |
| CRP | C-Reactive Protein |
| DM | Diabetes Mellitus |
| DPP4i | Dipeptidyl-Peptidase-4 inhibitors |
| ERK | Extracellular-signal-regulated Kinase |
| eNOS | Endothelial Nitric Oxide Synthase |
| ET-1 | Endothelin 1 |
| FOXO1 | Forkhead Box O1 |
| GABA | Gamma-Aminobutyric acid |
| GLP-1 RA | Glucagon-like Peptide-1 Receptor Agonists |
| GLUT-4 | Glucose Transporter 4 |
| hCRP | High-Sensitivity CRP |
| HMGB 1 | High Mobility Group Box chromosomal protein 1 |
| IRS | Insulin Receptor Substrate |
| ICAM1 | Intercellular Adhesion Molecule 1 |
| IL-1β | Interleukin-1Beta |
| IL-6 | Interleukin-6 |
| JNK | C-Jun N-terminal Kinase |
| KLF2 | Krüppel-Like Factor 2 |
| KLF4 | Krüppel-Like Factor 4 |
| mTOR | Mammalian Target of Rapamycin |
| MAPK | Mitogen-Activated Protein Kinase |
| MCP1 | Monocyte Chemoattractant Protein 1 |
| miRNAs | MicroRNAs |
| NADPH | Nicotinamide Adenine Dinucleotide Phosphate |
| NF-κB | Nuclear Factor-KappaB |
| NO | Nitric Oxide |
| NOX | NADPH Oxidase |
| Nrf2 | E2-related factor 2 |
| PDE-5i | Phosphodiesterase-5 inhibitors |
| PET-CT | Positron emission tomography—computed tomography |
| PI3K | Phosphatidylinositol 3 Kinase |
| PKC | Protein Kinase C |
| ROS | Reactive Oxygen Species |
| RAGE | Receptor for AGEs |
| SGLT2i | Sodium Glucose Cotransporter 2 inhibitors |
| SIRT1 | Sirtuin 1 |
| TXA2 | Thromboxane A2 |
| TNF-α | Tumour Necrosis Factor-A |
| VCAM1 | Vascular Cell Adhesion Molecule 1 |
| VEGF | Vascular Endothelial Growth Factor |
| VSMCs | Vascular Smooth Muscle Cells |
References
- Weir, C.B.; Jan, A. BMI Classification Percentile and Cut Off Points; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Obesity and Overweight. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 9 December 2021).
- Abarca-Gómez, L.; Abdeen, Z.A.; Hamid, Z.A.; Abu-Rmeileh, N.M.; Acosta-Cazares, B.; Acuin, C.; Adams, R.J.; Aekplakorn, W.; Afsana, K.; Aguilar-Salinas, C.A.; et al. Worldwide Trends in Body-Mass Index, Underweight, Overweight, and Obesity from 1975 to 2016: A Pooled Analysis of 2416 Population-Based Measurement Studies in 128·9 Million Children, Adolescents, and Adults. Lancet 2017, 390, 2627–2642. [Google Scholar] [CrossRef]
- Yanovski, J.A. Obesity: Trends in Underweight and Obesity-Scale of the Problem. Nat. Rev. Endocrinol. 2018, 14, 5–6. [Google Scholar] [CrossRef]
- Sung, H.; Siegel, R.L.; Torre, L.A.; Pearson-Stuttard, J.; Islami, F.; Fedewa, S.A.; Goding Sauer, A.; Shuval, K.; Gapstur, S.M.; Jacobs, E.J.; et al. Global Patterns in Excess Body Weight and the Associated Cancer Burden. CA Cancer J. Clin. 2018. [Google Scholar] [CrossRef]
- GBD 2015 Obesity Collaborators; Afshin, A.; Forouzanfar, M.H.; Reitsma, M.B.; Sur, P.; Estep, K.; Lee, A.; Marczak, L.; Mokdad, A.H.; Moradi-Lakeh, M.; et al. Health Effects of Overweight and Obesity in 195 Countries over 25 Years. N. Engl. J. Med. 2017, 377, 13–27. [Google Scholar] [CrossRef]
- Blüher, M. Obesity: Global Epidemiology and Pathogenesis. Nat. Rev. Endocrinol. 2019, 15, 288–298. [Google Scholar] [CrossRef]
- Schiaffino, S. Muscle Fiber Type Diversity Revealed by Anti-Myosin Heavy Chain Antibodies. FEBS J. 2018, 285, 3688–3694. [Google Scholar] [CrossRef]
- Umek, N.; Horvat, S.; Cvetko, E. Skeletal Muscle and Fiber Type-Specific Intramyocellular Lipid Accumulation in Obese Mice. Bosn. J. Basic Med. Sci. 2021, 21. [Google Scholar] [CrossRef]
- Thiebaud, D.; Jacot, E.; DeFronzo, R.A.; Maeder, E.; Jequier, E.; Felber, J.P. The Effect of Graded Doses of Insulin on Total Glucose Uptake, Glucose Oxidation, and Glucose Storage in Man. Diabetes 1982, 31, 957–963. [Google Scholar] [CrossRef] [PubMed]
- Czech, M.P. Insulin Action and Resistance in Obesity and Type 2 Diabetes. Nat. Med. 2017, 23, 804–814. [Google Scholar] [CrossRef] [PubMed]
- Levy, B.I.; Schiffrin, E.L.; Mourad, J.J.; Agostini, D.; Vicaut, E.; Safar, M.E.; Struijker-Boudier, H.A.J. Impaired Tissue Perfusion: A Pathology Common to Hypertension, Obesity, and Diabetes Mellitus. Circulation 2008, 118, 968–976. [Google Scholar] [CrossRef] [PubMed]
- Serné, E.H.; DeJongh, R.T.; Eringa, E.C.; Ijzerman, R.G.; DeBoer, M.P.; Stehouwer, C.D.A. Microvascular Dysfunction: Causative Role in the Association between Hypertension, Insulin Resistance and the Metabolic Syndrome? Essays Biochem. 2006, 42, 163–176. [Google Scholar] [CrossRef] [PubMed]
- De Boer, M.P.; Meijer, R.I.; Wijnstok, N.J.; Jonk, A.M.; Houben, A.J.; Stehouwer, C.D.; Smulders, Y.M.; Eringa, E.C.; Serné, E.H. Microvascular Dysfunction: A Potential Mechanism in the Pathogenesis of Obesity-Associated Insulin Resistance and Hypertension. Microcirculation 2012, 19, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Jonk, A.M.; Houben, A.J.H.M.; de Jongh, R.T.; Serné, E.H.; Schaper, N.C.; Stehouwer, C.D.A. Microvascular Dysfunction in Obesity: A Potential Mechanism in the Pathogenesis of Obesity-Associated Insulin Resistance and Hypertension. Physiology 2007, 22, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Boillot, A.; Zoungas, S.; Mitchell, P.; Klein, R.; Klein, B.; Ikram, M.K.; Klaver, C.; Wang, J.J.; Gopinath, B.; Tai, E.S.; et al. Obesity and the Microvasculature: A Systematic Review and Meta-Analysis. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Virdis, A.; Masi, S.; Colucci, R.; Chiriacò, M.; Uliana, M.; Puxeddu, I.; Bernardini, N.; Blandizzi, C.; Taddei, S. Microvascular Endothelial Dysfunction in Patients with Obesity. Curr. Hypertens. Rep. 2019, 21, 32. [Google Scholar] [CrossRef]
- van der Heijden, D.J.; van Leeuwen, M.A.H.; Janssens, G.N.; Lenzen, M.J.; van de Ven, P.M.; Eringa, E.C.; van Royen, N. Body Mass Index Is Associated With Microvascular Endothelial Dysfunction in Patients With Treated Metabolic Risk Factors and Suspected Coronary Artery Disease. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Villalobos-Labra, R.; Subiabre, M.; Toledo, F.; Pardo, F.; Sobrevia, L. Endoplasmic Reticulum Stress and Development of Insulin Resistance in Adipose, Skeletal, Liver, and Foetoplacental Tissue in Diabesity. Mol. Asp. Med. 2019, 66, 49–61. [Google Scholar] [CrossRef]
- Muris, D.M.J.; Houben, A.J.H.M.; Schram, M.T.; Stehouwer, C.D.A. Microvascular Dysfunction: An Emerging Pathway in the Pathogenesis of Obesity-Related Insulin Resistance. Rev. Endocr. Metab. Disord. 2013, 14, 29–38. [Google Scholar] [CrossRef]
- Stehouwer, C.D.A. Microvascular Dysfunction and Hyperglycemia: A Vicious Cycle with Widespread Consequences. Diabetes 2018, 67, 1729–1741. [Google Scholar] [CrossRef]
- Horton, W.B.; Barrett, E.J. Microvascular Dysfunction in Diabetes Mellitus and Cardiometabolic Disease. Endocr. Rev. 2021, 42, 29–55. [Google Scholar] [CrossRef]
- Krentz, A.J.; Clough, G.; Byrne, C.D. Interactions between Microvascular and Macrovascular Disease in Diabetes: Pathophysiology and Therapeutic Implications. Diabetes Obes. Metab. 2007, 9, 781–791. [Google Scholar] [CrossRef] [PubMed]
- De Jongh, R.T.; Serné, E.H.; Ijzerman, R.G.; de Vries, G.; Stehouwer, C.D.A. Impaired Microvascular Function in Obesity: Implications for Obesity-Associated Microangiopathy, Hypertension, and Insulin Resistance. Circulation 2004, 109, 2529–2535. [Google Scholar] [CrossRef] [PubMed]
- Karaca, Ü.; Schram, M.T.; Houben, A.J.H.M.; Muris, D.M.J.; Stehouwer, C.D.A. Microvascular Dysfunction as a Link between Obesity, Insulin Resistance and Hypertension. Diabetes Res. Clin. Pract. 2014, 103, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Casanova, F.; Adingupu, D.D.; Adams, F.; Gooding, K.M.; Looker, H.C.; Aizawa, K.; Dove, F.; Elyas, S.; Belch, J.J.F.; Gates, P.E.; et al. The Impact of Cardiovascular Co-Morbidities and Duration of Diabetes on the Association between Microvascular Function and Glycaemic Control. Cardiovasc. Diabetol. 2017, 16. [Google Scholar] [CrossRef]
- Levy, B.I.; Ambrosio, G.; Pries, A.R.; Struijker-Boudier, H.A.J. Microcirculation in Hypertension: A New Target for Treatment? Circulation 2001, 104, 735–740. [Google Scholar] [CrossRef]
- Latroche, C.; Gitiaux, C.; Chrétien, F.; Desguerre, I.; Mounie, R.; Chazaud, B. Skeletal Muscle Microvasculature: A Highly Dynamic Lifeline. Physiology 2015, 30, 417–427. [Google Scholar] [CrossRef]
- Ding, Y.; Zhou, Y.; Ling, P.; Feng, X.; Luo, S.; Zheng, X.; Little, P.J.; Xu, S.; Weng, J. Metformin in Cardiovascular Diabetology: A Focused Review of Its Impact on Endothelial Function. Theranostics 2021, 11, 9376–9396. [Google Scholar] [CrossRef]
- Vanhoutte, P.M.; Shimokawa, H.; Feletou, M.; Tang, E.H.C. Endothelial Dysfunction and Vascular Disease-a 30th Anniversary Update. Acta Physiol. (Oxf. Engl.) 2017, 219, 22–96. [Google Scholar] [CrossRef]
- Tejero, J.; Shiva, S.; Gladwin, M.T. Sources of Vascular Nitric Oxide and Reactive Oxygen Species and Their Regulation. Physiol. Rev. 2019, 99, 311–379. [Google Scholar] [CrossRef]
- Zeng, G.; Nystrom, F.H.; Ravichandran, L.V.; Cong, L.N.; Kirby, M.; Mostowski, H.; Quon, M.J. Roles for Insulin Receptor, PI3-Kinase, and Akt in Insulin-Signaling Pathways Related to Production of Nitric Oxide in Human Vascular Endothelial Cells. Circulation 2000, 101, 1539–1545. [Google Scholar] [CrossRef]
- Zaborska, K.E.; Wareing, M.; Austin, C. Comparisons between Perivascular Adipose Tissue and the Endothelium in Their Modulation of Vascular Tone. Br. J. Pharmacol. 2017, 174, 3388–3397. [Google Scholar] [CrossRef]
- Daiber, A.; Steven, S.; Weber, A.; Shuvaev, V.V.; Muzykantov, V.R.; Laher, I.; Li, H.; Lamas, S.; Münzel, T. Targeting Vascular (Endothelial) Dysfunction. Br. J. Pharmacol. 2017, 174, 1591–1619. [Google Scholar] [CrossRef]
- Janus, A.; Szahidewicz-Krupska, E.; Mazur, G.; Doroszko, A. Insulin Resistance and Endothelial Dysfunction Constitute a Common Therapeutic Target in Cardiometabolic Disorders. Mediat. Inflamm. 2016, 2016. [Google Scholar] [CrossRef]
- Chadderdon, S.M.; Belcik, J.T.; Bader, L.; Kievit, P.; Grove, K.L.; Lindner, J.R. Vasoconstrictor Eicosanoids and Impaired Microvascular Function in Inactive and Insulin-Resistant Primates. Int. J. Obes. 2016, 40, 1600–1603. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Shulman, G.I. The Pathogenesis of Insulin Resistance: Integrating Signaling Pathways and Substrate Flux. J. Clin. Investig. 2016, 126, 12–22. [Google Scholar] [CrossRef]
- Barrett, E.J.; Liu, Z.; Khamaisi, M.; King, G.L.; Klein, R.; Klein, B.E.K.; Hughes, T.M.; Craft, S.; Freedman, B.I.; Bowden, D.W.; et al. Diabetic Microvascular Disease: An Endocrine Society Scientific Statement. J. Clin. Endocrinol. Metab. 2017, 102, 4343–4410. [Google Scholar] [CrossRef] [PubMed]
- Williams, I.M.; Valenzuela, F.A.; Kahl, S.D.; Ramkrishna, D.; Mezo, A.R.; Young, J.D.; Sam Wells, K.; Wasserman, D.H. Insulin Exits Skeletal Muscle Capillaries by Fluid-Phase Transport. J. Clin. Investig. 2018, 128, 699–714. [Google Scholar] [CrossRef] [PubMed]
- Baron, A.D. Cardiovascular Actions of Insulin in Humans. Implications for Insulin Sensitivity and Vascular Tone. Bailliere’s Clin. Endocrinol. Metab. 1993, 7, 961–987. [Google Scholar] [CrossRef]
- Baron, A.D.; Steinberg, H.; Brechtel, G.; Johnson, A. Skeletal Muscle Blood Flow Independently Modulates Insulin-Mediated Glucose Uptake. Am. J. Physiol. 1994, 266. [Google Scholar] [CrossRef]
- Laakso, M.; Edelman, S.V.; Brechtel, G.; Baron, A.D. Decreased Effect of Insulin to Stimulate Skeletal Muscle Blood Flow in Obese Man. A Novel Mechanism for Insulin Resistance. J. Clin. Investig. 1990, 85, 1844–1852. [Google Scholar] [CrossRef]
- Clark, A.D.H.; Barrett, E.J.; Rattigan, S.; Wallis, M.G.; Clark, M.G. Insulin Stimulates Laser Doppler Signal by Rat Muscle in Vivo, Consistent with Nutritive Flow Recruitment. Clin. Sci. 2001, 100, 283–290. [Google Scholar] [CrossRef]
- Zhang, L.; Vincent, M.A.; Richards, S.M.; Clerk, L.H.; Rattigan, S.; Clark, M.G.; Barrett, E.J. Insulin Sensitivity of Muscle Capillary Recruitment in vivo. Diabetes 2004, 53, 447–453. [Google Scholar] [CrossRef]
- Vincent, M.A.; Clerk, L.H.; Lindner, J.R.; Klibanov, A.L.; Clark, M.G.; Rattigan, S.; Barrett, E.J. Microvascular Recruitment Is an Early Insulin Effect That Regulates Skeletal Muscle Glucose Uptake in vivo. Diabetes 2004, 53, 1418–1423. [Google Scholar] [CrossRef]
- Shulman, G.I. Cellular Mechanisms of Insulin Resistance. J. Clin. Investig. 2000, 106, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Wasserman, D.H.; Wang, T.J.; Brown, N.J. The Vasculature in Prediabetes. Circ. Res. 2018, 122, 1135–1150. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.Y.; Lin, Y.W.; Clemont, A.; Feener, E.P.; Hein, K.D.; Igarashi, M.; Yamauchi, T.; White, M.F.; King, G.L. Characterization of Selective Resistance to Insulin Signaling in the Vasculature of Obese Zucker (Fa/Fa) Rats. J. Clin. Investig. 1999, 104, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Martens, R.J.H.; Henry, R.M.A.; Houben, A.J.H.M.; van der Kallen, C.J.H.; Kroon, A.A.; Schalkwijk, C.G.; Schram, M.T.; Sep, S.J.S.; Schaper, N.C.; Dagnelie, P.C.; et al. Capillary Rarefaction Associates with Albuminuria: The Maastricht Study. J. Am. Soc. Nephrol. 2016, 27, 3748–3757. [Google Scholar] [CrossRef]
- Martens, R.J.H.; Houben, A.J.H.M.; Kooman, J.P.; Berendschot, T.T.J.M.; Dagnelie, P.C.; van der Kallen, C.J.H.; Kroon, A.A.; Leunissen, K.M.L.; van der Sande, F.M.; Schaper, N.C.; et al. Microvascular Endothelial Dysfunction Is Associated with Albuminuria: The Maastricht Study. J. Hypertens. 2018, 36, 1178–1187. [Google Scholar] [CrossRef]
- Williams, I.M.; McClatchey, P.M.; Bracy, D.P.; Bonner, J.S.; Valenzuela, F.A.; Wasserman, D.H. Transendothelial Insulin Transport Is Impaired in Skeletal Muscle Capillaries of Obese Male Mice. Obesity 2020, 28, 303–314. [Google Scholar] [CrossRef]
- Krogh, A. The Number and Distribution of Capillaries in Muscles with Calculations of the Oxygen Pressure Head Necessary for Supplying the Tissue. J. Physiol. 1919, 52, 409–415. [Google Scholar] [CrossRef]
- Hepple, R.T.; Mathieu-Costello, O. Estimating the Size of the Capillary-to-Fiber Interface in Skeletal Muscle: A Comparison of Methods. J. Appl. Physiol. 2001, 91, 2150–2156. [Google Scholar] [CrossRef]
- Eržen, I.; Janáček, J.; Kreft, M.; Kubínová, L.; Cvetko, E. Capillary Network Morphometry of Pig Soleus Muscle Significantly Changes in 24 Hours After Death. J. Histochem. Cytochem. 2018, 66, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Schaad, L.; Hlushchuk, R.; Barré, S.; Gianni-Barrera, R.; Haberthür, D.; Banfi, A.; Djonov, V. Correlative Imaging of the Murine Hind Limb Vasculature and Muscle Tissue by MicroCT and Light Microscopy. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Umek, N.; Horvat, S.; Cvetko, E.; Kreft, M.; Janáček, J.; Kubínová, L.; Stopar Pintarič, T.; Eržen, I. 3D Analysis of Capillary Network in Skeletal Muscle of Obese Insulin-Resistant Mice. Histochem. Cell Biol. 2019, 152, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Dresner, A.; Laurent, D.; Marcucci, M.; Griffin, M.E.; Dufour, S.; Cline, G.W.; Slezak, L.A.; Andersen, D.K.; Hundal, R.S.; Rothman, D.L.; et al. Effects of Free Fatty Acids on Glucose Transport and IRS-1-Associated Phosphatidylinositol 3-Kinase Activity. J. Clin. Investig. 1999, 103, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.W.; Zhang, L.; Youker, K.; Zhang, M.X.; Wang, J.; LeMaire, S.A.; Coselli, J.S.; Shen, Y.H. Free Fatty Acids Inhibit Insulin Signaling-Stimulated Endothelial Nitric Oxide Synthase Activation through Upregulating PTEN or Inhibiting Akt Kinase. Diabetes 2006, 55, 2301–2310. [Google Scholar] [CrossRef]
- García-Prieto, C.F.; Hernández-Nuño, F.; del Rio, D.; Ruiz-Hurtado, G.; Aránguez, I.; Ruiz-Gayo, M.; Somoza, B.; Fernández-Alfonso, M.S. High-Fat Diet Induces Endothelial Dysfunction through a down-Regulation of the Endothelial AMPK-PI3K-Akt-ENOS Pathway. Mol. Nutr. Food Res. 2015, 59, 520–532. [Google Scholar] [CrossRef]
- Sorop, O.; Olver, T.D.; van DeWouw, J.; Heinonen, I.; van Duin, R.W.; Duncker, D.J.; Merkus, D. The Microcirculation: A Key Player in Obesity-Associated Cardiovascular Disease. Cardiovasc. Res. 2017, 113, 1035–1045. [Google Scholar] [CrossRef]
- Meijer, R.I.; Serné, E.H.; Korkmaz, H.I.; van der Peet, D.L.; de Boer, M.P.; Niessen, H.W.M.; van Hinsbergh, V.W.M.; Yudkin, J.S.; Smulders, Y.M.; Eringa, E.C. Insulin-Induced Changes in Skeletal Muscle Microvascular Perfusion Are Dependent upon Perivascular Adipose Tissue in Women. Diabetologia 2015, 58, 1907–1915. [Google Scholar] [CrossRef]
- Inoguchi, T.; Li, P.; Umeda, F.; Yu, H.Y.; Kakimoto, M.; Imamura, M.; Aoki, T.; Etoh, T.; Hashimoto, T.; Naruse, M.; et al. High Glucose Level and Free Fatty Acid Stimulate Reactive Oxygen Species Production through Protein Kinase C--Dependent Activation of NAD(P)H Oxidase in Cultured Vascular Cells. Diabetes 2000, 49, 1939–1945. [Google Scholar] [CrossRef]
- Savage, D.B.; Watson, L.; Carr, K.; Adams, C.; Brage, S.; Chatterjee, K.K.; Hodson, L.; Boesch, C.; Kemp, G.J.; Sleigh, A. Accumulation of Saturated Intramyocellular Lipid Is Associated with Insulin Resistance. J. Lipid Res. 2019, 60, 1323–1332. [Google Scholar] [CrossRef]
- Limberg, J.K.; Johansson, R.E.; Carter, K.J.; Peltonen, G.; Harrell, J.W.; Kellawan, J.M.; Eldridge, M.W.; Sebranek, J.J.; Walker, B.J.; Schrage, W.G. Preserved β-Adrenergic Mediated Vasodilation in Skeletal Muscle of Young Obese Adults despite Shifts in Cyclooxygenase and Nitric Oxide Synthase. Am. J. Physiol. Heart Circ. Physiol. 2021. [Google Scholar] [CrossRef]
- Limberg, J.K.; Morgan, B.J.; Schrage, W.G. Peripheral Blood Flow Regulation in Human Obesity and Metabolic Syndrome. Exerc. Sport Sci. Rev. 2016, 44, 116–122. [Google Scholar] [CrossRef]
- Limberg, J.K.; de Vita, M.D.; Blain, G.M.; Schrage, W.G. Muscle Blood Flow Responses to Dynamic Exercise in Young Obese Humans. J. Appl. Physiol. 2010, 108, 349–355. [Google Scholar] [CrossRef]
- Bender, S.B.; de Beer, V.J.; Tharp, D.L.; van Deel, E.D.; Bowles, D.K.; Duncker, D.J.; Laughlin, M.H.; Merkus, D. Reduced Contribution of Endothelin to the Regulation of Systemic and Pulmonary Vascular Tone in Severe Familial Hypercholesterolaemia. J. Physiol. 2014, 592, 1757–1769. [Google Scholar] [CrossRef]
- Karpoff, L.; Vinet, A.; Schuster, I.; Oudot, C.; Goret, L.; Dauzat, M.; Obert, P.; Perez-Martin, A. Abnormal Vascular Reactivity at Rest and Exercise in Obese Boys. Eur. J. Clin. Investig. 2009, 39, 94–102. [Google Scholar] [CrossRef]
- Vinet, A.; Karpoff, L.; Walther, G.; Startun, A.; Obert, P.; Goret, L.; Dauzat, M.; Perez-Martin, A. Vascular Reactivity at Rest and during Exercise in Middle-Aged Obese Men: Effects of Short-Term, Low-Intensity, Exercise Training. Int. J. Obes. 2011, 35, 820–828. [Google Scholar] [CrossRef]
- Agapitov, A.V.; Correia, M.L.G.; Sinkey, C.A.; Dopp, J.M.; Haynes, W.G. Impaired Skeletal Muscle and Skin Microcirculatory Function in Human Obesity. J. Hypertens. 2002, 20, 1401–1405. [Google Scholar] [CrossRef] [PubMed]
- Frisbee, J.C.; Goodwill, A.G.; Frisbee, S.J.; Butcher, J.T.; Brock, R.W.; Olfert, I.M.; DeVallance, E.R.; Chantler, P.D. Distinct Temporal Phases of Microvascular Rarefaction in Skeletal Muscle of Obese Zucker Rats. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1714–H1728. [Google Scholar] [CrossRef] [PubMed]
- Sjöstrand, M.; Gudbjörnsdottir, S.; Holmäng, A.; Lönn, L.; Strindberg, L.; Lönnroth, P. Delayed Transcapillary Transport of Insulin to Muscle Interstitial Fluid in Obese Subjects. Diabetes 2002, 51, 2742–2748. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Kubota, N.; Kumagai, H.; Yamaguchi, S.; Kozono, H.; Takahashi, T.; Inoue, M.; Itoh, S.; Takamoto, I.; Sasako, T.; et al. Impaired Insulin Signaling in Endothelial Cells Reduces Insulin-Induced Glucose Uptake by Skeletal Muscle. Cell Metab. 2011, 13, 294–307. [Google Scholar] [CrossRef]
- Ayala, J.E.; Bracy, D.P.; Julien, B.M.; Rottman, J.N.; Fueger, P.T.; Wasserman, D.H. Chronic Treatment with Sildenafil Improves Energy Balance and Insulin Action in High Fat-Fed Conscious Mice. Diabetes 2007, 56, 1025–1033. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, A.; Fraccarollo, D.; Pförtsch, S.; Flierl, U.; Vogt, C.; Pfrang, J.; Kobsar, A.; Renné, T.; Eigenthaler, M.; Ertl, G.; et al. Improvement of Vascular Function by Acute and Chronic Treatment with the PDE-5 Inhibitor Sildenafil in Experimental Diabetes Mellitus. Br. J. Pharmacol. 2008, 153, 886–893. [Google Scholar] [CrossRef] [PubMed]
- Chadderdon, S.M.; Belcik, J.T.; Bader, L.; Peters, D.M.; Kievit, P.; Alkayed, N.J.; Kaul, S.; Grove, K.L.; Lindner, J.R. Temporal Changes in Skeletal Muscle Capillary Responses and Endothelial-Derived Vasodilators in Obesity-Related Insulin Resistance. Diabetes 2016, 65, 2249–2257. [Google Scholar] [CrossRef]
- Lemaster, K.A.; Farid, Z.; Brock, R.W.; Shrader, C.D.; Goldman, D.; Jackson, D.N.; Frisbee, J.C. Altered Post-Capillary and Collecting Venular Reactivity in Skeletal Muscle with Metabolic Syndrome. J. Physiol. 2017, 595, 5159–5174. [Google Scholar] [CrossRef] [PubMed]
- Bonner, J.S.; Lantier, L.; Hasenour, C.M.; James, F.D.; Bracy, D.P.; Wasserman, D.H. Muscle-Specific Vascular Endothelial Growth Factor Deletion Induces Muscle Capillary Rarefaction Creating Muscle Insulin Resistance. Diabetes 2013, 62, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Messa, G.A.M.; Piasecki, M.; Hurst, J.; Hill, C.; Tallis, J.; Degens, H. The Impact of a High-Fat Diet in Mice Is Dependent on Duration and Age, and Differs between Muscles. J. Exp. Biol. 2020, 223. [Google Scholar] [CrossRef]
- Eriksson, K.F.; Saltin, B.; Lindgarde, F. Increased Skeletal Muscle Capillary Density Precedes Diabetes Development in Men with Impaired Glucose Tolerance. A 15-Year Follow-Up. Diabetes 1994, 43, 805–808. [Google Scholar] [CrossRef]
- O’Reilly, J.; Ono-Moore, K.D.; Chintapalli, S.V.; Rutkowsky, J.M.; Tolentino, T.; Lloyd, K.C.K.; Olfert, I.M.; Adams, S.H. Sex Differences in Skeletal Muscle Revealed through Fiber Type, Capillarity, and Transcriptomics Profiling in Mice. Physiol. Rep. 2021, 9. [Google Scholar] [CrossRef]
- Huxley, V.H.; Kemp, S.S. Sex-Specific Characteristics of the Microcirculation. Adv. Exp. Med. Biol. 2018, 1065, 307–328. [Google Scholar] [CrossRef]
- Sidsworth, D.A.; Sellers, S.L.; Reutens-Hernandez, J.P.; Dunn, E.A.; Gray, S.L.; Payne, G.W. Impact of Sex on Microvascular Reactivity in a Murine Model of Diet-Induced Obesity and Insulin Resistance. Heliyon 2021, 7. [Google Scholar] [CrossRef]
- de Vriese, A.S.; Verbeuren, T.J.; van de Voorde, J.; Lameire, N.H.; Vanhoutte, P.M. Endothelial Dysfunction in Diabetes. Br. J. Pharmacol. 2000, 130, 963–974. [Google Scholar] [CrossRef]
- Scioli, M.G.; Storti, G.; D’Amico, F.; Rodríguez Guzmán, R.; Centofanti, F.; Doldo, E.; Céspedes Miranda, E.M.; Orlandi, A. Oxidative Stress and New Pathogenetic Mechanisms in Endothelial Dysfunction: Potential Diagnostic Biomarkers and Therapeutic Targets. J. Clin. Med. 2020, 9, 1995. [Google Scholar] [CrossRef]
- Kaur, R.; Kaur, M.; Singh, J. Endothelial Dysfunction and Platelet Hyperactivity in Type 2 Diabetes Mellitus: Molecular Insights and Therapeutic Strategies. Cardiovasc. Diabetol. 2018, 17. [Google Scholar] [CrossRef] [PubMed]
- Dhananjayan, R.; Koundinya, K.S.S.; Malati, T.; Kutala, V.K. Endothelial Dysfunction in Type 2 Diabetes Mellitus. Indian J. Clin. Biochem. IJCB 2016, 31, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Dimmeler, S.; Fleming, I.; Fisslthaler, B.; Hermann, C.; Busse, R.; Zeiher, A.M. Activation of Nitric Oxide Synthase in Endothelial Cells by Akt-Dependent Phosphorylation. Nature 1999, 399, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Ke, B.; Shen, W.; Fang, X.; Wu, Q. The NLPR3 Inflammasome and Obesity-Related Kidney Disease. J. Cell. Mol. Med. 2018, 22, 16–24. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, L.; Pitzer, A.L.; Li, X.; Li, P.L.; Zhang, Y. Contribution of Redox-Dependent Activation of Endothelial Nlrp3 Inflammasomes to Hyperglycemia-Induced Endothelial Dysfunction. J. Mol. Med. 2016, 94, 1335–1347. [Google Scholar] [CrossRef]
- Wang, L.; Chen, Y.; Li, X.; Zhang, Y.; Gulbins, E.; Zhang, Y. Enhancement of Endothelial Permeability by Free Fatty Acid through Lysosomal Cathepsin B-Mediated Nlrp3 Inflammasome Activation. Oncotarget 2016, 7, 73229–73241. [Google Scholar] [CrossRef]
- Las, G.; Serada, S.B.; Wikstrom, J.D.; Twig, G.; Shirihai, O.S. Fatty Acids Suppress Autophagic Turnover in β-Cells. J. Biol. Chem. 2011, 286, 42534. [Google Scholar] [CrossRef]
- Mameli, E.; Martello, A.; Caporali, A. Autophagy at the Interface of Endothelial Cell Homeostasis and Vascular Disease. FEBS J. 2021. [Google Scholar] [CrossRef]
- Tsushima, Y.; Nishizawa, H.; Tochino, Y.; Nakatsuji, H.; Sekimoto, R.; Nagao, H.; Shirakura, T.; Kato, K.; Imaizumi, K.; Takahashi, H.; et al. Uric Acid Secretion from Adipose Tissue and Its Increase in Obesity. J. Biol. Chem. 2013, 288, 27138–27149. [Google Scholar] [CrossRef]
- Niu, Y.; Tang, Q.; Zhao, X.; Zhao, X.; Mao, X.; Sheng, J.; Cai, W.; Feng, Y. Obesity-Induced Insulin Resistance Is Mediated by High Uric Acid in Obese Children and Adolescents. Front. Endocrinol. 2021, 12, 773820. [Google Scholar] [CrossRef]
- Liu, F.; Chen, S.; Zhao, W.; Chen, M.; Ke, J.; Zhang, Z.; Lu, J.; Li, L. Urine Uric Acid Excretion Levels Are Positively Associated with Obesity and Abdominal Obesity in Type 2 Diabetes Patients without Chronic Kidney Disease. Diabetes Metab. Syndr. Obes. Targets Ther. 2021, 14, 4691–4703. [Google Scholar] [CrossRef]
- Cai, W.; Duan, X.M.; Liu, Y.; Yu, J.; Tang, Y.L.; Liu, Z.L.; Jiang, S.; Zhang, C.P.; Liu, J.Y.; Xu, J.X. Uric Acid Induces Endothelial Dysfunction by Activating the HMGB1/RAGE Signaling Pathway. BioMed Res. Int. 2017, 2017, 4391920. [Google Scholar] [CrossRef] [PubMed]
- Johnson, F.K.; Peyton, K.J.; Liu, X.M.; Azam, M.A.; Shebib, A.R.; Johnson, R.A.; Durante, W. Arginase Promotes Endothelial Dysfunction and Hypertension in Obese Rats. Obesity 2015, 23, 383–390. [Google Scholar] [CrossRef]
- Chung, J.H.; Moon, J.; Lee, Y.S.; Chung, H.K.; Lee, S.M.; Shin, M.J. Arginase Inhibition Restores Endothelial Function in Diet-Induced Obesity. Biochem. Biophys. Res. Commun. 2014, 451, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Xia, N.; Horke, S.; Habermeier, A.; Closs, E.I.; Reifenberg, G.; Gericke, A.; Mikhed, Y.; Münzel, T.; Daiber, A.; Förstermann, U.; et al. Uncoupling of Endothelial Nitric Oxide Synthase in Perivascular Adipose Tissue of Diet-Induced Obese Mice. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Bhatta, A.; Yao, L.; Xu, Z.; Toque, H.A.; Chen, J.; Atawia, R.T.; Fouda, A.Y.; Bagi, Z.; Lucas, R.; Caldwell, R.B.; et al. Obesity-Induced Vascular Dysfunction and Arterial Stiffening Requires Endothelial Cell Arginase 1. Cardiovasc. Res. 2017, 113, 1664–1676. [Google Scholar] [CrossRef]
- el Assar, M.; Angulo, J.; Santos-Ruiz, M.; Ruiz de Adana, J.C.; Pindado, M.L.; Sánchez-Ferrer, A.; Hernández, A.; Rodríguez-Mañas, L. Asymmetric Dimethylarginine (ADMA) Elevation and Arginase up-Regulation Contribute to Endothelial Dysfunction Related to Insulin Resistance in Rats and Morbidly Obese Humans. J. Physiol. 2016, 594, 3045–3060. [Google Scholar] [CrossRef]
- Masi, S.; Colucci, R.; Duranti, E.; Nannipieri, M.; Anselmino, M.; Ippolito, C.; Tirotta, E.; Georgiopoulos, G.; Garelli, F.; Nericcio, A.; et al. Aging Modulates the Influence of Arginase on Endothelial Dysfunction in Obesity. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2474–2483. [Google Scholar] [CrossRef] [PubMed]
- Taniyama, Y.; Hitomi, H.; Shah, A.; Alexander, R.W.; Griendling, K.K. Mechanisms of Reactive Oxygen Species-Dependent Downregulation of Insulin Receptor Substrate-1 by Angiotensin II. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1142–1147. [Google Scholar] [CrossRef] [PubMed]
- Hitomi, H.; Kiyomoto, H.; Nishiyama, A.; Hara, T.; Moriwaki, K.; Kaifu, K.; Ihara, G.; Fujita, Y.; Ugawa, T.; Kohno, M. Aldosterone Suppresses Insulin Signaling via the Downregulation of Insulin Receptor Substrate-1 in Vascular Smooth Muscle Cells. Hypertension 2007, 50, 750–755. [Google Scholar] [CrossRef]
- Mazak, I.; Fiebeler, A.; Muller, D.N.; Park, J.K.; Shagdarsuren, E.; Lindschau, C.; Dechend, R.; Viedt, C.; Pilz, B.; Haller, H.; et al. Aldosterone Potentiates Angiotensin II-Induced Signaling in Vascular Smooth Muscle Cells. Circulation 2004, 109, 2792–2800. [Google Scholar] [CrossRef]
- Sherajee, S.J.; Fujita, Y.; Rafiq, K.; Nakano, D.; Mori, H.; Masaki, T.; Hara, T.; Kohno, M.; Nishiyama, A.; Hitomi, H. Aldosterone Induces Vascular Insulin Resistance by Increasing Insulin-like Growth Factor-1 Receptor and Hybrid Receptor. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; DeMarco, V.G.; Sowers, J.R. Insulin Resistance and Hyperinsulinaemia in Diabetic Cardiomyopathy. Nat. Rev. Endocrinol. 2016, 12, 144–153. [Google Scholar] [CrossRef]
- Ait-Aissa, K.; Nguyen, Q.M.; Gabani, M.; Kassan, A.; Kumar, S.; Choi, S.K.; Gonzalez, A.A.; Khataei, T.; Sahyoun, A.M.; Chen, C.; et al. MicroRNAs and Obesity-Induced Endothelial Dysfunction: Key Paradigms in Molecular Therapy. Cardiovasc. Diabetol. 2020, 19. [Google Scholar] [CrossRef]
- Caporali, A.; Meloni, M.; Völlenkle, C.; Bonci, D.; Sala-Newby, G.B.; Addis, R.; Spinetti, G.; Losa, S.; Masson, R.; Baker, A.H.; et al. Deregulation of MicroRNA-503 Contributes to Diabetes Mellitus-Induced Impairment of Endothelial Function and Reparative Angiogenesis after Limb Ischemia. Circulation 2011, 123, 282–291. [Google Scholar] [CrossRef]
- Murphy, K.; O’Donovan, A.N.; Caplice, N.M.; Ross, R.P.; Stanton, C. Exploring the Gut Microbiota and Cardiovascular Disease. Metabolites 2021, 11, 493. [Google Scholar] [CrossRef]
- Zhang, B.; Wang, X.; Xia, R.; Li, C. Gut Microbiota in Coronary Artery Disease: A Friend or Foe? Biosci. Rep. 2020, 40. [Google Scholar] [CrossRef]
- Malik, M.; Suboc, T.M.; Tyagi, S.; Salzman, N.; Wang, J.; Ying, R.; Tanner, M.J.; Kakarla, M.; Baker, J.E.; Widlansky, M.E. Lactobacillus Plantarum 299v Supplementation Improves Vascular Endothelial Function and Reduces Inflammatory Biomarkers in Men with Stable Coronary Artery Disease. Circ. Res. 2018, 123, 1091–1102. [Google Scholar] [CrossRef] [PubMed]
- Rovella, V.; Rodia, G.; di Daniele, F.; Cardillo, C.; Campia, U.; Noce, A.; Candi, E.; Della-Morte, D.; Tesauro, M. Association of Gut Hormones and Microbiota with Vascular Dysfunction in Obesity. Nutrients 2021, 13, 613. [Google Scholar] [CrossRef]
- Vikram, A.; Kim, Y.R.; Kumar, S.; Li, Q.; Kassan, M.; Jacobs, J.S.; Irani, K. Vascular MicroRNA-204 Is Remotely Governed by the Microbiome and Impairs Endothelium-Dependent Vasorelaxation by Downregulating Sirtuin1. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Nirmalkar, K.; Murugesan, S.; Pizano-Zárate, M.L.; Villalobos-Flores, L.E.; García-González, C.; Morales-Hernández, R.M.; Nuñez-Hernández, J.A.; Hernández-Quiroz, F.; Romero-Figueroa, M.D.S.; Hernández-Guerrero, C.; et al. Gut Microbiota and Endothelial Dysfunction Markers in Obese Mexican Children and Adolescents. Nutrients 2018, 10, 2009. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.; Litwin, N.; van Ark, H.; Hartley, S.; Fischer, E.; Michell, K.; Vazquez, A.; Lee, D.; Trikha, S.R.; Wrigley, S.; et al. The Gut Microbiota Is Associated with Vascular Function and Blood Pressure Phenotypes in Overweight and Obese Middle-Aged/Older Adults (P21-024-19). Curr. Dev. Nutr. 2019, 3. [Google Scholar] [CrossRef]
- King, R.J.; Ajjan, R.A. Vascular Risk in Obesity: Facts, Misconceptions and the Unknown. Diabetes Vasc. Dis. Res. 2017, 14, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Eringa, E.C.; Stehouwer, C.D.A.; Walburg, K.; Clark, A.D.; van Nieuw Amerongen, G.P.; Westerhof, N.; Sipkema, P. Physiological Concentrations of Insulin Induce Endothelin-Dependent Vasoconstriction of Skeletal Muscle Resistance Arteries in the Presence of Tumor Necrosis Factor-Alpha Dependence on c-Jun N-Terminal Kinase. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Yuan, Y.; Yi, W.; Lau, W.B.; Wang, Y.; Wang, X.; Sun, Y.; Lopez, B.L.; Christopher, T.A.; Peterson, J.M.; et al. C1q/TNF-Related Proteins, a Family of Novel Adipokines, Induce Vascular Relaxation through the Adiponectin Receptor-1/AMPK/ENOS/Nitric Oxide Signaling Pathway. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2616–2623. [Google Scholar] [CrossRef]
- Houben, A.J.; Eringa, E.C.; Jonk, A.M.; Serne, E.H.; Smulders, Y.M.; Stehouwer, C.D. Perivascular Fat and the Microcirculation: Relevance to Insulin Resistance, Diabetes, and Cardiovascular Disease. Curr. Cardiovasc. Risk Rep. 2012, 6, 80–90. [Google Scholar] [CrossRef][Green Version]
- Sena, C.M.; Pereira, A.; Fernandes, R.; Letra, L.; Seiça, R.M. Adiponectin Improves Endothelial Function in Mesenteric Arteries of Rats Fed a High-Fat Diet: Role of Perivascular Adipose Tissue. Br. J. Pharmacol. 2017, 174, 3514–3526. [Google Scholar] [CrossRef]
- Serné, E.H.; de Jongh, R.T.; Eringa, E.C.; IJzerman, R.G.; Stehouwer, C.D.A. Microvascular Dysfunction. Hypertension 2007, 50, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Virdis, A.; Colucci, R.; Bernardini, N.; Blandizzi, C.; Taddei, S.; Masi, S. Microvascular Endothelial Dysfunction in Human Obesity: Role of TNF-α. J. Clin. Endocrinol. Metab. 2019, 104, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Schinzari, F.; Tesauro, M.; Cardillo, C. Endothelial and Perivascular Adipose Tissue Abnormalities in Obesity-Related Vascular Dysfunction: Novel Targets for Treatment. J. Cardiovasc. Pharmacol. 2017, 69, 360–368. [Google Scholar] [CrossRef]
- Murphy, M.P. How Mitochondria Produce Reactive Oxygen Species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Thiruvengadam, M.; Venkidasamy, B.; Subramanian, U.; Samynathan, R.; Ali Shariati, M.; Rebezov, M.; Girish, S.; Thangavel, S.; Dhanapal, A.R.; Fedoseeva, N.; et al. Bioactive Compounds in Oxidative Stress-Mediated Diseases: Targeting the NRF2/ARE Signaling Pathway and Epigenetic Regulation. Antioxidants 2021, 10, 1859. [Google Scholar] [CrossRef]
- Ramprasath, T.; Freddy, A.J.; Velmurugan, G.; Tomar, D.; Rekha, B.; Suvekbala, V.; Ramasamy, S. Context-Dependent Regulation of Nrf2/ARE Axis on Vascular Cell Function during Hyperglycemic Condition. Curr. Diabetes Rev. 2020, 16, 797–806. [Google Scholar] [CrossRef]
- Lennicke, C.; Cochemé, H.M. Redox Regulation of the Insulin Signalling Pathway. Redox Biol. 2021, 42. [Google Scholar] [CrossRef]
- Creager, M.A.; Lüscher, T.F.; Cosentino, F.; Beckman, J.A. Diabetes and Vascular Disease: Pathophysiology, Clinical Consequences, and Medical Therapy: Part I. Circulation 2003, 108, 1527–1532. [Google Scholar] [CrossRef]
- Inoguchi, T.; Sonta, T.; Tsubouchi, H.; Etoh, T.; Kakimoto, M.; Sonoda, N.; Sato, N.; Sekiguchi, N.; Kobayashi, K.; Sumimoto, H.; et al. Protein Kinase C-Dependent Increase in Reactive Oxygen Species (ROS) Production in Vascular Tissues of Diabetes: Role of Vascular NAD(P)H Oxidase. J. Am. Soc. Nephrol. 2003, 14. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.F.; Laitano, O. Regulation of NADPH Oxidases in Skeletal Muscle. Free Radic. Biol. Med. 2016, 98, 18–28. [Google Scholar] [CrossRef]
- Mahmoud, A.M.; Ali, M.M.; Miranda, E.R.; Mey, J.T.; Blackburn, B.K.; Haus, J.M.; Phillips, S.A. Nox2 Contributes to Hyperinsulinemia-Induced Redox Imbalance and Impaired Vascular Function. Redox Biol. 2017, 13, 288–300. [Google Scholar] [CrossRef]
- Cully, T.R.; Rodney, G.G. Nox4–RyR1–Nox2: Regulators of Micro-Domain Signaling in Skeletal Muscle. Redox Biol. 2020, 36. [Google Scholar] [CrossRef]
- Nishikawa, T.; Edelstein, D.; Du, X.L.; Yamagishi, S.I.; Matsumura, T.; Kaneda, Y.; Yorek, M.A.; Beebe, D.; Oates, P.J.; Hammes, H.P.; et al. Normalizing Mitochondrial Superoxide Production Blocks Three Pathways of Hyperglycaemic Damage. Nature 2000, 404, 787–790. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, X.; Ying, L.; Dou, D.; Li, Y.; Bai, Y.; Liu, J.; Liu, L.; Feng, H.; Yu, X.; et al. CIMP Synthesized by SGC as a Mediator of Hypoxic Contraction of Coronary Arteries. Am. J. Physiol. Heart Circ. Physiol. 2014, 307. [Google Scholar] [CrossRef] [PubMed]
- Meza, C.A.; la Favor, J.D.; Kim, D.H.; Hickner, R.C. Endothelial Dysfunction: Is There a Hyperglycemia-Induced Imbalance of NOX and NOS? Int. J. Mol. Sci. 2019, 20, 3775. [Google Scholar] [CrossRef]
- Kang, L.; Ayala, J.E.; Lee-Young, R.S.; Zhang, Z.; James, F.D.; Neufer, P.D.; Pozzi, A.; Zutter, M.M.; Wasserman, D.H. Diet-Induced Muscle Insulin Resistance Is Associated with Extracellular Matrix Remodeling and Interaction with Integrin A2β 1in Mice. Diabetes 2011, 60, 416–426. [Google Scholar] [CrossRef]
- Kang, L.; Lantier, L.; Kennedy, A.; Bonner, J.S.; Mayes, W.H.; Bracy, D.P.; Bookbinder, L.H.; Hasty, A.H.; Thompson, C.B.; Wasserman, D.H. Hyaluronan Accumulates with High-Fat Feeding and Contributes to Insulin Resistance. Diabetes 2013, 62, 1888–1896. [Google Scholar] [CrossRef] [PubMed]
- Berria, R.; Wang, L.; Richardson, D.K.; Finlayson, J.; Belfort, R.; Pratipanawatr, T.; de Filippis, E.A.; Kashyap, S.; Mandarino, L.J. Increased Collagen Content in Insulin-Resistant Skeletal Muscle. Am. J. Physiol. -Endocrinol. Metab. 2006, 290, 560–565. [Google Scholar] [CrossRef]
- Fogelstrand, P.; Borén, J. Treatment of Hyaluronan Accumulation Ameliorates High-Fat Diet-Induced Insulin Resistance in Mice. Diabetes 2013, 62, 1816–1817. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kodama, K.; Toda, K.; Morinaga, S.; Yamada, S.; Butte, A.J. Anti-CD44 Antibody Treatment Lowers Hyperglycemia and Improves Insulin Resistance, Adipose Inflammation, and Hepatic Steatosis in Diet-Induced Obese Mice. Diabetes 2015, 64, 867–875. [Google Scholar] [CrossRef]
- Saunders, K.H.; Shukla, A.P.; Igel, L.I.; Aronne, L.J. Obesity: When to Consider Medication. J. Fam. Pract. 2017, 66, 608–617. [Google Scholar] [PubMed]
- Gadde, K.M.; Martin, C.K.; Berthoud, H.R.; Heymsfield, S.B. Obesity: Pathophysiology and Management. J. Am. Coll. Cardiol. 2018, 71, 69–84. [Google Scholar] [CrossRef]
- Robinson, E.; Boyland, E.; Chisholm, A.; Harrold, J.; Maloney, N.G.; Marty, L.; Mead, B.R.; Noonan, R.; Hardman, C.A. Obesity, Eating Behavior and Physical Activity during COVID-19 Lockdown: A Study of UK Adults. Appetite 2021, 156. [Google Scholar] [CrossRef] [PubMed]
- Deschasaux-Tanguy, M.; Druesne-Pecollo, N.; Esseddik, Y.; de Edelenyi, F.S.; Allès, B.; Andreeva, V.A.; Baudry, J.; Charreire, H.; Deschamps, V.; Egnell, M.; et al. Diet and Physical Activity during the Coronavirus Disease 2019 (COVID-19) Lockdown (March-May 2020): Results from the French NutriNet-Santé Cohort Study. Am. J. Clin. Nutr. 2021, 113, 924–938. [Google Scholar] [CrossRef]
- Rodríguez, J.E.; Campbell, K.M. Past, Present, and Future of Pharmacologic Therapy in Obesity. Prim. Care 2016, 43, 61–67. [Google Scholar] [CrossRef]
- Khera, R.; Murad, M.H.; Chandar, A.K.; Dulai, P.S.; Wang, Z.; Prokop, L.J.; Loomba, R.; Camilleri, M.; Singh, S. Association of Pharmacological Treatments for Obesity Withweight Loss and Adverse Events a Systematic Review and Meta-Analysis. JAMA-J. Am. Med. Assoc. 2016, 315, 2424–2434. [Google Scholar] [CrossRef] [PubMed]
- Tak, Y.J.; Lee, S.Y. Long-Term Efficacy and Safety of Anti-Obesity Treatment: Where Do We Stand? Curr. Obes. Rep. 2021, 10, 14–30. [Google Scholar] [CrossRef]
- Daneschvar, H.L.; Aronson, M.D.; Smetana, G.W. FDA-Approved Anti-Obesity Drugs in the United States. Am. J. Med. 2016, 129, 879.e1–879.e6. [Google Scholar] [CrossRef]
- Sharretts, J.; Galescu, O.; Gomatam, S.; Andraca-Carrera, E.; Hampp, C.; Yanoff, L. Cancer Risk Associated with Lorcaserin-The FDA’s Review of the CAMELLIA-TIMI 61 Trial. N. Engl. J. Med. 2020, 383, 1000–1002. [Google Scholar] [CrossRef]
- Coulter, A.A.; Rebello, C.J.; Greenway, F.L. Centrally Acting Agents for Obesity: Past, Present, and Future. Drugs 2018, 78, 1113–1132. [Google Scholar] [CrossRef]
- Narayanaswami, V.; Dwoskin, L.P. Obesity: Current and Potential Pharmacotherapeutics and Targets. Pharmacol. Ther. 2017, 170, 116–147. [Google Scholar] [CrossRef] [PubMed]
- Bougoulia, M.; Triantos, A.; Koliakos, G. Effect of Weight Loss with or without Orlistat Treatment on Adipocytokines, Inflammation, and Oxidative Markers in Obese Women. Hormones 2006, 5, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.J.; Wang, P.W.; Liu, R.T.; Tung, S.C.; Chien, W.Y.; Chen, J.F.; Chen, C.H.; Kuo, M.C.; Hu, Y.H. Orlistat for Obesity: Benefits beyond Weight Loss. Diabetes Res. Clin. Pract. 2005, 67, 78–83. [Google Scholar] [CrossRef]
- Vorsanger, M.H.; Subramanyam, P.; Weintraub, H.S.; Lamm, S.H.; Underberg, J.A.; Gianos, E.; Goldberg, I.J.; Schwartzbard, A.Z. Cardiovascular Effects of the New Weight Loss Agents. J. Am. Coll. Cardiol. 2016, 68, 849–859. [Google Scholar] [CrossRef] [PubMed]
- Gadde, K.M.; Allison, D.B.; Ryan, D.H.; Peterson, C.A.; Troupin, B.; Schwiers, M.L.; Day, W.W. Effects of Low-Dose, Controlled-Release, Phentermine plus Topiramate Combination on Weight and Associated Comorbidities in Overweight and Obese Adults (CONQUER): A Randomised, Placebo-Controlled, Phase 3 Trial. Lancet 2011, 377, 1341–1352. [Google Scholar] [CrossRef]
- von Scholten, B.J.; Persson, F.; Rosenlund, S.; Eugen-Olsen, J.; Pielak, T.; Faber, J.; Hansen, T.W.; Rossing, P. Effects of Liraglutide on Cardiovascular Risk Biomarkers in Patients with Type 2 Diabetes and Albuminuria: A Sub-Analysis of a Randomized, Placebo-Controlled, Double-Blind, Crossover Trial. Diabetes Obes. Metab. 2017, 19, 901–905. [Google Scholar] [CrossRef]
- Zobel, E.H.; Ripa, R.S.; von Scholten, B.J.; Rotbain Curovic, V.; Kjaer, A.; Hansen, T.W.; Rossing, P.; Størling, J. Effect of Liraglutide on Expression of Inflammatory Genes in Type 2 Diabetes. Sci. Rep. 2021, 11. [Google Scholar] [CrossRef]
- Zobel, E.H.; Ripa, R.S.; von Scholten, B.J.; Curovic, V.R.; Diaz, L.J.; Hansen, T.W.; Rossing, P.; Kjaer, A. Effect of Liraglutide on Vascular Inflammation Evaluated by [64 Cu] DOTATATE. Diagnostics 2021, 11, 1431. [Google Scholar] [CrossRef]
- Savchenko, L.G.; Digtiar, N.I.; Selikhova, L.G.; Kaidasheva, E.I.; Shlykova, O.A.; Vesnina, L.E.; Kaidashev, I.P. Liraglutide Exerts an Anti-Inflammatory Action in Obese Patients with Type 2 Diabetes. Rom. J. Intern. Med. Rev. Roum. Med. Interne 2019, 57, 233–240. [Google Scholar] [CrossRef]
- Ripa, R.S.; Zobel, E.H.; von Scholten, B.J.; Jensen, J.K.; Binderup, T.; Diaz, L.J.; Curovic, V.R.; Hansen, T.W.; Rossing, P.; Kjaer, A. Effect of Liraglutide on Arterial Inflammation Assessed as [18 F] FDG Uptake in Patients With Type 2 Diabetes: A Randomized, Double-Blind, Placebo-Controlled Trial. Circulation. Cardiovasc. Imaging 2021, 14, 632–644. [Google Scholar] [CrossRef]
- Smits, M.M.; Tonneijck, L.; Muskiet, M.H.A.; Hoekstra, T.; Kramer, M.H.H.; Diamant, M.; Serné, E.H.; van Raalte, D.H. GLP-1-Based Therapies Have No Microvascular Effects in Type 2 Diabetes Mellitus: An Acute and 12-Week Randomized, Double-Blind, Placebo-Controlled Trial. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2125–2132. [Google Scholar] [CrossRef] [PubMed]
- Heidari, B.; Lerman, A.; Lalia, A.Z.; Lerman, L.O.; Chang, A.Y. Effect of Metformin on Microvascular Endothelial Function in Polycystic Ovary Syndrome. Mayo Clin. Proc. 2019, 94, 2455–2466. [Google Scholar] [CrossRef]
- Andrews, M.; Soto, N.; Arredondo, M. [Effect of Metformin on the Expression of Tumor Necrosis Factor-α, Toll like Receptors 2/4 and C Reactive Protein in Obese Type-2 Diabetic Patients]. Rev. Med. Chile 2012, 140, 1377–1382. [Google Scholar] [CrossRef] [PubMed]
- Schiapaccassa, A.; Maranhão, P.A.; de Souza, M.D.G.C.; Panazzolo, D.G.; Nogueira Neto, J.F.; Bouskela, E.; Kraemer-Aguiar, L.G. 30-Days Effects of Vildagliptin on Vascular Function, Plasma Viscosity, Inflammation, Oxidative Stress, and Intestinal Peptides on Drug-Naïve Women with Diabetes and Obesity: A Randomized Head-to-Head Metformin-Controlled Study. Diabetol. Metab. Syndr. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Schiappacassa, A.; Maranhão, P.A.; de Souza, M.D.G.C.; Panazzolo, D.G.; Neto, J.F.N.; Bouskela, E.; Kraemer-Aguiar, L.G. Acute Effects of Metformin and Vildagliptin after a Lipid-Rich Meal on Postprandial Microvascular Reactivity in Patients with Type 2 Diabetes and Obesity: A Randomized Trial. J. Clin. Med. 2020, 9, 3228. [Google Scholar] [CrossRef]
- Kitao, N.; Miyoshi, H.; Furumoto, T.; Ono, K.; Nomoto, H.; Miya, A.; Yamamoto, C.; Inoue, A.; Tsuchida, K.; Manda, N.; et al. The Effects of Vildagliptin Compared with Metformin on Vascular Endothelial Function and Metabolic Parameters: A Randomized, Controlled Trial (Sapporo Athero-Incretin Study 3). Cardiovasc. Diabetol. 2017, 16. [Google Scholar] [CrossRef] [PubMed]
- Petrie, J.R.; Chaturvedi, N.; Ford, I.; Brouwers, M.C.G.J.; Greenlaw, N.; Tillin, T.; Hramiak, I.; Hughes, A.D.; Jenkins, A.J.; Klein, B.E.K.; et al. Cardiovascular and Metabolic Effects of Metformin in Patients with Type 1 Diabetes (REMOVAL): A Double-Blind, Randomised, Placebo-Controlled Trial. Lancet. Diabetes Endocrinol. 2017, 5, 597–609. [Google Scholar] [CrossRef]
- Jax, T.; Stirban, A.; Terjung, A.; Esmaeili, H.; Berk, A.; Thiemann, S.; Chilton, R.; Eynatten, M.; Marx, N. A Randomised, Active- and Placebo-Controlled, Three-Period Crossover Trial to Investigate Short-Term Effects of the Dipeptidyl Peptidase-4 Inhibitor Linagliptin on Macro- and Microvascular Endothelial Function in Type 2 Diabetes. Cardiovasc. Diabetol. 2017, 16. [Google Scholar] [CrossRef]
- Gabriel, R.; Abdelkader, N.B.; Acosta, T.; Gilis-Januszewska, A.; Gómez-Huelgas, R.; Makrilakis, K.; Kamenov, Z.; Paulweber, B.; Satman, I.; Djordjevic, P.; et al. Early Prevention of Diabetes Microvascular Complications in People with Hyperglycaemia in Europe. EPREDICE Randomized Trial. Study Protocol, Recruitment and Selected Baseline Data. PLoS ONE 2020, 15. [Google Scholar] [CrossRef]
- Kelly, A.S.; Bergenstal, R.M.; Gonzalez-Campoy, J.M.; Katz, H.; Bank, A.J. Effects of Exenatide vs. Metformin on Endothelial Function in Obese Patients with Pre-Diabetes: A Randomized Trial. Cardiovasc. Diabetol. 2012, 11. [Google Scholar] [CrossRef]
- Hamidi, V.; Riggs, K.; Zhu, L.; Bermudez Saint Andre, K.; Westby, C.; Coverdale, S.; Dursteler, A.; Wang, H.; Miller, C.; Taegtmeyer, H.; et al. Acute Exenatide Therapy Attenuates Postprandial Vasodilation in Humans with Prediabetes: A Randomized Controlled Trial. Metab. Syndr. Relat. Disord. 2020, 18, 225–233. [Google Scholar] [CrossRef]
- Thomas, M.C.; Cherney, D.Z.I. The Actions of SGLT2 Inhibitors on Metabolism, Renal Function and Blood Pressure. Diabetologia 2018, 61, 2098–2107. [Google Scholar] [CrossRef]
- Sun, X.; Han, F.; Lu, Q.; Li, X.; Ren, D.; Zhang, J.; Han, Y.; Xiang, Y.K.; Li, J. Empagliflozin Ameliorates Obesity-Related Cardiac Dysfunction by Regulating Sestrin2-Mediated AMPK-MTOR Signaling and Redox Homeostasis in High-Fat Diet-Induced Obese Mice. Diabetes 2020, 69, 1292–1305. [Google Scholar] [CrossRef]
- Tanaka, A.; Shimabukuro, M.; Machii, N.; Teragawa, H.; Okada, Y.; Shima, K.R.; Takamura, T.; Taguchi, I.; Hisauchi, I.; Toyoda, S.; et al. Secondary Analyses to Assess the Profound Effects of Empagliflozin on Endothelial Function in Patients with Type 2 Diabetes and Established Cardiovascular Diseases: The Placebo-Controlled Double-Blind Randomized Effect of Empagliflozin on Endothelial Function in Cardiovascular High Risk Diabetes Mellitus: Multi-Center Placebo-Controlled Double-Blind Randomized Trial. J. Diabetes Investig. 2020, 11, 1551–1563. [Google Scholar] [CrossRef]
- Zemel, M.B.; Kolterman, O.; Rinella, M.; Vuppalanchi, R.; Flores, O.; Barritt, A.S.; Siddiqui, M.; Chalasani, N. Randomized Controlled Trial of a Leucine-Metformin-Sildenafil Combination (NS-0200) on Weight and Metabolic Parameters. Obesity 2019, 27, 59–67. [Google Scholar] [CrossRef]
- Rebello, C.J.; Zemel, M.B.; Kolterman, O.; Fleming, G.A.; Greenway, F.L. Leucine and Sildenafil Combination Therapy Reduces Body Weight and Metformin Enhances the Effect at Low Dose: A Randomized Controlled Trial. Am. J. Ther. 2021, 28, e1–e13. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, S.P.; Egginton, S.; Madsen, M.; Hansen, J.B.; Munch, G.D.W.; Iepsen, U.W.; Åkerström, T.; Pedersen, B.K.; Hellsten, Y. Alpha Adrenergic Receptor Blockade Increases Capillarization and Fractional O 2 Extraction and Lowers Blood Flow in Contracting Human Skeletal Muscle. Acta Physiol. (Oxf. Engl.) 2017, 221, 32–43. [Google Scholar] [CrossRef]
- Dunford, E.C.; Leclair, E.; Aiken, J.; Mandel, E.R.; Haas, T.L.; Birot, O.; Riddell, M.C. The Effects of Voluntary Exercise and Prazosin on Capillary Rarefaction and Metabolism in Streptozotocin-Induced Diabetic Male Rats. J. Appl. Physiol. 2017, 122, 492. [Google Scholar] [CrossRef]
- Abuissa, H.; Jones, P.G.; Marso, S.P.; O’Keefe, J.H. Angiotensin-Converting Enzyme Inhibitors or Angiotensin Receptor Blockers for Prevention of Type 2 Diabetes: A Meta-Analysis of Randomized Clinical Trials. J. Am. Coll. Cardiol. 2005, 46, 821–826. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.; Nheu, L.; Dai, A.; Guo, Z.; Komesaroff, P. Effects of Four Medicinal Herbs on Human Vascular Endothelial Cells in Culture. Int. J. Cardiol. 2008, 128, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Lv, P.; Tong, X.; Peng, Q.; Liu, Y.; Jin, H.; Liu, R.; Sun, W.; Pan, B.; Zheng, L.; Huang, Y. Treatment with the Herbal Medicine, Naoxintong Improves the Protective Effect of High-Density Lipoproteins on Endothelial Function in Patients with Type 2 Diabetes. Mol. Med. Rep. 2016, 13, 2007–2016. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Khodadadi, S.; Zabihi, N.A.; Niazmand, S.; Abbasnezhad, A.; Mahmoudabady, M.; Rezaee, S.A. Teucrium Polium Improves Endothelial Dysfunction by Regulating ENOS and VCAM-1 Genes Expression and Vasoreactivity in Diabetic Rat Aorta. Biomed. Pharmacother. Biomed. Pharmacother. 2018, 103, 1526–1530. [Google Scholar] [CrossRef]
- Parsamanesh, N.; Asghari, A.; Sardari, S.; Tasbandi, A.; Jamialahmadi, T.; Xu, S.; Sahebkar, A. Resveratrol and Endothelial Function: A Literature Review. Pharmacol. Res. 2021, 170. [Google Scholar] [CrossRef]
- Minozzo, B.R.; Fernandes, D.; Beltrame, F.L. Phenolic Compounds as Arginase Inhibitors: New Insights Regarding Endothelial Dysfunction Treatment. Planta Med. 2018, 84, 277–295. [Google Scholar] [CrossRef] [PubMed]
- Noce, A.; di Lauro, M.; di Daniele, F.; Zaitseva, A.P.; Marrone, G.; Borboni, P.; di Daniele, N. Natural Bioactive Compounds Useful in Clinical Management of Metabolic Syndrome. Nutrients 2021, 13, 630. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Wang, X.; Feng, W.; Liu, Q.; Zhou, S.; Liu, Q.; Cai, L. The Gut Microbiota and Its Interactions with Cardiovascular Disease. Microb. Biotechnol. 2020, 13, 637–656. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ugwoke, C.K.; Cvetko, E.; Umek, N. Skeletal Muscle Microvascular Dysfunction in Obesity-Related Insulin Resistance: Pathophysiological Mechanisms and Therapeutic Perspectives. Int. J. Mol. Sci. 2022, 23, 847. https://doi.org/10.3390/ijms23020847
Ugwoke CK, Cvetko E, Umek N. Skeletal Muscle Microvascular Dysfunction in Obesity-Related Insulin Resistance: Pathophysiological Mechanisms and Therapeutic Perspectives. International Journal of Molecular Sciences. 2022; 23(2):847. https://doi.org/10.3390/ijms23020847
Chicago/Turabian StyleUgwoke, Chiedozie Kenneth, Erika Cvetko, and Nejc Umek. 2022. "Skeletal Muscle Microvascular Dysfunction in Obesity-Related Insulin Resistance: Pathophysiological Mechanisms and Therapeutic Perspectives" International Journal of Molecular Sciences 23, no. 2: 847. https://doi.org/10.3390/ijms23020847
APA StyleUgwoke, C. K., Cvetko, E., & Umek, N. (2022). Skeletal Muscle Microvascular Dysfunction in Obesity-Related Insulin Resistance: Pathophysiological Mechanisms and Therapeutic Perspectives. International Journal of Molecular Sciences, 23(2), 847. https://doi.org/10.3390/ijms23020847