
Computational Design of Miniproteins as SARS-CoV-2 Therapeutic Inhibitors



Abstract
:1. Introduction
2. Results
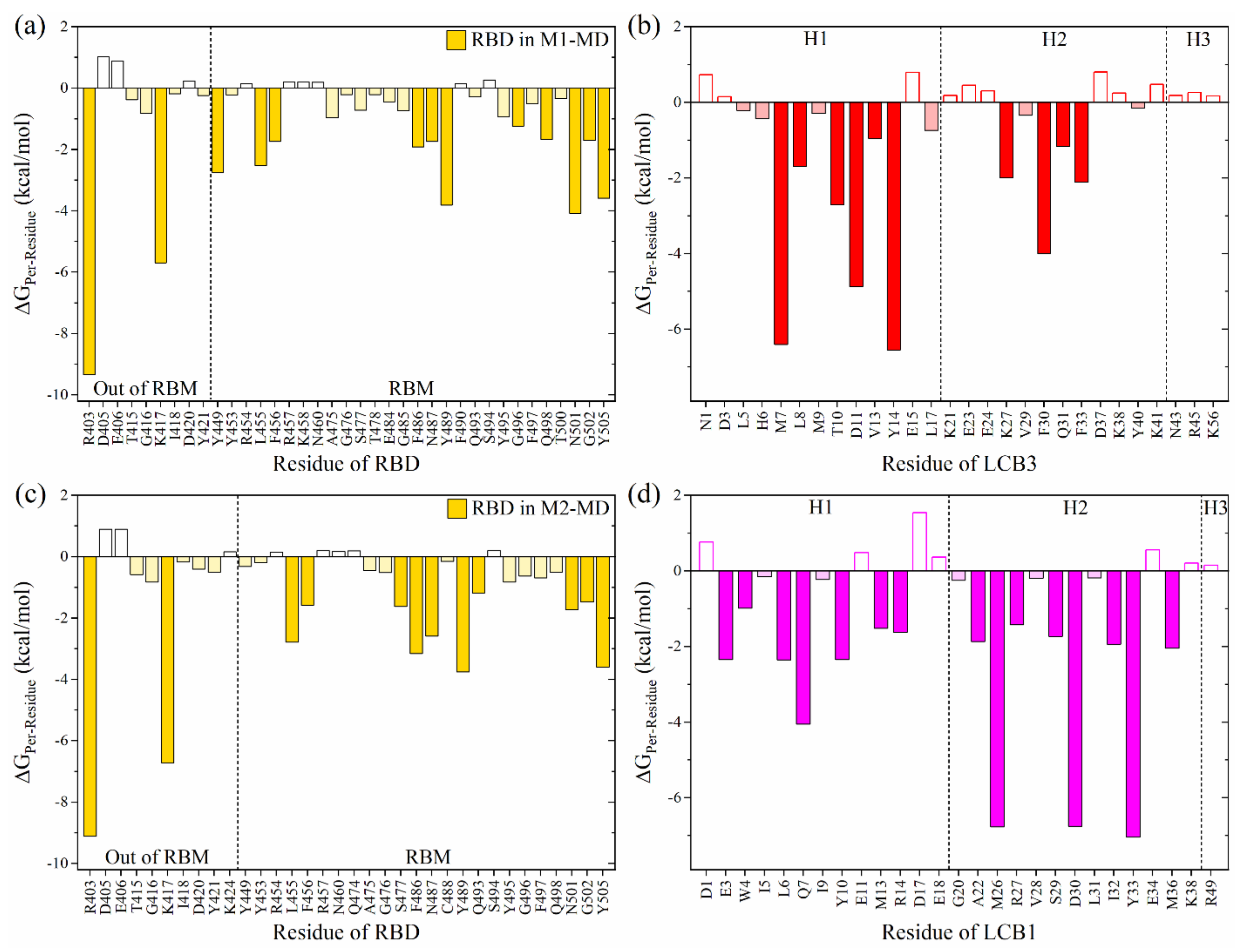
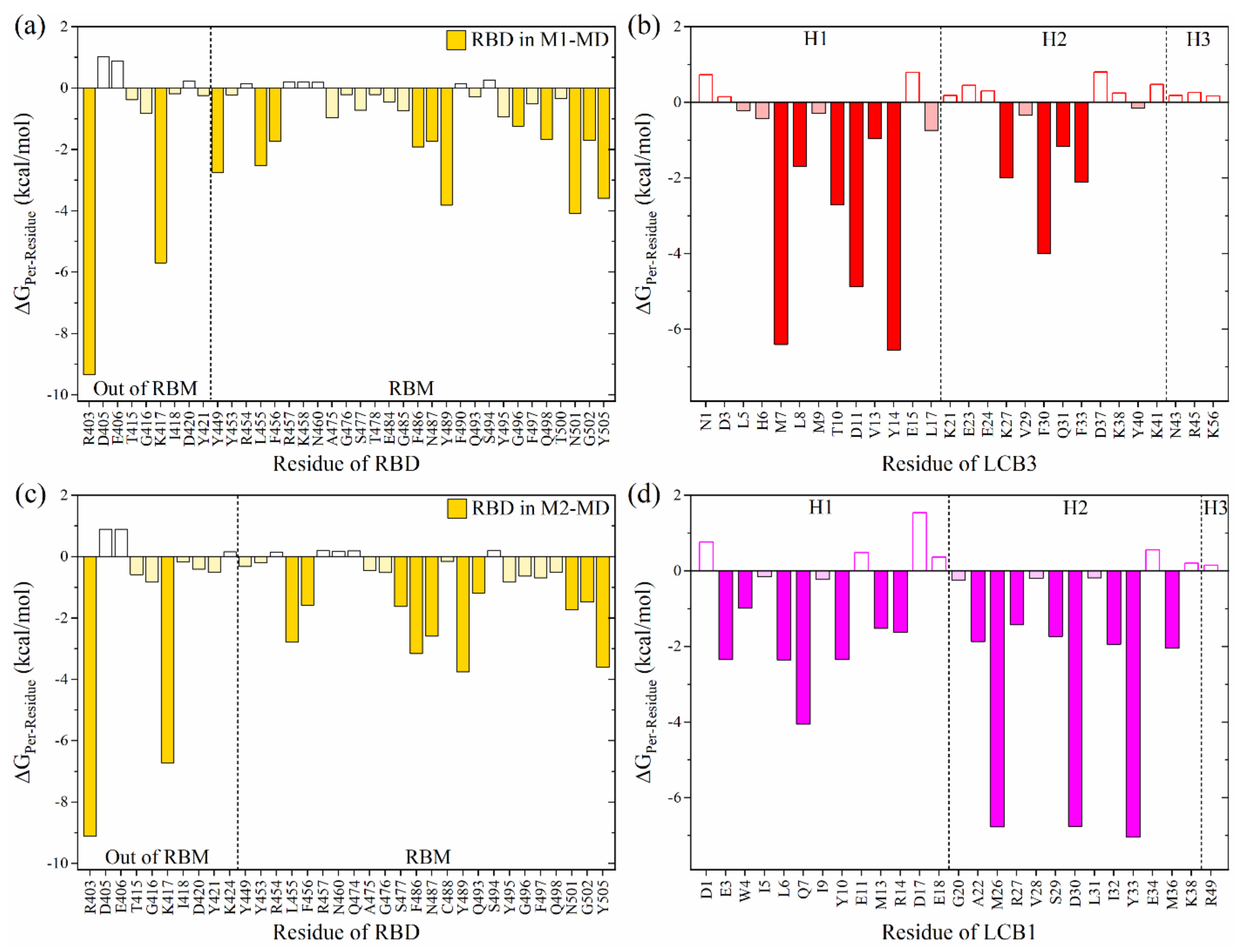
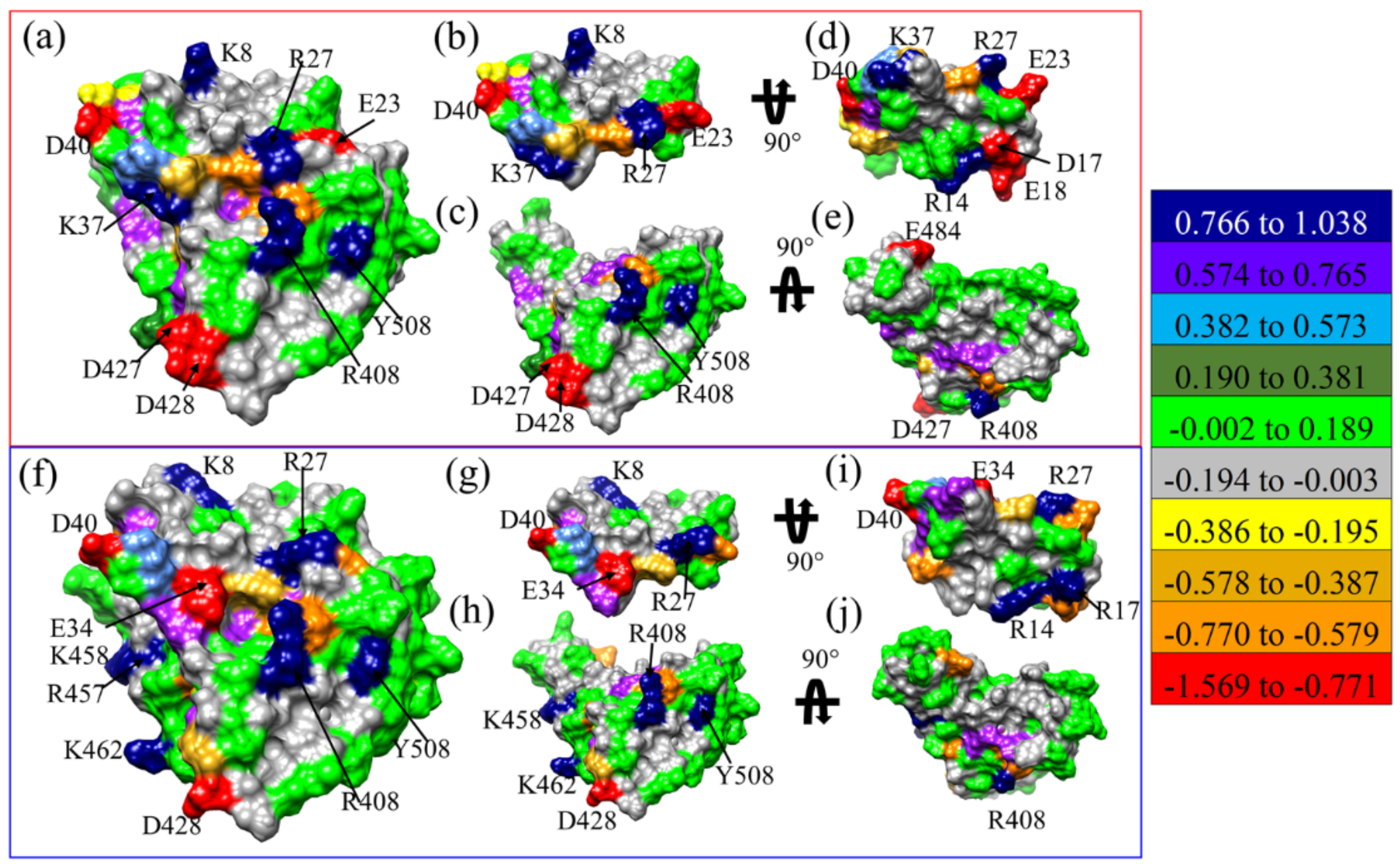
2.1. Binding of LCB1 and LCB3 to RBD SARS-CoV-2
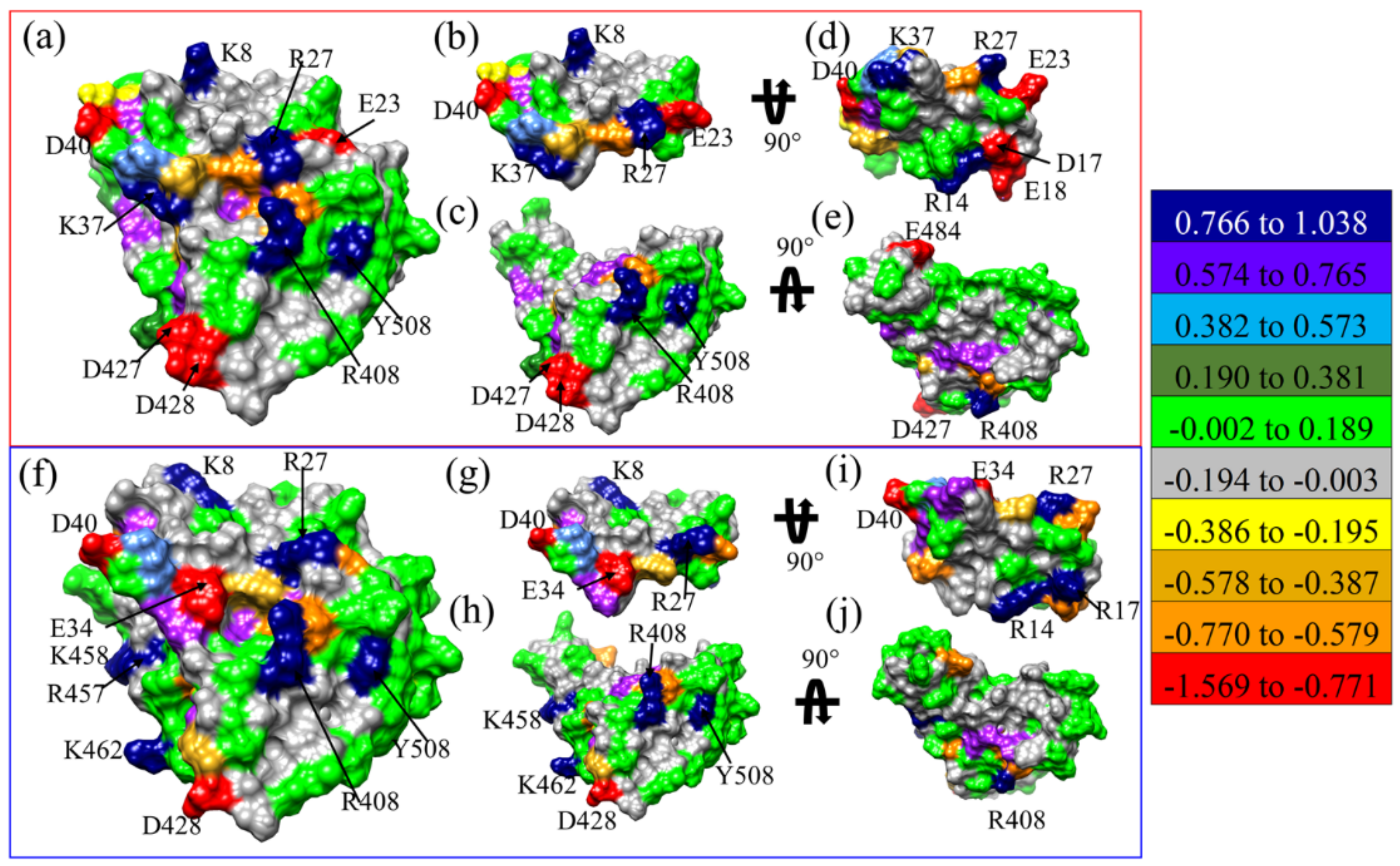
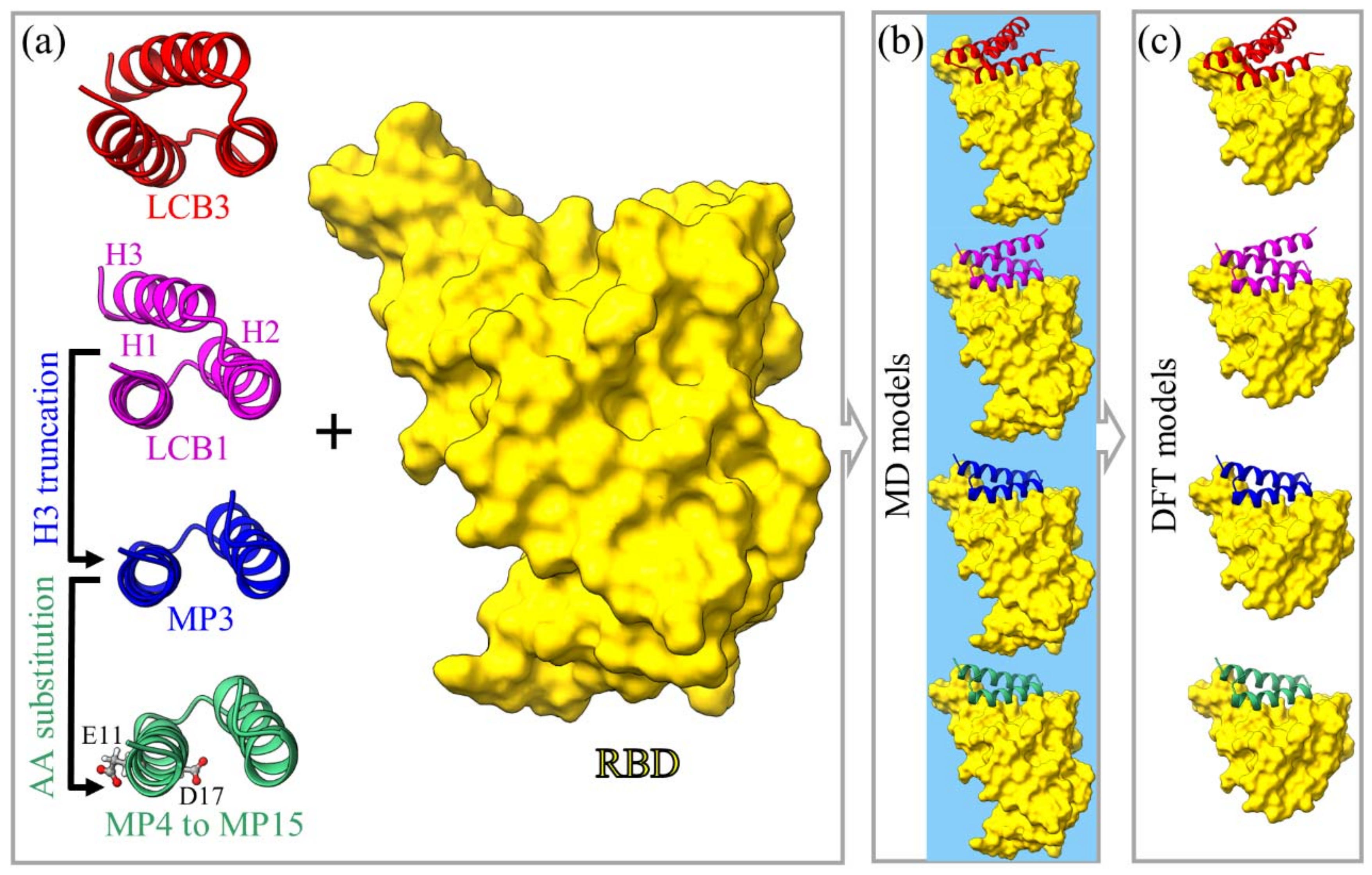
2.2. Structural Modification on LCB1
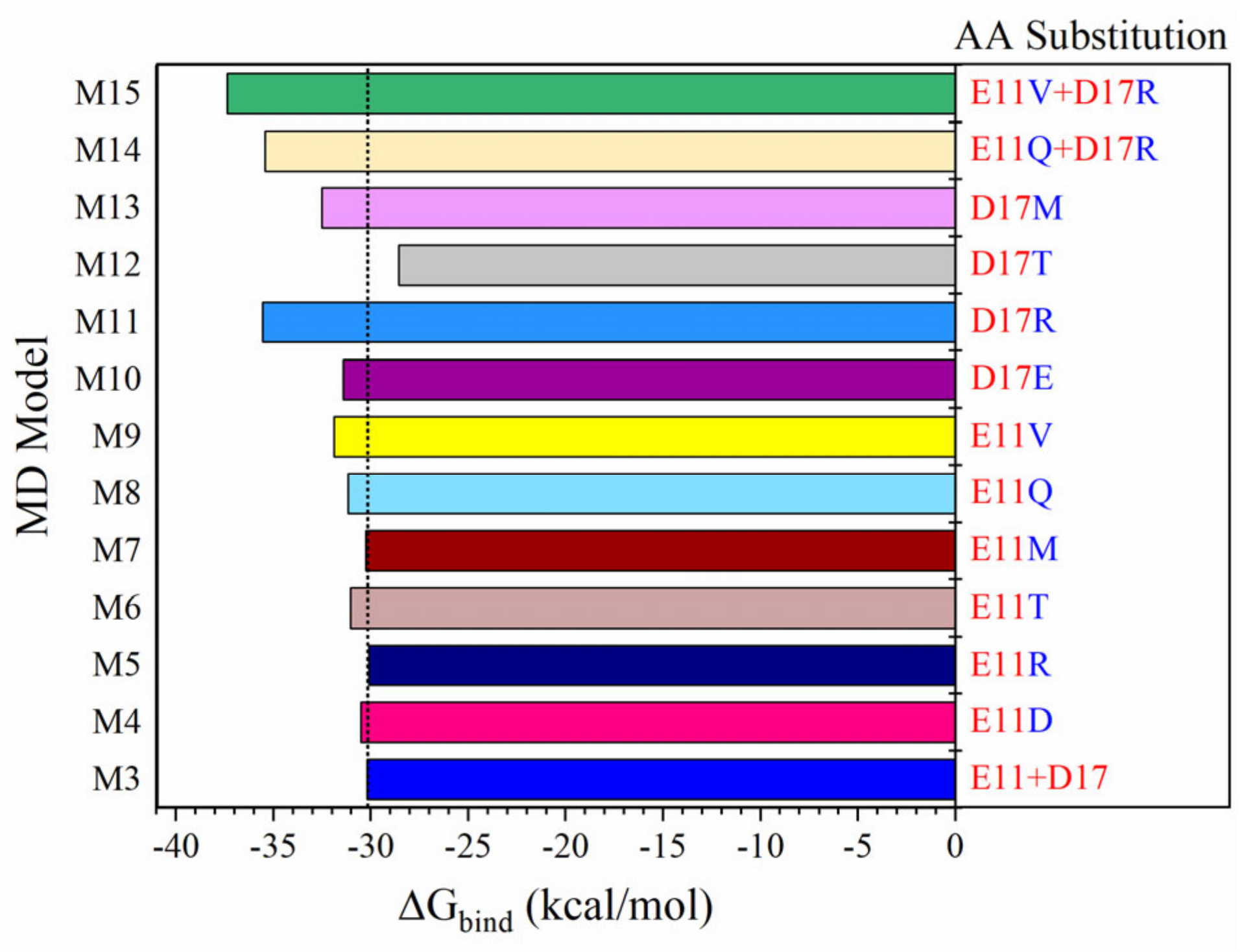
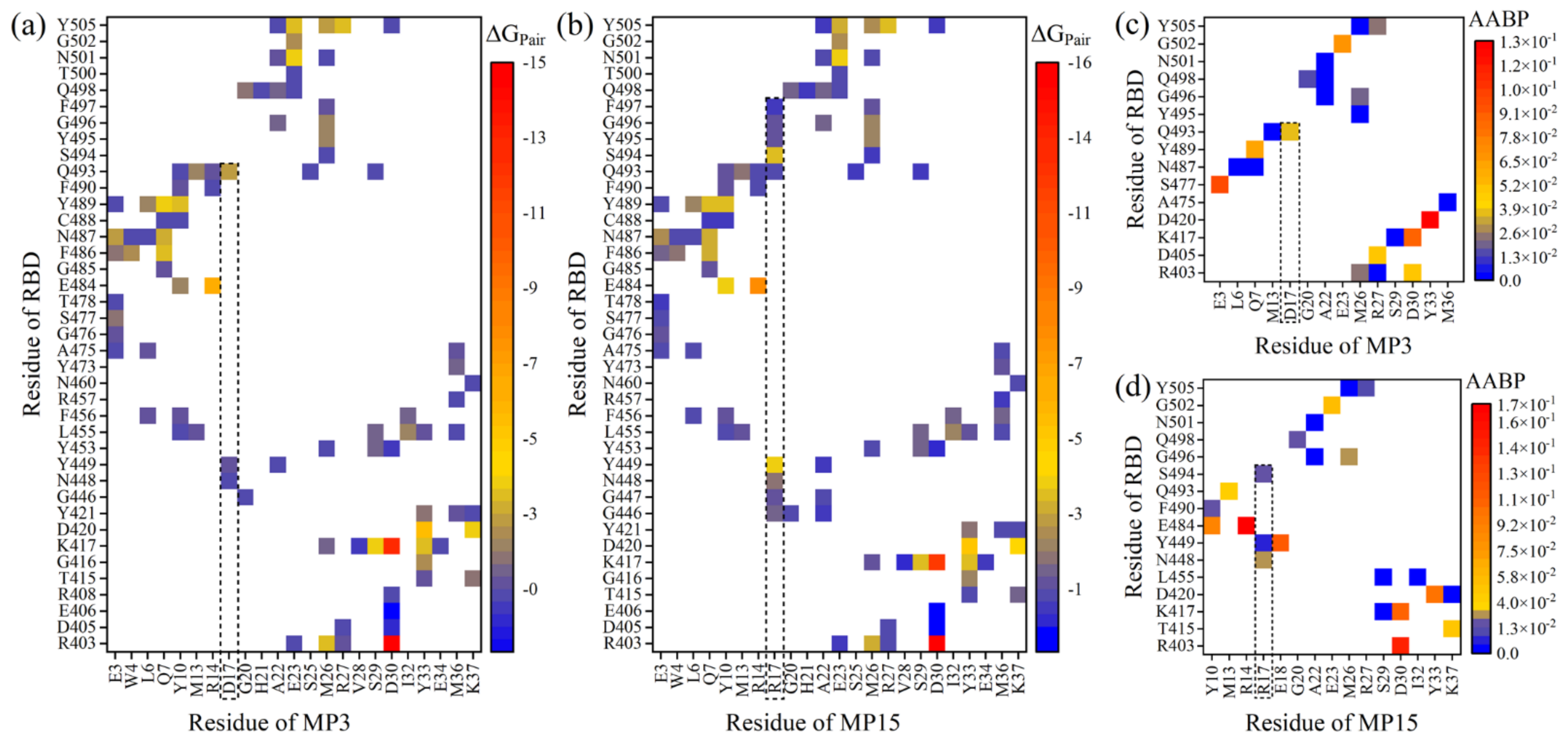
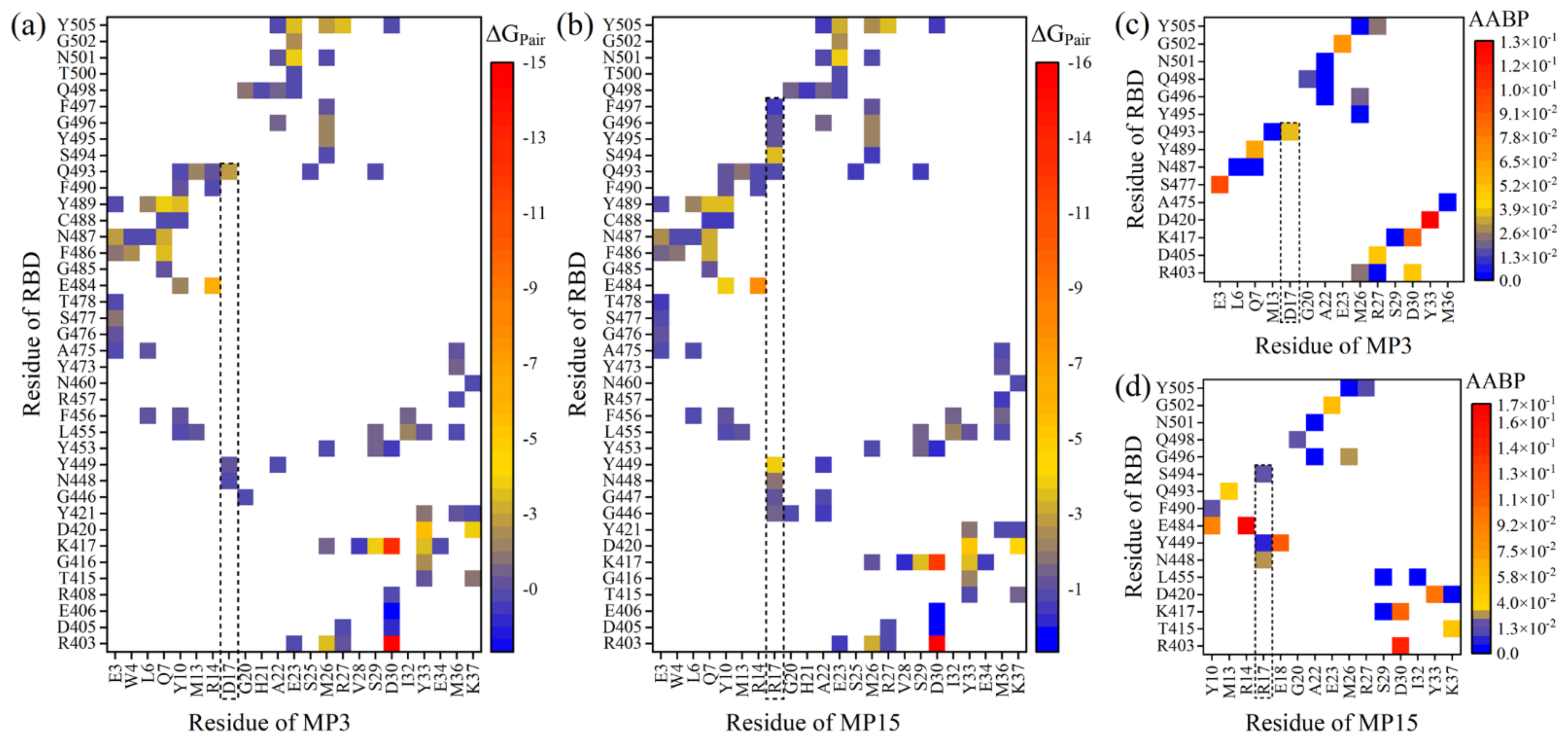
2.3. Amino Acid Substitutions on MP3
3. Discussion
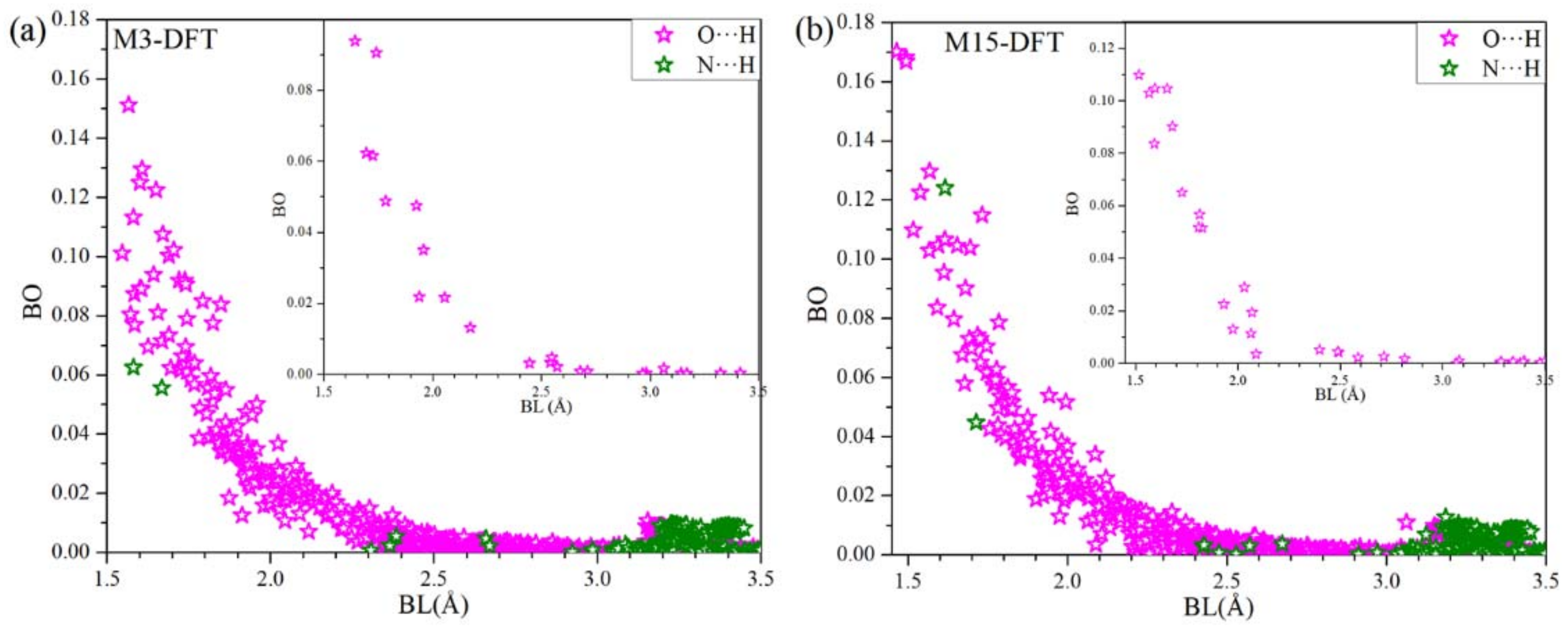
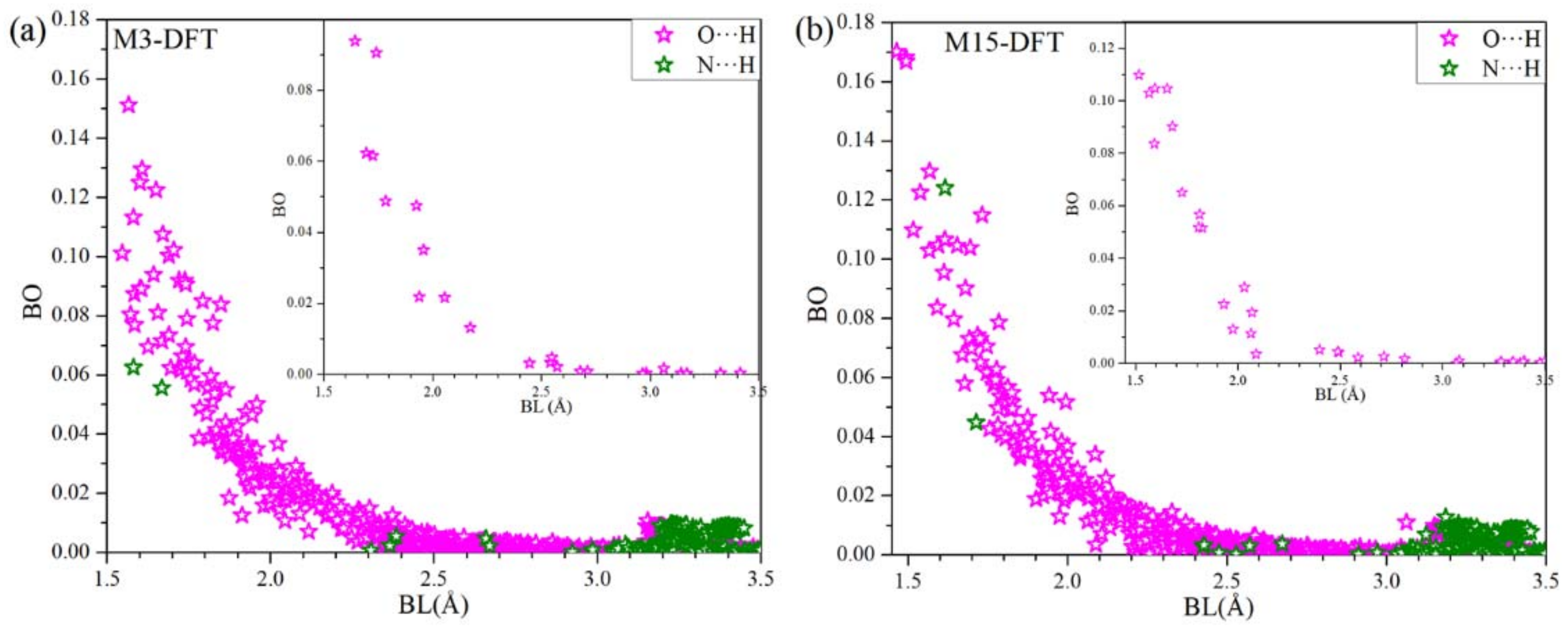
3.1. DFT Results
3.2. Combination of MD and DFT Results
3.3. Potential Connection to Experimental Verifications
4. Materials and Methods
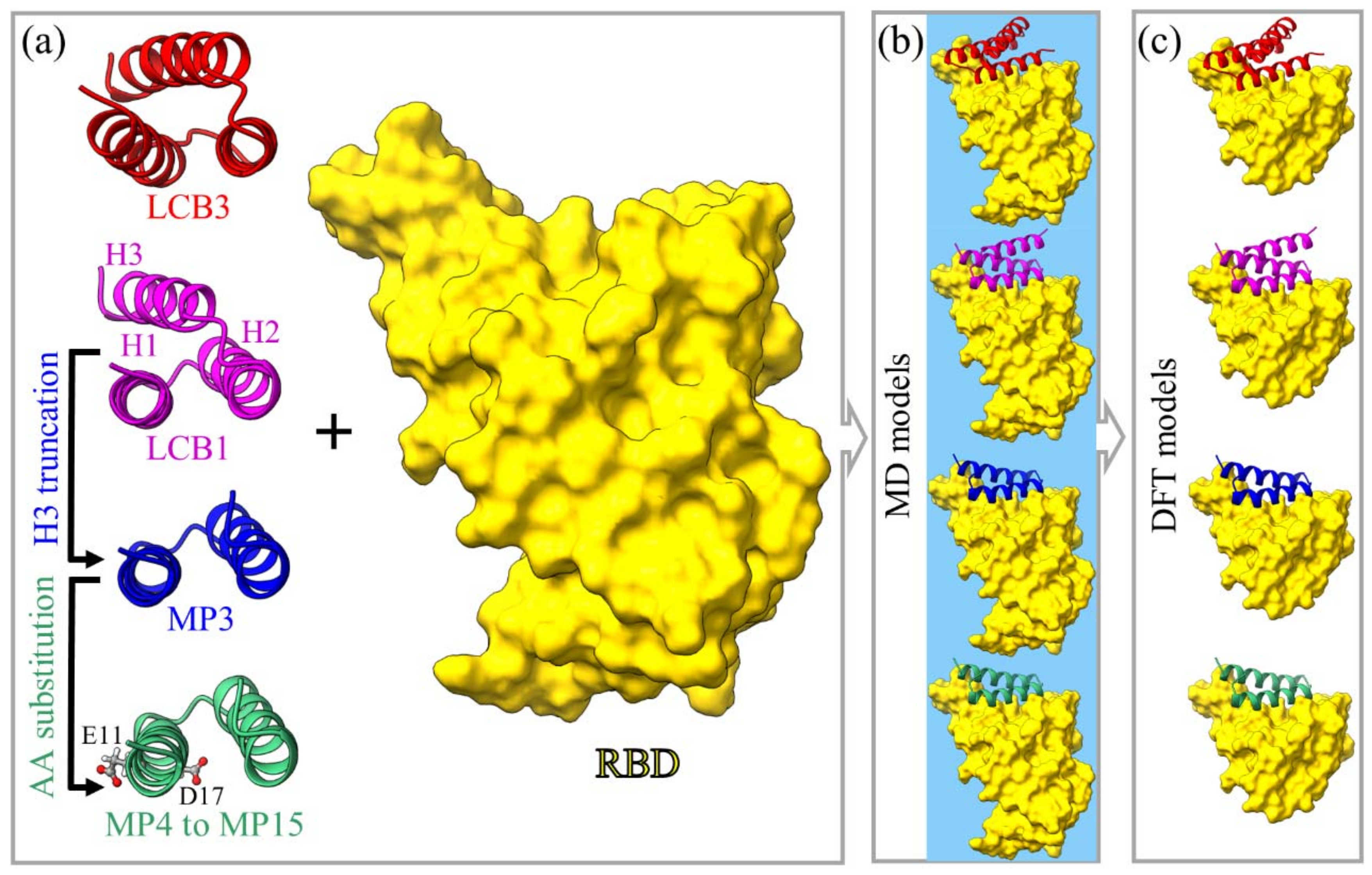
4.1. Model Construction
- i.
- The initial models for LCB1:RBD and LCB3:RBD complexes are similar to [20] but without an extra AA in RBD (195 vs. 194 AAs of 6M0J) and with protonated cysteine residues. The structure comparison tool in the UCSF Chimera software [49] is used to align the RBD in 6M0J with LCB1:RBD or LCB3:RBD of [20]. The RBDs are then replaced by the ones from the 6M0J.
- ii.
- Explicit H atoms are added to the saved structure using LEaP module in AMBER [26].
- iii.
- iv.
- The same procedures are adopted to generate the new design for the other minibinder:RBD complex. The details of these constructions are illustrated in Figure 6, Table 2, and Table S2 in Supplementary Materials.
4.1.1. Models M1 and M2
4.1.2. Model M3
4.1.3. Models M4 to M15
4.2. Methods
5. Conclusions
- i.
- From detailed and systematic MD simulations at the microsecond time scale, the complete energetic profile and interaction spectrum of the miniprotein:RBD complexes have been obtained, suggesting that either the original miniproteins (LCB3 or LCB1) or the designed ones, obtained from the LCB1, can compete for ACE2 binding due to their high binding affinity with RBD and selectivity to occupy the binding site of ACE2 on RBD.
- ii.
- Truncation of the alpha-helix 3 (H3) of LCB1 results in the development of a small candidate (MP3) with a better binding profile to RBD. Additionally, amino acid substitutions at residues 11 and 17 of MP3 enhance its binding more, especially D17R. The D17R substitution shows significant change in PC, which could be the reason behind their enhanced binding.
- iii.
- Since this work is limited to only the RBD of wild-type SARS-CoV-2, we plan to investigate the ability of the best candidate from this study (MP15) to inhibit the RBD of existing SARS-CoV-2 variants, particularly Omicron RBD.
- iv.
- Because of the computational demands, amino acid substitutions of MP3 are restricted to a few AAs at only two sites (11 and 17), which do not cover every residue of MP3 and potential substituted of the 20 AAs at each one.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jackson, L.A.; Anderson, E.J.; Rouphael, N.G.; Roberts, P.C.; Makhene, M.; Coler, R.N.; McCullough, M.P.; Chappell, J.D.; Denison, M.R.; Stevens, L.J.; et al. An mRNA Vaccine against SARS-CoV-2—Preliminary Report. N. Engl. J. Med. 2020, 383, 1920–1931. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.-H.; Patel, N.; Haupt, R.; Zhou, H.; Weston, S.; Hammond, H.; Logue, J.; Portnoff, A.D.; Norton, J.; Guebre-Xabier, M.; et al. SARS-CoV-2 spike glycoprotein vaccine candidate NVX-CoV2373 immunogenicity in baboons and protection in mice. Nat. Commun. 2021, 12, 372. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.B.; Kanevsky, I.; Che, Y.; Swanson, K.A.; Muik, A.; Vormehr, M.; Kranz, L.M.; Walzer, K.C.; Hein, S.; Güler, A.; et al. BNT162b vaccines protect rhesus macaques from SARS-CoV-2. Nature 2021, 592, 283–289. [Google Scholar] [CrossRef]
- Panda, P.K.; Arul, M.N.; Patel, P.; Verma, S.K.; Luo, W.; Rubahn, H.-G.; Mishra, Y.K.; Suar, M.; Ahuja, R. Structure-based drug designing and immunoinformatics approach for SARS-CoV-2. Sci. Adv. 2020, 6, eabb8097. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.-F.; Sun, C.; Zhuang, Z.; Yuan, R.-Y.; Zheng, Q.; Li, J.-P.; Zhou, P.-P.; Chen, X.-C.; Liu, Z.; Zhang, X.; et al. Rapid Development of SARS-CoV-2 Spike Protein Receptor-Binding Domain Self-Assembled Nanoparticle Vaccine Candidates. ACS Nano 2021, 15, 2738–2752. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6, Erratum in Cell 2020, 183, 1735. [Google Scholar] [CrossRef]
- Wang, G.; Yang, M.-L.; Duan, Z.-L.; Liu, F.-L.; Jin, L.; Long, C.-B.; Zhang, M.; Tang, X.-P.; Xu, L.; Li, Y.-C.; et al. Dalbavancin binds ACE2 to block its interaction with SARS-CoV-2 spike protein and is effective in inhibiting SARS-CoV-2 infection in animal models. Cell Res. 2021, 31, 17–24. [Google Scholar] [CrossRef]
- Xiang, Y.; Nambulli, S.; Xiao, Z.; Liu, H.; Sang, Z.; Duprex, W.P.; Schneidman-Duhovny, D.; Zhang, C.; Shi, Y. Versatile and multivalent nanobodies efficiently neutralize SARS-CoV-2. Science 2020, 370, 1479–1484. [Google Scholar] [CrossRef]
- Wang, C.; Li, W.; Drabek, D.; Okba, N.M.A.; van Haperen, R.; Osterhaus, A.D.M.E.; van Kuppeveld, F.J.M.; Haagmans, B.L.; Grosveld, F.; Bosch, B.-J. A human monoclonal antibody blocking SARS-CoV-2 infection. Nat. Commun. 2020, 11, 2251. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wang, P.; Nair, M.S.; Yu, J.; Rapp, M.; Wang, Q.; Luo, Y.; Chan, J.F.; Sahi, V.; Figueroa, A.; et al. Potent neutralizing antibodies against multiple epitopes on SARS-CoV-2 spike. Nature 2020, 584, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, P.J.M.; Caniels, T.G.; van der Straten, K.; Snitselaar, J.L.; Aldon, Y.; Bangaru, S.; Torres, J.L.; Okba, N.M.A.; Claireaux, M.; Kerster, G.; et al. Potent neutralizing antibodies from COVID-19 patients define multiple targets of vulnerability. Science 2020, 369, 643–650. [Google Scholar] [CrossRef]
- LaRue, R.C.; Xing, E.; Kenney, A.D.; Zhang, Y.; Tuazon, J.A.; Li, J.; Yount, J.S.; Li, P.-K.; Sharma, A. Rationally Designed ACE2-Derived Peptides Inhibit SARS-CoV-2. Bioconjug. Chem. 2021, 32, 215–223. [Google Scholar] [CrossRef]
- Han, Y.; Král, P. Computational Design of ACE2-Based Peptide Inhibitors of SARS-CoV-2. ACS Nano 2020, 14, 5143–5147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamson, C.S.; Chibale, K.; Goss, R.J.M.; Jaspars, M.; Newman, D.J.; Dorrington, R.A. Antiviral Drug Discovery: Preparing for the next Pandemic. Chem. Soc. Rev. 2021, 50, 3647–3655. [Google Scholar] [CrossRef]
- Vanpatten, S.; He, M.; Altiti, A.; Cheng, K.F.; Ghanem, M.H.; Al-Abed, Y. Evidence Supporting the Use of Peptides and Peptidomimetics as Potential SARS-CoV-2 (COVID-19) Therapeutics. Future Med. Chem. 2020, 12, 1647–1656. [Google Scholar] [CrossRef] [PubMed]
- Marovich, M.; Mascola, J.R.; Cohen, M.S. Monoclonal Antibodies for Prevention and Treatment of COVID-19. JAMA 2020, 324, 131–132. [Google Scholar] [CrossRef]
- Arvin, A.M.; Fink, K.; Schmid, M.A.; Cathcart, A.; Spreafico, R.; Havenar-Daughton, C.; Lanzavecchia, A.; Corti, D.; Virgin, H.W. A Perspective on Potential Antibody-Dependent Enhancement of SARS-CoV-2. Nature 2020, 584, 353–363. [Google Scholar] [CrossRef]
- Baum, A.; Fulton, B.O.; Wloga, E.; Copin, R.; Pascal, K.E.; Russo, V.; Giordano, S.; Lanza, K.; Negron, N.; Ni, M.; et al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science 2020, 369, 1014–1018. [Google Scholar] [CrossRef]
- Cao, L.; Goreshnik, I.; Coventry, B.; Case, J.B.; Miller, L.; Kozodoy, L.; Chen, R.E.; Carter, L.; Walls, A.C.; Park, Y.-J.; et al. De novo design of picomolar SARS-CoV-2 miniprotein inhibitors. Science 2020, 370, 426–431. [Google Scholar] [CrossRef]
- Matrajt, L.; Eaton, J.; Leung, T.; Brown, E.R. Vaccine optimization for COVID-19: Who to vaccinate first? Sci. Adv. 2021, 7, eabf1374. [Google Scholar] [CrossRef] [PubMed]
- Kyriakidis, N.C.; López-Cortés, A.; González, E.V.; Grimaldos, A.B.; Prado, E.O. SARS-CoV-2 Vaccines Strategies: A Comprehensive Review of Phase 3 Candidates. NPJ Vaccines 2021, 6, 28. [Google Scholar] [CrossRef] [PubMed]
- Hacisuleyman, E.; Hale, C.; Saito, Y.; Blachere, N.E.; Bergh, M.; Conlon, E.G.; Schaefer-Babajew, D.J.; DaSilva, J.; Muecksch, F.; Gaebler, C.; et al. Vaccine Breakthrough Infections with SARS-CoV-2 Variants. N. Engl. J. Med. 2021, 384, 2212–2218. [Google Scholar] [CrossRef] [PubMed]
- Linsky, T.W.; Vergara, R.; Codina, N.; Nelson, J.W.; Walker, M.J.; Su, W.; Barnes, C.O.; Hsiang, T.-Y.; Esser-Nobis, K.; Yu, K.; et al. De novo design of potent and resilient hACE2 decoys to neutralize SARS-CoV-2. Science 2020, 370, eabe0075. [Google Scholar] [CrossRef] [PubMed]
- Schütz, D.; Ruiz-Blanco, Y.B.; Münch, J.; Kirchhoff, F.; Sanchez-Garcia, E.; Müller, J.A. Peptide and Peptide-Based Inhibitors of SARS-CoV-2 Entry. Adv. Drug Deliv. Rev. 2020, 167, 47–65. [Google Scholar] [CrossRef]
- Pearlman, D.A.; Case, D.A.; Caldwell, J.W.; Ross, W.S.; Cheatham, T.E., III; DeBolt, S.; Ferguson, D.; Seibel, G.; Kollman, P. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun. 1995, 91, 1–41. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initiomolecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes forab initiototal-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Ching, W.Y.; Rulis, P. Electronic Structure Methods for Complex Materials: The Orthogonalized Linear Combination of Atomic Orbitals; Oxford University Press: Oxford, UK, 2012. [Google Scholar]
- Jawad, B.; Adhikari, P.; Podgornik, R.; Ching, W.-Y. Key Interacting Residues between RBD of SARS-CoV-2 and ACE2 Receptor: Combination of Molecular Dynamics Simulation and Density Functional Calculation. J. Chem. Inf. Model. 2021, 61, 4425–4441. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, M.; Gao, J. Enhanced receptor binding of SARS-CoV-2 through networks of hydrogen-bonding and hydrophobic interactions. Proc. Natl. Acad. Sci. USA 2020, 117, 13967–13974. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Hilton, S.K.; Ellis, D.; Crawford, K.H.; Dingens, A.S.; Navarro, M.J.; Bowen, J.E.; Tortorici, M.A.; Walls, A.C.; et al. Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding. Cell 2020, 182, 1295–1310.e20. [Google Scholar] [CrossRef] [PubMed]
- Laurini, E.; Marson, D.; Aulic, S.; Fermeglia, A.; Pricl, S. Computational Mutagenesis at the SARS-CoV-2 Spike Protein/Angiotensin-Converting Enzyme 2 Binding Interface: Comparison with Experimental Evidence. ACS Nano 2021, 15, 6929–6948. [Google Scholar] [CrossRef]
- Adhikary, P.; Kandel, S.; Mamani, U.; Mustafa, B.; Hao, S.; Qiu, J.; Fetse, J.; Liu, Y.; Ibrahim, N.M.; Li, Y.; et al. Discovery of Small Anti-ACE2 Peptides to Inhibit SARS-CoV-2 Infectivity. Adv. Ther. 2021, 4, 2100087. [Google Scholar] [CrossRef] [PubMed]
- Muttenthaler, M.; King, G.F.; Adams, D.J.; Alewood, P.F. Trends in peptide drug discovery. Nat. Rev. Drug Discov. 2021, 20, 309–325. [Google Scholar] [CrossRef]
- Pierce, B.G.; Wiehe, K.; Hwang, H.; Kim, B.-H.; Vreven, T.; Weng, Z. ZDOCK server: Interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics 2014, 30, 1771–1773. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Hebditch, M.; Carballo-Amador, M.A.; Charonis, S.; Curtis, R.; Warwicker, J. Protein–Sol: A web tool for predicting protein solubility from sequence. Bioinformatics 2017, 33, 3098–3100. [Google Scholar] [CrossRef] [Green Version]
- Pucci, F.; Kwasigroch, J.M.; Rooman, M. SCooP: An accurate and fast predictor of protein stability curves as a function of temperature. Bioinformatics 2017, 33, 3415–3422. [Google Scholar] [CrossRef]
- Senn, H.M.; Thiel, W. QM/MM Methods for Biomolecular Systems. Angew. Chem.-Int. Ed. 2009, 48, 1198–1229. [Google Scholar] [CrossRef]
- Sousa, S.F.; Ribeiro, A.J.M.; Neves, R.P.P.; Bras, N.; Cerqueira, N.M.F.S.A.; Fernandes, P.; Ramos, M.J. Application of quantum mechanics/molecular mechanics methods in the study of enzymatic reaction mechanisms. WIREs Comput. Mol. Sci. 2017, 7, e1281. [Google Scholar] [CrossRef]
- Cui, Q.; Pal, T.; Xie, L. Biomolecular QM/MM Simulations: What Are Some of the “Burning Issues”? J. Phys. Chem. B 2021, 125, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, S.A.; Dror, R.O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef] [Green Version]
- Ratcliff, L.E.; Mohr, S.; Huhs, G.; Deutsch, T.; Masella, M.; Genovese, L. Challenges in large scale quantum mechanical calculations. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2017, 7, e1290. [Google Scholar] [CrossRef] [Green Version]
- Adhikari, P.; Ching, W.-Y. Amino acid interacting network in the receptor-binding domain of SARS-CoV-2 spike protein. RSC Adv. 2020, 10, 39831–39841. [Google Scholar] [CrossRef]
- Liu, H.; Zhao, Z.; Zhang, L.; Li, Y.; Jain, A.; Barve, A.; Jin, W.; Liu, Y.; Fetse, J.; Cheng, K. Discovery of low-molecular weight anti-PD-L1 peptides for cancer immunotherapy. J. Immunother. Cancer 2019, 7, 270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Huang, C.C.; Ferrin, T.E. Tools for integrated sequence-structure analysis with UCSF Chimera. BMC Bioinform. 2006, 7, 339. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Shapovalov, M.V.; Dunbrack, R.L. A Smoothed Backbone-Dependent Rotamer Library for Proteins Derived from Adaptive Kernel Density Estimates and Regressions. Structure 2011, 19, 844–858. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Miller, I.B.R.; McGee, J.T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Jawad, B.; Poudel, L.; Podgornik, R.; Steinmetz, N.F.; Ching, W.-Y. Molecular mechanism and binding free energy of doxorubicin intercalation in DNA. Phys. Chem. Chem. Phys. 2019, 21, 3877–3893. [Google Scholar] [CrossRef]
- Jawad, B.; Poudel, L.; Podgornik, R.; Ching, W.-Y. Thermodynamic dissection of the intercalation binding process of doxorubicin to dsDNA with implications of ionic and solvent effects. J. Phys. Chem. B 2020, 124, 7803–7818. [Google Scholar] [CrossRef]
- Leelananda, S.P.; Lindert, S. Computational methods in drug discovery. Beilstein J. Org. Chem. 2016, 12, 2694–2718. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, B.; Gerber, P.; Schulz-Gasch, T.; Stahl, M. Validation and Use of the MM-PBSA Approach for Drug Discovery. J. Med. Chem. 2005, 48, 4040–4048. [Google Scholar] [CrossRef]
- Lionta, E.; Spyrou, G.; Vassilatis, D.K.; Cournia, Z. Structure-Based Virtual Screening for Drug Discovery: Principles, Applications and Recent Advances. Curr. Top. Med. Chem. 2014, 14, 1923–1938. [Google Scholar] [CrossRef]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef]
- Durrant, J.D.; McCammon, J.A. Molecular dynamics simulations and drug discovery. BMC Biol. 2011, 9, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borhani, D.W.; Shaw, D.E. The future of molecular dynamics simulations in drug discovery. J. Comput.-Aided Mol. Des. 2012, 26, 15–26. [Google Scholar] [CrossRef] [Green Version]
- Schames, J.R.; Henchman, R.H.; Siegel, J.S.; Sotriffer, C.A.; Ni, A.H.; McCammon, J.A. Discovery of a Novel Binding Trench in HIV Integrase. J. Med. Chem. 2004, 47, 1879–1881. [Google Scholar] [CrossRef]
- Summa, V.; Petrocchi, A.; Bonelli, F.; Crescenzi, B.; Donghi, M.; Ferrara, M.; Fiore, F.; Gardelli, C.; Gonzalez Paz, O.; Hazuda, D.J.; et al. Discovery of Raltegravir, a Potent, Selective Orally Bioavailable HIV-Integrase Inhibitor for the Treatment of HIV-AIDS Infection. J. Med. Chem. 2008, 51, 5843–5855. [Google Scholar] [CrossRef]
- Athanasiou, C.; Cournia, Z. From Computers to Bedside: Computational Chemistry Contributing to FDA Approval. In Biomolecular Simulations in Structure-Based Drug Discovery; John Wiley & Sons: New York, NY, USA, 2018; pp. 163–203. [Google Scholar] [CrossRef]
- Adhikari, P.; Li, N.; Shin, M.; Steinmetz, N.F.; Twarock, R.; Podgornik, R.; Ching, W.-Y. Intra- and intermolecular atomic-scale interactions in the receptor binding domain of SARS-CoV-2 spike protein: Implication for ACE2 receptor binding. Phys. Chem. Chem. Phys. 2020, 22, 18272–18283. [Google Scholar] [CrossRef]
- Ching, W.-Y.; Adhikari, P.; Jawad, B.; Podgornik, R. Ultra-large-scale ab initio quantum chemical computation of bio-molecular systems: The case of spike protein of SARS-CoV-2 virus. Comput. Struct. Biotechnol. J. 2021, 19, 1288–1301. [Google Scholar] [CrossRef]
- Adhikari, P.; Podgornik, R.; Jawad, B.; Ching, W.-Y. First-Principles Simulation of Dielectric Function in Biomolecules. Materials 2021, 14, 5774. [Google Scholar] [CrossRef]
- Baral, K.; Adhikari, P.; Jawad, B.; Podgornik, R.; Ching, W.-Y. Solvent Effect on the Structure and Properties of RGD Peptide (1FUV) at Body Temperature (310 K) Using Ab Initio Molecular Dynamics. Polymers 2021, 13, 3434. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, P.; Jawad, B.; Rao, P.; Podgornik, R.; Ching, W. Delta Variant with P681R Critical Mutation Revealed by Ultra-Large Atomic-Scale Ab Initio Simulation: Implications for the Fundamentals of Biomolecular Interactions. bioRxiv 2021, 1–18. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Energy | M1-MD (SEM) | M2-MD (SEM) | ΔΔG(M2-M1) |
|---|---|---|---|
| ΔEvdW | −93.26 (0.13) | −91.32 (0.10) | 1.95 |
| ΔEele | −492.67 (0.62) | −516.55 (0.58) | −23.88 |
| ΔEMM | −585.93 (0.66) | −607.86 (0.59) | −21.93 |
| ΔGGB | 527.00 (0.57) | 540.09 (0.55) | 13.09 |
| ΔGSA | −14.36 (0.02) | −13.86 (0.01) | 0.50 |
| ΔGsol | 512.63 (0.57) | 526.22 (0.55) | 13.59 |
| ΔGele 1 | 34.32 (0.142) | 23.54 (0.12) | −10.78 |
| −TΔS | −47.82 (1.50) | −52.91 (1.05) | −5.09 |
| ΔGbind | −25.48 (0.60) | −28.73 (0.51) | −3.25 |
| Model 1 | Total Atoms Number | MP | MP Sequence |
|---|---|---|---|
| M1-MD | 34099 | MP1 (LCB3) | NDDELHMLMTDLVYEALHFAKDEEIKKRVFQLFELADKAYKNNDRQKLEKVVEELKELLERLLS |
| M1(a)-DFT | 2773 | ||
| M1(b)-DFT | 2773 | ||
| M2-MD | 33961 | MP2 (LCB1) | DKEWILQKIYEIMRLLDELGHAEASMRVSDLIYEFMKKGD ERLLEEAERLLEEVER |
| M2-DFT | 2635 | ||
| M3-MD | 33678 | MP3 | DKEWILQKIYEIMRLLDELGHAEASMRVSDLIYEFMKKGD |
| M3-DFT | 2352 | ||
| M4-MD | 33675 | MP4 | DKEWILQKIYEIMRLLDELGHAEASMRVSDLIYEFMKKGD |
| M5-MD | 33687 | MP5 | DKEWILQKIYRIMRLLDELGHAEASMRVSDLIYEFMKKGD |
| M6-MD | 33676 | MP6 | DKEWILQKIYDIMRLLDELGHAEASMRVSDLIYEFMKKGD |
| M7-MD | 33679 | MP7 | DKEWILQKIYRIMRLLDELGHAEASMRVSDLIYEFMKKGD |
| M8-MD | 33679 | MP8 | DKEWILQKIYTIMRLLDELGHAEASMRVSDLIYEFMKKGD |
| M9-MD | 33678 | MP9 | DKEWILQKIYMIMRLLDELGHAEASMRVSDLIYEFMKKGD |
| M10-MD | 33681 | MP10 | DKEWILQKIYQIMRLLDELGHAEASMRVSDLIYEFMKKGD |
| M11-MD | 33690 | MP11 | DKEWILQKIYVIMRLLDELGHAEASMRVSDLIYEFMKKGD |
| M12-MD | 33679 | MP12 | DKEWILQKIYEIMRLLEELGHAEASMRVSDLIYEFMKKGD |
| M13-MD | 33682 | MP13 | DKEWILQKIYEIMRLLRELGHAEASMRVSDLIYEFMKKGD |
| M14-MD | 33693 | MP14 | DKEWILQKIYEIMRLLTELGHAEASMRVSDLIYEFMKKGD |
| M15-MD | 33692 | MP15 | DKEWILQKIYEIMRLLMELGHAEASMRVSDLIYEFMKKGD |
| M15-DFT | 2362 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jawad, B.; Adhikari, P.; Cheng, K.; Podgornik, R.; Ching, W.-Y. Computational Design of Miniproteins as SARS-CoV-2 Therapeutic Inhibitors. Int. J. Mol. Sci. 2022, 23, 838. https://doi.org/10.3390/ijms23020838
Jawad B, Adhikari P, Cheng K, Podgornik R, Ching W-Y. Computational Design of Miniproteins as SARS-CoV-2 Therapeutic Inhibitors. International Journal of Molecular Sciences. 2022; 23(2):838. https://doi.org/10.3390/ijms23020838
Chicago/Turabian StyleJawad, Bahaa, Puja Adhikari, Kun Cheng, Rudolf Podgornik, and Wai-Yim Ching. 2022. "Computational Design of Miniproteins as SARS-CoV-2 Therapeutic Inhibitors" International Journal of Molecular Sciences 23, no. 2: 838. https://doi.org/10.3390/ijms23020838
APA StyleJawad, B., Adhikari, P., Cheng, K., Podgornik, R., & Ching, W.-Y. (2022). Computational Design of Miniproteins as SARS-CoV-2 Therapeutic Inhibitors. International Journal of Molecular Sciences, 23(2), 838. https://doi.org/10.3390/ijms23020838