
CpG Island Methylator Phenotype—A Hope for the Future or a Road to Nowhere?


Abstract
1. Introduction
2. DNA Methylation
3. Discovery of CpG Island Methylator Phenotype (CIMP)
4. Pre-Microarray Era—Lack of CIMP-Specific Markers
5. Microarray and TCGA Era
6. Post-TCGA Era
7. Divergent Routes to CIMP
8. CIMP and Targeted Therapy
9. Conclusions and Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Peixoto, P.; Cartron, P.F.; Serandour, A.A.; Hervouet, E. From 1957 to nowadays: A brief history of epigenetics. Int. J. Mol. Sci. 2020, 21, 7571. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, M.V.C.; Bourc’his, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef] [PubMed]
- Majchrzak-Celińska, A.; Warych, A.; Szoszkiewicz, M. Novel Approaches to Epigenetic Therapies: From Drug Combinations to Epigenetic Editing. Genes 2021, 12, 208. [Google Scholar] [CrossRef] [PubMed]
- Hughes, L.A.; Melotte, V.; de Schrijver, J.; Maat, M.; Smit, V.T.H.B.M.; Bovée, J.V.M.G.; French, P.G.; Brandt, P.A.; Schouten, L.J.; Meyer, T.; et al. The CpG island methylator phenotype: What’s in a name? Cancer Res. 2013, 73, 5858–5868. [Google Scholar] [CrossRef]
- Linnekamp, J.F.; Butter, R.; Spijker, R.; Medema, J.P.; van Laarhoven, H.W.M. Clinical and biological effects of demethylating agents on solid tumours—A systematic review. Cancer Treat. Rev. 2017, 54, 10–23. [Google Scholar] [CrossRef]
- Urbano, A.; Smith, J.; Weeks, R.J.; Chatterjee, A. Gene-specific targeting of DNA methylation in the mammalian genome. Cancers 2019, 11, 1515. [Google Scholar] [CrossRef] [PubMed]
- Koch, A.; Joosten, S.C.; Feng, Z.; de Ruijter, T.C.; Draht, M.X.; Melotte, V.; Smits, K.M.; Veeck, J.; Herman, J.G.; Van Neste, L.; et al. Analysis of DNA methylation in cancer: Location revisited. Nat. Rev. Clin. Oncol. 2018, 15, 459–466. [Google Scholar] [CrossRef]
- Lee, J.E.; Kim, M.Y. Cancer epigenetics: Past, present and future. Semin. Cancer Biol. 2021; in press. [Google Scholar]
- Nishiyama, A.; Nakanishi, M. Navigating the DNA methylation landscape of cancer. Trends Genet. 2021, 37, 1012–1027. [Google Scholar] [CrossRef]
- Blum, R. Stepping inside the realm of epigenetic modifiers. Biomol. Concepts 2015, 6, 119–136. [Google Scholar] [CrossRef]
- Kulis, M.; Esteller, M. DNA methylation and cancer. Adv. Genet. 2010, 70, 27–56. [Google Scholar]
- Issa, J.P.; Shen, L.; Toyota, M. CIMP, at last. Gastroenterology 2005, 129, 1121–1124. [Google Scholar] [CrossRef]
- Frommer, M.; McDonald, L.E.; Millar, D.S.; Collis, C.M.; Watt, F.; Grigg, G.W.; Molloy, P.L.; Paul, C.L. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. USA 1992, 89, 1827–1831. [Google Scholar] [CrossRef]
- Herman, J.G.; Graff, J.R.; Myöhänen, S.; Nelkin, B.D.; Baylin, S.B. Methylation-specific PCR: A novel PCR assay for methylation status of CpG islands. Proc. Natl. Acad. Sci. USA 1996, 93, 9821–9826. [Google Scholar] [CrossRef]
- Xiong, Z.; Laird, P.W. COBRA: A sensitive and quantitative DNA methylation assay. Nucleic Acids Res. 1997, 25, 2532–2534. [Google Scholar] [CrossRef]
- Harrison, A.; Parle-McDermott, A. DNA methylation: A timeline of methods and applications. Front. Genet. 2011, 2, 74. [Google Scholar] [CrossRef]
- Akama, T.O.; Okazaki, Y.; Ito, M.; Okuizumi, H.; Konno, H.; Muramatsu, M.; Plass, C.; Held, W.A.; Hayashizaki, Y. Restriction landmark genomic scanning (RLGS-M)-based genome-wide scanning of mouse liver tumors for alterations in DNA methylation status. Cancer Res. 1997, 57, 3294–3299. [Google Scholar]
- Gonzalgo, M.L.; Liang, G.; Spruck, C.H.; Zingg, J.M.; Rideout, W.M.; Jones, P.A. Identification and characterization of differentially methylated regions of genomic DNA by methylation-sensitive arbitrarily primed PCR. Cancer Res. 1997, 57, 594–599. [Google Scholar] [PubMed]
- Toyota, M.; Ho, C.; Ahuja, N.; Jair, K.W.; Li, Q.; Ohe-Toyota, M.; Baylin, S.B.; Issa, J.P. Identification of differentially methylated sequences in colorectal cancer by methylated CpG island amplification. Cancer Res. 1999, 59, 2307–2312. [Google Scholar]
- Toyota, M.; Ahuja, N.; Ohe-Toyota, M.; Herman, J.G.; Baylin, S.B.; Issa, J.P. CpG island methylator phenotype in colorectal cancer. Proc. Natl. Acad. Sci. USA 1999, 96, 8681–8686. [Google Scholar] [CrossRef]
- Toyota, M.; Ahuja, N.; Suzuki, H.; Itoh, F.; Ohe-Toyota, M.; Imai, K.; Baylin, S.B.; Issa, J.P. Aberrant methylation in gastric cancer associated with the CpG island methylator phenotype. Cancer Res. 1999, 59, 5438–5442. [Google Scholar]
- Toyota, M.; Ohe-Toyota, M.; Ahuja, N.; Issa, J.P. Distinct genetic profiles in colorectal tumors with or without the CpG island methylator phenotype. Proc. Natl. Acad. Sci. USA 2000, 97, 710–715. [Google Scholar] [CrossRef]
- Issa, J.P. The epigenetics of colorectal cancer. Ann. N. Y. Acad. Sci. 2000, 910, 140–153. [Google Scholar] [CrossRef] [PubMed]
- Issa, J.P. CpG island methylator phenotype in cancer. Nat. Rev. Cancer 2004, 4, 988–993. [Google Scholar] [CrossRef]
- Bender, C.M.; Pao, M.M.; Jones, P.A. Inhibition of DNA methylation by 5-aza-2′-deoxycytidine suppresses the growth of human tumor cell lines. Cancer Res. 1998, 58, 95–101. [Google Scholar]
- Esteller, M.; Corn, P.G.; Baylin, S.B.; Herman, J.G. A gene hypermethylation profile of human cancer. Cancer Res. 2001, 61, 3225–3229. [Google Scholar]
- Toyota, M.; Kopecky, K.J.; Toyota, M.O.; Jair, K.W.; Willman, C.L.; Issa, J.P. Methylation profiling in acute myeloid leukemia. Blood 2001, 97, 2823–2829. [Google Scholar] [CrossRef]
- Strathdee, G.; Appleton, K.; Illand, M.; Millan, D.W.; Sargent, J.; Paul, J.; Brown, R. Primary ovarian carcinomas display multiple methylator phenotypes involving known tumor suppressor genes. Am. J. Pathol. 2001, 158, 1121–1127. [Google Scholar] [CrossRef]
- Yanagawa, N.; Tamura, G.; Oizumi, H.; Takahashi, N.; Shimazaki, Y.; Motoyama, T. Promoter hypermethylation of tumor suppressor and tumor-related genes in non-small cell lung cancers. Cancer Sci. 2003, 94, 589–592. [Google Scholar] [CrossRef]
- Ueki, T.; Toyota, M.; Sohn, T.; Yeo, C.J.; Issa, J.P.; Hruban, R.H.; Goggins, M. Hypermethylation of multiple genes in pancreatic adenocarcinoma. Cancer Res. 2000, 60, 1835–1839. [Google Scholar]
- Shen, L.; Ahuja, N.; Shen, Y.; Habib, N.A.; Toyota, M.; Rashid, A.; Issa, J.P. DNA methylation and environmental exposures in human hepatocellular carcinoma. J. Natl. Cancer Inst. 2002, 94, 755–761. [Google Scholar] [CrossRef]
- Eads, C.A.; Lord, R.V.; Wickramasinghe, K.; Long, T.I.; Kurumboor, S.K.; Bernstein, L.; Peters, J.H.; DeMeester, S.R.; DeMeester, T.R.; Skinner, K.A.; et al. Epigenetic patterns in the progression of esophageal adenocarcinoma. Cancer Res. 2001, 61, 3410–3418. [Google Scholar]
- Maruyama, R.; Toyooka, S.; Toyooka, K.O.; Harada, K.; Virmani, A.K.; Zöchbauer-Müller, S.; Farinas, A.J.; Vakar-Lopez, F.; Minna, J.D.; Sagalowsky, A.; et al. Aberrant promoter methylation profile of bladder cancer and its relationship to clinicopathological features. Cancer Res. 2001, 61, 8659–8663. [Google Scholar] [PubMed]
- Yamashita, K.; Dai, T.; Dai, Y.; Yamamoto, F.; Perucho, M. Genetics supersedes epigenetics in colon cancer phenotype. Cancer Cell 2003, 4, 121–131. [Google Scholar] [CrossRef]
- Anacleto, C.; Leopoldino, A.M.; Rossi, B.; Soares, F.A.; Lopes, A.; Rocha, J.C.; Caballero, O.; Camargo, A.A.; Simpson, A.J.; Pena, S.D. Colorectal cancer “methylator phenotype”: Fact or artifact? Neoplasia 2005, 7, 331–335. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kurdyukov, S.; Bullock, M. DNA methylation analysis: Choosing the right method. Biology 2016, 5, 3. [Google Scholar] [CrossRef]
- Eads, C.A.; Danenberg, K.D.; Kawakami, K.; Saltz, L.B.; Blake, C.; Shibata, D.; Danenberg, P.V.; Laird, P. MethyLight: A high-throughput assay to measure DNA methylation. Nucleic Acids Res. 2000, 28, E32. [Google Scholar] [CrossRef] [PubMed]
- Weisenberger, D.J.; Siegmund, K.D.; Campan, M.; Young, J.; Long, T.I.; Faasse, M.A.; Kang, G.H.; Widschwendter, M.; Weener, D.; Buchanan, D.; et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat. Genet. 2006, 38, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Kawasaki, T.; Kirkner, G.J.; Ohnishi, M.; Fuchs, C.S. 18q loss of heterozygosity in microsatellite stable colorectal cancer is correlated with CpG island methylator phenotype-negative (CIMP-0) and inversely with CIMP-low and CIMP-high. BMC Cancer 2007, 7, 72. [Google Scholar] [CrossRef]
- Ogino, S.; Kawasaki, T.; Nosho, K.; Ohnishi, M.; Suemoto, Y.; Kirkner, G.J.; Fuchs, C.S. LINE-1 hypomethylation is inversely associated with microsatellite instability and CpG island methylator phenotype in colorectal cancer. Int. J. Cancer 2008, 122, 2767–2773. [Google Scholar] [CrossRef]
- Ogino, S.; Kawasaki, T.; Kirkner, G.J.; Kraft, P.; Loda, M.; Fuchs, C.S. Evaluation of markers for CpG island methylator phenotype (CIMP) in colorectal cancer by a large population-based sample. J. Mol. Diagn. 2007, 9, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Samowitz, W.S.; Albertsen, H.; Herrick, J.; Levin, T.R.; Sweeney, C.; Murtaugh, M.A.; Wolff, R.K.; Slattery, M.L. Evaluation of a large, population-based sample supports a CpG island methylator phenotype in colon cancer. Gastroenterology 2005, 129, 837–845. [Google Scholar] [CrossRef]
- Kim, J.H.; Shin, S.H.; Kwon, H.J.; Cho, N.Y.; Kang, G.H. Prognostic implications of CpG island hypermethylator phenotype in colorectal cancers. Virchows Arch. 2009, 455, 485–494. [Google Scholar] [CrossRef]
- Juo, Y.Y.; Johnston, F.M.; Zhang, D.Y.; Juo, H.H.; Wang, H.; Pappou, E.P.; Yu, T.; Easwaran, H.; Baylin, S.; Engeland, M.; et al. Prognostic value of CpG island methylator phenotype among colorectal cancer patients: A systematic review and meta-analysis. Ann. Oncol. 2014, 25, 2314–2327. [Google Scholar] [CrossRef]
- Hazra, A.; Fuchs, C.S.; Kawasaki, T.; Kirkner, G.J.; Hunter, D.J.; Ogino, S. Germline polymorphisms in the one-carbon metabolism pathway and DNA methylation in colorectal cancer. Cancer Causes Control. 2010, 21, 331–345. [Google Scholar] [CrossRef]
- Karpinski, P.; Myszka, A.; Ramsey, D.; Kielan, W.; Sasiadek, M.M. Detection of viral DNA sequences in sporadic colorectal cancers in relation to CpG island methylation and methylator phenotype. Tumour Biol. 2011, 32, 653–659. [Google Scholar] [CrossRef]
- Curtin, K.; Slattery, M.L.; Ulrich, C.M.; Bigler, J.; Levin, T.R.; Wolff, R.K.; Albertsen, H.; Potter, J.D.; Samowitz, W.S. Genetic polymorphisms in one-carbon metabolism: Associations with CpG island methylator phenotype (CIMP) in colon cancer and the modifying effects of diet. Carcinogenesis 2007, 28, 1672–1679. [Google Scholar] [CrossRef]
- Goel, A.; Li, M.S.; Nagasaka, T.; Shin, S.K.; Fuerst, F.; Ricciardiello, L.; Wasserman, L.; Boland, C.R. Association of JC virus T-antigen expression with the methylator phenotype in sporadic colorectal cancers. Gastroenterology 2006, 130, 1950–1961. [Google Scholar] [CrossRef] [PubMed]
- Nosho, K.; Shima, K.; Kure, S.; Irahara, N.; Baba, Y.; Chen, L.; Kirkner, G.J.; Fuchs, C.S.; Ogino, S. JC virus T-antigen in colorectal cancer is associated with p53 expression and chromosomal instability, independent of CpG island methylator phenotype. Neoplasia 2009, 11, 87–95. [Google Scholar] [CrossRef]
- Chang, M.S.; Uozaki, H.; Chong, J.M.; Ushiku, T.; Sakuma, K.; Ishikawa, S.; Hino, R.; Barua, R.R.; Iwasaki, Y.; Arai, K.; et al. CpG island methylation status in gastric carcinoma with and without infection of Epstein-Barr virus. Clin. Cancer Res. 2006, 12, 2995–3002. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Karpinski, P.; Myszka, A.; Ramsey, D.; Misiak, B.; Gil, J.; Laczmanska, I.; Grzebieniak, Z.; Sebzda, T.; Smigiel, R.; Stembalska, A.; et al. Polymorphisms in methyl-group metabolism genes and risk of sporadic colorectal cancer with relation to the CpG island methylator phenotype. Cancer Epidemiol. 2010, 34, 338–344. [Google Scholar] [CrossRef] [PubMed][Green Version]
- De Vogel, S.; Wouters, K.A.; Gottschalk, R.W.; van Schooten, F.J.; de Goeij, A.F.; de Bruïne, A.P.; Goldbohm, R.A.; van den Brandt, P.A.; Weijenberg, M.P.; van Engeland, M. Genetic variants of methyl metabolizing enzymes and epigenetic regulators: Associations with promoter CpG island hypermethylation in colorectal cancer. Cancer Epidemiol. Biomark. Prev. 2009, 18, 3086–3096. [Google Scholar] [CrossRef]
- Thomas, R.; Trapani, D.; Goodyer-Sait, L.; Tomkova, M.; Fernandez-Rozadilla, C.; Sahnane, N.; Woolley, C.; Davis, H.; Chegwidden, L.; Kriaucionis, S.; et al. The polymorphic variant rs1800734 influences methylation acquisition and allele-specific TFAP4 binding in the MLH1 promoter leading to differential mRNA expression. Sci. Rep. 2019, 9, 13463. [Google Scholar] [CrossRef] [PubMed]
- Samowitz, W.S.; Curtin, K.; Wolff, R.K.; Albertsen, H.; Sweeney, C.; Caan, B.J.; Ulrich, C.M.; Potter, J.D.; Slattery, M.L. The MLH1 -93 G>A promoter polymorphism and genetic and epigenetic alterations in colon cancer. Genes Chromosomes Cancer 2008, 47, 835–844. [Google Scholar] [CrossRef]
- Yagi, K.; Akagi, K.; Hayashi, H.; Nagae, G.; Tsuji, S.; Isagawa, T.; Midorikawa, Y.; Nishimura, Y.; Sakamoto, H.; Seto, Y.; et al. Three DNA methylation epigenotypes in human colorectal cancer. Clin. Cancer Res. 2010, 16, 21–33. [Google Scholar] [CrossRef]
- Kaneda, A.; Yagi, K. Two groups of DNA methylation markers to classify colorectal cancer into three epigenotypes. Cancer Sci. 2011, 102, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Toyota, M.; Kondo, Y.; Lin, E.; Zhang, L.; Guo, Y.; Hernandez, N.S.; Chen, X.; Ahmed, S.; Konishi, K.; et al. Integrated genetic and epigenetic analysis identifies three different subclasses of colon cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 18654–18659. [Google Scholar] [CrossRef]
- Jass, J.R. Molecular heterogeneity of colorectal cancer: Implications for cancer control. Surg. Oncol. 2007, 16, S7–S9. [Google Scholar] [CrossRef]
- Ogino, S.; Goel, A. Molecular classification and correlates in colorectal cancer. J. Mol. Diagn. 2008, 10, 13–27. [Google Scholar] [CrossRef]
- Arnold, C.N.; Sosnowski, A.; Schmitt-Gräff, A.; Arnold, R.; Blum, H.E. Analysis of molecular pathways in sporadic neuroendocrine tumors of the gastro-entero-pancreatic system. Int. J. Cancer 2007, 120, 2157–2164. [Google Scholar] [CrossRef]
- Abe, M.; Ohira, M.; Kaneda, A.; Yagi, Y.; Yamamoto, S.; Kitano, Y.; Takato, T.; Nakagawara, A.; Ushijima, T. CpG island methylator phenotype is a strong determinant of poor prognosis in neuroblastomas. Cancer Res. 2005, 65, 828–834. [Google Scholar]
- Kraszewska, M.D.; Dawidowska, M.; Larmonie, N.S.; Kosmalska, M.; Sędek, Ł.; Szczepaniak, M.; Grzeszczak, W.; Langerak, A.W.; Szczepański, T.; Witt, M.; et al. DNA methylation pattern is altered in childhood T-cell acute lymphoblastic leukemia patients as compared with normal thymic subsets: Insights into CpG island methylator phenotype in T-ALL. Leukemia 2012, 26, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Zhang, C.; Zhao, J.; Wang, C.; Xu, Y.; Han, Z.; Jiang, G.; Guo, X.; Li, R.; Bu, X.; et al. Correlation of CpG island methylator phenotype with poor prognosis in hepatocellular carcinoma. Exp. Mol. Pathol. 2010, 88, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.K.; Brown, A.; Garrett, E.; Bornman, D.; Fackler, M.J.; Sukumar, S.; Herman, J.G.; Gabrielson, E. Hypermethylation in histologically distinct classes of breast cancer. Clin. Cancer Res. 2004, 10, 5998–6005. [Google Scholar] [CrossRef] [PubMed]
- Merhavi, E.; Cohen, Y.; Avraham, B.C.; Frenkel, S.; Chowers, I.; Pe’er, J.; Goldenberg-Cohen, N. Promoter methylation status of multiple genes in uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2007, 48, 4403–4406. [Google Scholar] [CrossRef][Green Version]
- Cohen, Y.; Merhavi-Shoham, E.; Avraham, R.B.; Frenkel, S.; Pe’er, J.; Goldenberg-Cohen, N. Hypermethylation of CpG island loci of multiple tumor suppressor genes in retinoblastoma. Exp. Eye Res. 2008, 86, 201–206. [Google Scholar] [CrossRef]
- Buhard, O.; Cattaneo, F.; Wong, Y.F.; Yim, S.F.; Friedman, E.; Flejou, J.F.; Duval, A.; Hamelin, R. Multipopulation analysis of polymorphisms in five mononucleotide repeats used to determine the microsatellite instability status of human tumors. J. Clin. Oncol. 2006, 24, 241–251. [Google Scholar] [CrossRef]
- Gebhard, C.; Schwarzfischer, L.; Pham, T.H.; Schilling, E.; Klug, M.; Andreesen, R.; Rehli, M. Genome-wide profiling of CpG methylation identifies novel targets of aberrant hypermethylation in myeloid leukemia. Cancer Res. 2006, 66, 6118–6128. [Google Scholar] [CrossRef]
- Weng, Y.I.; Huang, T.H.; Yan, P.S. Methylated DNA immunoprecipitation and microarray-based analysis: Detection of DNA methylation in breast cancer cell lines. Methods Mol. Biol. 2009, 590, 165–176. [Google Scholar]
- Bibikova, M.; Lin, Z.; Zhou, L.; Chudin, E.; Garcia, E.W.; Wu, B.; Doucet, D.; Thomas, N.J.; Wang, Y.; Vollmer, E.; et al. High-throughput DNA methylation profiling using universal bead arrays. Genome Res. 2006, 16, 383–393. [Google Scholar] [CrossRef]
- Bibikova, M.; Le, J.; Barnes, B.; Saedinia-Melnyk, S.; Zhou, L.; Shen, R.; Gunderson, K.L. Genome-wide DNA methylation profiling using Infinium® assay. Epigenomics 2009, 1, 177–200. [Google Scholar] [CrossRef]
- Sandoval, J.; Heyn, H.; Moran, S.; Serra-Musach, J.; Pujana, M.A.; Bibikova, M.; Esteller, M. Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics 2011, 6, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Pidsley, R.; Zotenko, E.; Peters, T.J.; Lawrence, M.G.; Risbridger, G.P.; Molloy, P.; Van Djik, S.; Muhlhausler, B.; Stirzaker, C.; Clark, S.J. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 2016, 17, 208. [Google Scholar] [CrossRef] [PubMed]
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. 2015, 19, A68–A77. [Google Scholar] [CrossRef] [PubMed]
- Arora, A.; Olshen, A.B.; Seshan, V.E.; Shen, R. Pan-cancer identification of clinically relevant genomic subtypes using outcome-weighted integrative clustering. Genome Med. 2020, 12, 110. [Google Scholar] [CrossRef]
- Hoadley, K.A.; Yau, C.; Wolf, D.M.; Cherniack, A.D.; Tamborero, D.; Ng, S.; Leiserson, M.D.; Niu, B.; McLellan, M.D.; Uzunangelov, V.; et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell 2014, 158, 929–944. [Google Scholar] [CrossRef]
- Weisenberger, D.J. Characterizing DNA methylation alterations from The Cancer Genome Atlas. J. Clin. Investig. 2014, 124, 17–23. [Google Scholar] [CrossRef]
- Sweeney, T.E.; Chen, A.C.; Gevaert, O. Combined mapping of multiple clUsteriNg ALgorithms (COMMUNAL): A robust method for selection of cluster number, K. Sci. Rep. 2015, 5, 16971. [Google Scholar] [CrossRef]
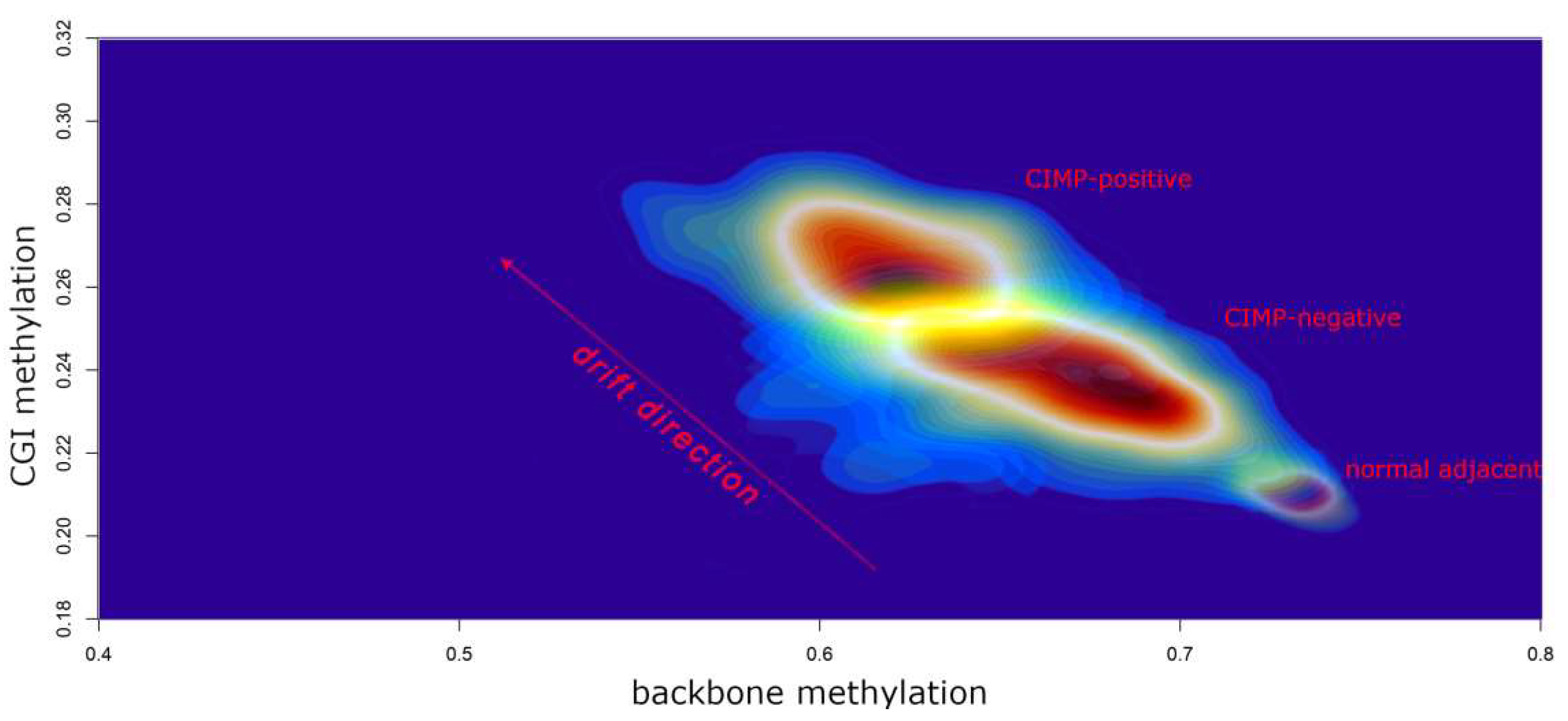
- Karpinski, P.; Pesz, K.; Sasiadek, M.M. Pan-cancer analysis reveals presence of pronounced DNA methylation drift in CpG island methylator phenotype clusters. Epigenomics 2017, 9, 1341–1352. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. The molecular taxonomy of primary prostate cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014, 507, 315–322. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A.G.; Pashtan, I.; Shen, R.; Benz, C.C.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar]
- Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.; Hoadley, K.; Triche, T.J.; Laird, P.W.; Baty, J.D.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar]
- The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Integrated genomic characterization of papillary thyroid carcinoma. Cell 2014, 159, 676–690. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [PubMed]
- Brat, D.J.; Verhaak, R.G.; Aldape, K.D.; Yung, W.K.; Salama, S.R.; Cooper, L.A.; Rheinbay, E.; Miller, C.R.; Vitucci, M.; Morozova, O.; et al. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N. Engl. J. Med. 2015, 372, 2481–2498. [Google Scholar]
- Linehan, W.M.; Spellman, P.T.; Ricketts, C.J.; Creighton, C.J.; Fei, S.S.; Davis, C.; Wheeler, D.A.; Murray, B.A.; Schmidt, L.; Vocke, C.D.; et al. Comprehensive molecular characterization of papillary renal-cell carcinoma. N. Engl. J. Med. 2016, 374, 135–145. [Google Scholar] [PubMed]
- Zheng, S.; Cherniack, A.D.; Dewal, N.; Moffitt, R.A.; Danilova, L.; Murray, B.A.; Lerario, A.M.; Else, T.; Knijnenburg, T.A.; Ciriello, G.; et al. Comprehensive pan-genomic characterization of adrenocortical carcinoma. Cancer Cell 2016, 29, 723–736. [Google Scholar] [CrossRef] [PubMed]
- Cherniack, A.D.; Shen, H.; Walter, V.; Stewart, C.; Murray, B.A.; Bowlby, R.; Hu, X.; Ling, S.; Soslow, R.A.; Broaddus, R.R.; et al. Integrated molecular characterization of uterine carcinosarcoma. Cancer Cell 2017, 31, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Farshidfar, F.; Zheng, S.; Gingras, M.C.; Newton, Y.; Shih, J.; Robertson, A.G.; Hinoue, T.; Hoadley, K.A.; Gibb, E.A.; Roszik, J.; et al. Integrative genomic analysis of cholangiocarcinoma identifies distinct IDH-mutant molecular profiles. Cell Rep. 2017, 18, 2780–2794. [Google Scholar] [CrossRef]
- CGARNEa; The Cancer Genome Atlas Network. Integrated genomic and molecular characterization of cervical cancer. Nature 2017, 543, 378–384. [Google Scholar] [CrossRef]
- CGARNEa; The Cancer Genome Atlas Network. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell 2017, 169, 1327–1341.e23. [Google Scholar] [CrossRef] [PubMed]
- Robertson, A.G.; Shih, J.; Yau, C.; Gibb, E.A.; Oba, J.; Mungall, K.L.; Hess, J.M.; Uzunangelov, V.; Walter, V.; Danilova, L.; et al. Integrative analysis identifies four molecular and clinical subsets in uveal melanoma. Cancer Cell 2017, 32, 204–220.e15. [Google Scholar] [CrossRef] [PubMed]
- CGARNEa; The Cancer Genome Atlas Network. Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell 2017, 32, 185–203.e13. [Google Scholar] [CrossRef] [PubMed]
- CGARNEa; The Cancer Genome Atlas Network. Comprehensive and integrated genomic characterization of adult soft tissue sarcomas. Cell 2017, 171, 950–965.e28. [Google Scholar] [CrossRef] [PubMed]
- Radovich, M.; Pickering, C.R.; Felau, I.; Ha, G.; Zhang, H.; Jo, H.; Hoadley, K.A.; Anur, P.; Zhang, J.; McLellan, M.; et al. The integrated genomic landscape of thymic epithelial tumors. Cancer Cell 2018, 33, 244–258.e10. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Shih, J.; Hollern, D.P.; Wang, L.; Bowlby, R.; Tickoo, S.K.; Thorsson, V.; Mungall, A.J.; Newton, Y.; Hegde, A.M.; et al. Integrated molecular characterization of testicular germ cell tumors. Cell Rep. 2018, 23, 3392–3406. [Google Scholar] [CrossRef] [PubMed]
- Hmeljak, J.; Sanchez-Vega, F.; Hoadley, K.A.; Shih, J.; Stewart, C.; Heiman, D.; Tarpey, P.; Danilova, L.; Drill, E.; Gibb, E.A.; et al. Integrative molecular characterization of malignant pleural mesothelioma. Cancer Discov. 2018, 8, 1548–1565. [Google Scholar] [CrossRef]
- CGARNEa; The Cancer Genome Atlas Network. Integrated genomic characterization of oesophageal carcinoma. Nature 2017, 541, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Widschwendter, M.; Teschendorff, A.E. A comparison of feature selection and classification methods in DNA methylation studies using the Illumina Infinium platform. BMC Bioinform. 2012, 13, 59. [Google Scholar] [CrossRef]
- Gevaert, O.; Tibshirani, R.; Plevritis, S.K. Pancancer analysis of DNA methylation-driven genes using MethylMix. Genome Biol. 2015, 16, 17. [Google Scholar] [CrossRef]
- Brennan, K.; Koenig, J.L.; Gentles, A.J.; Sunwoo, J.B.; Gevaert, O. Identification of an atypical etiological head and neck squamous carcinoma subtype featuring the CpG island methylator phenotype. EBioMedicine 2017, 17, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Moarii, M.; Reyal, F.; Vert, J.P. Integrative DNA methylation and gene expression analysis to assess the universality of the CpG island methylator phenotype. Hum. Genom. 2015, 9, 26. [Google Scholar] [CrossRef]
- Sánchez-Vega, F.; Gotea, V.; Margolin, G.; Elnitski, L. Pan-cancer stratification of solid human epithelial tumors and cancer cell lines reveals commonalities and tissue-specific features of the CpG island methylator phenotype. Epigenetics Chromatin 2015, 8, 14. [Google Scholar] [CrossRef]
- Karpinski, P.; Patai, A.V.; Hap, W.; Kielan, W.; Laczmanska, I.; Sasiadek, M.M. Multilevel omic data clustering reveals variable contribution of methylator phenotype to integrative cancer subtypes. Epigenomics 2018, 10, 1289–1299. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.K.; Ramnarayanan, K.; Zhu, F.; Srivastava, S.; Xu, C.; Tan, A.L.K.; Lee, M.; Tay, S.; Das, K.; Xing, M.; et al. Genomic and epigenomic profiling of high-risk intestinal metaplasia reveals molecular determinants of progression to gastric cancer. Cancer Cell 2018, 33, 137–150.e5. [Google Scholar] [CrossRef]
- Verburg, N.; Barthel, F.P.; Anderson, K.J.; Johnson, K.C.; Koopman, T.; Yaqub, M.M.; Hoekstra, O.S.; Lammertsma, A.A.; Barkhof, F.; Pouwels, P.J.W.; et al. Spatial concordance of DNA methylation classification in diffuse glioma. Neuro-Oncology 2021. [Google Scholar] [CrossRef] [PubMed]
- Van Lanschot, M.C.J.; Carvalho, B.; Rausch, C.; Snaebjornsson, P.; van Engeland, M.; Kuipers, E.J.; Stoker, J.; Tutein Nolthenius, C.J.; Dekker, E.; Meijer, G.A. Molecular profiling of longitudinally observed small colorectal polyps: A cohort study. EBioMedicine 2019, 39, 292–300. [Google Scholar] [CrossRef]
- Nosho, K.; Irahara, N.; Shima, K.; Kure, S.; Kirkner, G.J.; Schernhammer, E.S.; Hazra, A.; Hunter, D.J.; Quackenbush, J.; Spiegelman, D.; et al. Comprehensive biostatistical analysis of CpG island methylator phenotype in colorectal cancer using a large population-based sample. PLoS ONE 2008, 3, e3698. [Google Scholar] [CrossRef] [PubMed]
- Simons, C.C.; Hughes, L.A.; Smits, K.M.; Khalid-de Bakker, C.A.; de Bruïne, A.P.; Carvalho, B.; Meijer, G.A.; Schouten, L.J.; van den Brandt, P.A.; Weijenberg, M.P.; et al. A novel classification of colorectal tumors based on microsatellite instability, the CpG island methylator phenotype and chromosomal instability: Implications for prognosis. Ann. Oncol. 2013, 24, 2048–2056. [Google Scholar] [CrossRef]
- Flatin, B.T.B.; Vedeld, H.M.; Pinto, R.; Langerud, J.; Lind, G.E.; Lothe, R.A.; Sveen, A.; Jeanmougin, M. Multiregional assessment of CIMP in primary colorectal cancers: Phenotype concordance but marker variability. Int. J. Cancer 2021, 148, 1652–1657. [Google Scholar] [CrossRef] [PubMed]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef] [PubMed]
- Noushmehr, H.; Weisenberger, D.J.; Diefes, K.; Phillips, H.S.; Pujara, K.; Berman, B.P.; Pan, F.; Pelloski, C.E.; Sulman, E.P.; Bhat, K.P.; et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 2010, 17, 510–522. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Duncan, C.G.; Barwick, B.G.; Jin, G.; Rago, C.; Kapoor-Vazirani, P.; Powell, D.R.; Chi, J.T.; Bigner, D.D.; Vertino, P.M.; Yan, H. A heterozygous IDH1R132H/WT mutation induces genome-wide alterations in DNA methylation. Genome Res. 2012, 22, 2339–2355. [Google Scholar] [CrossRef] [PubMed]
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.A.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.; Lu, C.; Ward, P.S.; et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Tang, K.; Liang, T.Y.; Zhang, W.Z.; Li, J.Y.; Wang, W.; Hu, H.M.; Li, M.Y.; Wang, H.Q.; He, X.Z.; et al. The comparison of clinical and biological characteristics between IDH1 and IDH2 mutations in gliomas. J. Exp. Clin. Cancer Res. 2016, 35, 86. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Xiao, M.T.; Liu, L.X.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Raineri, S.; Mellor, J. Linking metabolism and epigenetics. Front. Genet. 2018, 9, 493. [Google Scholar] [CrossRef] [PubMed]
- Kelly, A.D.; Kroeger, H.; Yamazaki, J.; Taby, R.; Neumann, F.; Yu, S.; Lee, J.T.; Patel, B.; Li, Y.; He, R.; et al. A CpG island methylator phenotype in acute myeloid leukemia independent of IDH mutations and associated with a favorable outcome. Leukemia 2017, 31, 2011–2019. [Google Scholar] [CrossRef]
- Sasaki, M.; Knobbe, C.B.; Munger, J.C.; Lind, E.F.; Brenner, D.; Brüstle, A.; Harris, I.S.; Holmes, R.; Wakeham, A.; Haight, J.; et al. IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature 2012, 488, 656–659. [Google Scholar] [CrossRef]
- Lee, K.; Song, Y.S.; Shin, Y.; Wen, X.; Kim, Y.; Cho, N.Y.; Bae, J.M.; Kang, G.H. Intrahepatic cholangiocarcinomas with IDH1/2 mutation-associated hypermethylation at selective genes and their clinicopathological features. Sci. Rep. 2020, 10, 15820. [Google Scholar] [CrossRef]
- Nicolle, R.; Ayadi, M.; Gomez-Brouchet, A.; Armenoult, L.; Banneau, G.; Elarouci, N.; Tallegas, M.; Decouvelaere, A.V.; Aubert, S.; Rédini, F.; et al. Integrated molecular characterization of chondrosarcoma reveals critical determinants of disease progression. Nat. Commun. 2019, 10, 4622. [Google Scholar] [CrossRef] [PubMed]
- Lauss, M.; Ringnér, M.; Karlsson, A.; Harbst, K.; Busch, C.; Geisler, J.; Lønning, P.E.; Staaf, J.; Jönsson, G. DNA methylation subgroups in melanoma are associated with proliferative and immunological processes. BMC Med. Genom. 2015, 8, 73. [Google Scholar] [CrossRef]
- Unruh, D.; Zewde, M.; Buss, A.; Drumm, M.R.; Tran, A.N.; Scholtens, D.M.; Horbinski, C. Methylation and transcription patterns are distinct in IDH mutant gliomas compared to other IDH mutant cancers. Sci. Rep. 2019, 9, 8946. [Google Scholar] [CrossRef] [PubMed]
- Stanland, L.J.; Luftig, M.A. The role of EBV-induced hypermethylation in gastric cancer tumorigenesis. Viruses 2020, 12, 1222. [Google Scholar] [CrossRef] [PubMed]
- Costa, N.R.; Gil da Costa, R.M.; Medeiros, R. A viral map of gastrointestinal cancers. Life Sci. 2018, 199, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Matsusaka, K.; Funata, S.; Fukuyo, M.; Seto, Y.; Aburatani, H.; Fukayama, M.; Kaneda, A. Epstein-Barr virus infection induces genome-wide de novo DNA methylation in non-neoplastic gastric epithelial cells. J. Pathol. 2017, 242, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Funata, S.; Matsusaka, K.; Yamanaka, R.; Yamamoto, S.; Okabe, A.; Fukuyo, M.; Aburatani, H.; Fukayama, M.; Kaneda, A. Histone modification alteration coordinated with acquisition of promoter DNA methylation during Epstein-Barr virus infection. Oncotarget 2017, 8, 55265–55279. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Jia, K.; Lv, H.; Wang, S.Q.; Wu, Y.; Lei, H.; Chen, X. EBV-positive gastric cancer: Current knowledge and future perspectives. Front. Oncol. 2020, 10, 583463. [Google Scholar] [CrossRef]
- Issa, J.P. Aging and epigenetic drift: A vicious cycle. J. Clin. Investig. 2014, 124, 24–29. [Google Scholar] [CrossRef]
- Zheng, S.C.; Widschwendter, M.; Teschendorff, A.E. Epigenetic drift, epigenetic clocks and cancer risk. Epigenomics 2016, 8, 705–719. [Google Scholar] [CrossRef]
- Tao, Y.; Kang, B.; Petkovich, D.A.; Bhandari, Y.R.; In, J.; Stein-O’Brien, G.; Kong, X.; Xie, W.; Zachos, N.; Maegawa, S.; et al. Aging-like spontaneous epigenetic silencing facilitates Wnt activation, stemness, and BrafV600E-induced tumorigenesis. Cancer Cell 2019, 35, 315–328.e6. [Google Scholar] [CrossRef]
- Scala, G.; Federico, A.; Palumbo, D.; Cocozza, S.; Greco, D. DNA sequence context as a marker of CpG methylation instability in normal and cancer tissues. Sci. Rep. 2020, 10, 1721. [Google Scholar] [CrossRef] [PubMed]
- Bates, S.E. Epigenetic therapies for cancer. N. Engl. J. Med. 2020, 383, 650–663. [Google Scholar] [CrossRef] [PubMed]
- Egger, G.; Liang, G.; Aparicio, A.; Jones, P.A. Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004, 429, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, W.; Cao, P. Advances in CpG island methylator phenotype colorectal cancer therapies. Front. Oncol. 2021, 11, 629390. [Google Scholar] [CrossRef]
- Duan, Y.C.; Zhang, S.J.; Shi, X.J.; Jin, L.F.; Yu, T.; Song, Y.; Guan, Y.Y. Research progress of dual inhibitors targeting crosstalk between histone epigenetic modulators for cancer therapy. Eur. J. Med. Chem. 2021, 222, 113588. [Google Scholar] [CrossRef]
- Golub, D.; Iyengar, N.; Dogra, S.; Wong, T.; Bready, D.; Tang, K.; Modrek, A.S.; Placantonakis, D.G. Mutant isocitrate dehydrogenase inhibitors as targeted cancer therapeutics. Front. Oncol. 2019, 9, 417. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, T.A.; Mukherjee, J.; Viswanath, P.; Ohba, S.; Ronen, S.M.; Bjerkvig, R.; Pieper, R.O. Rapid conversion of mutant IDH1 from driver to passenger in a model of human gliomagenesis. Mol. Cancer Res. 2016, 14, 976–983. [Google Scholar] [CrossRef]
- Wouters, B.J. Targeting IDH1 and IDH2 mutations in acute myeloid leukemia: Emerging options and pending questions. Hemasphere 2021, 5, e583. [Google Scholar] [CrossRef] [PubMed]
- Konteatis, Z.; Artin, E.; Nicolay, B.; Straley, K.; Padyana, A.K.; Jin, L.; Chen, Y.; Narayaraswamy, R.; Tong, S.; Wang, F.; et al. Vorasidenib (AG-881): A first-in-class, brain-penetrant dual inhibitor of mutant IDH1 and 2 for treatment of glioma. ACS Med. Chem. Lett. 2020, 11, 101–107. [Google Scholar] [CrossRef]
- Zhu, A.X.; Macarulla, T.; Javle, M.M.; Kelley, R.K.; Lubner, S.J.; Adeva, J.; Cleary, J.M.; Catenacci, D.V.T.; Borad, M.J.; Bridgewater, J.A.; et al. Final overall survival efficacy results of ivosidenib for patients with advanced Cholangiocarcinoma with IDH1 mutation: The phase 3 randomized clinical ClarIDHy trial. JAMA Oncol. 2021, 7, 1669–1677. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The immune landscape of cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef] [PubMed]
- Roth, M.T.; Das, S. Pembrolizumab in unresectable or metastatic MSI-high colorectal cancer: Safety and efficacy. Expert Rev. Anticancer Ther. 2021, 21, 229–238. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer | Abbreviation | Illumina Platform | CIMP or Highly Methylated Cluster | Probes Selection | No. of Probes Selected | Tumor Purity Addresed | Clustering | Clustering Method | Ref. | Publication Year |
|---|---|---|---|---|---|---|---|---|---|---|
| Breast adenocarcinoma | BRCA | 27K and 450K | Yes | most variable (highest standard deviations) | 574 | no | NA | RPMM clustering | [81] | 2012 |
| Prostate adenocarcinoma | PRAD | 450K | Yes | cancer specific hypermethylation (beta value > 0.3) | 5000 | yes | binary distance clustering/Ward’s method for linkage | hierarchical clustering | [82] | 2015 |
| Bladder urothelial carcinoma | BLCA | 450K | Yes | cancer specific/promoter and CpG island associated (beta value > 0.3) | 31,249 | yes | binary distance clustering/Ward’s method for linkage | hierarchical clustering | [83] | 2014 |
| Ovarian serous cystadenocarcinoma | OV | 27K | Not reported | most variable (highest standard deviations) | 858 | no | K-means clustering/Euclidean distance | consensus clustering | [84] | 2011 |
| Colorectal adenocarcinoma | COAD and READ | 27K | Yes | most variable (highest standard deviations) | 2758 | no | NA | RPMM clustering | [85] | 2012 |
| Lung adenocarcinoma | LUAD | 27K and 450K | Yes | promoter and CpG island associated/1.0% of most variable | not provided | no | PAM clustering/Euclidean distance | consensus clustering | [86] | 2014 |
| Lung squamous cell carcinoma | LUSC | 27K and 450K | Not reported | most variable (highest standard deviations) | 8228 | no | PAM clustering/Euclidean distance | consensus clustering | [87] | 2012 |
| Uterine corpus endometrial carcinoma | UCEC | 27K and 450K | Yes | most variable (highest standard deviations) | 785 | no | NA | RPMM clustering | [88] | 2013 |
| Acute Myeloid Leukemia | AML | 450K | Yes | most variable (highest standard deviations) | 1000 | no | Euclidean distance clustering/Ward’s method for linkage | hierarchical clustering | [89] | 2013 |
| Glioblastoma | GBM | GoldenGate and 27K | Yes | most variable (highest standard deviations) | 370 | no | K-means clustering/Euclidean distance | consensus clustering | [90] | 2008 |
| Stomach adenocarcinoma | STAD | 27K and 450K | Yes | cancer specific hypermethylation | 1375 | yes | binary distance clustering/Ward’s method for linkage | consensus clustering | [51] | 2014 |
| Thyroid carcinoma | THCA | 450K | Yes | most variable 1.0% of probes | not provided | no | PAM clustering/Euclidean distance | consensus clustering | [91] | 2014 |
| Head and neck squamous cell carcinoma | HNSC | 450K | Yes | most variable 1.0% of probes | not provided | no | PAM clustering/Euclidean distance | consensus clustering | [92] | 2015 |
| Skin cutaneous melanoma | SKCM | 450K | Yes | most variable 1.0% of probes | not provided | no | PAM clustering/Euclidean distance | consensus clustering | [93] | 2015 |
| Brain lower grade glioma | LGG | 450K | Yes | cancer specific hypermethylation (beta value > 0.3) | 11,977 | yes | binary distanceclustering/Ward’s method for linkage | hierarchical clustering | [94] | 2015 |
| Kidney renal papillary cell carcinoma | KIRP | 27K and 450K | Yes | cancer specific hypermethylation/most variable | not provided | no | Ward’s method | hierarchical clustering | [95] | 2016 |
| Adrenocortical carcinoma | ACC | 450K | Yes | promoter and CpG island associated/1.0% of most variable | not provided | no | PAM clustering/Euclidean distance | consensus clustering | [96] | 2016 |
| Uterine carcinosarcoma | UCS | 450K | Yes | cancer specific hypermethylation/most variable | 5000 | no | Ward’s method for linkage | hierarchical clustering | [97] | 2017 |
| Cholangiocarcinoma | CHOL | 450K | Yes | cancer specific hypermethylation (beta value > 0.3) | 37,743 | yes | binary distance clustering/Ward’s method for linkage | hierarchical clustering | [98] | 2017 |
| Cervical cancer | CESC | 450K | Yes | promoter and CpG island associated/1.0% of most variable | not provided | no | PAM clustering/Euclidean distance | consensus clustering | [99] | 2017 |
| Hepatocellular carcinoma | HCC | 450K | Yes | cancer specific hypermethylation (beta value > 0.3) | 37,848 | yes | binary distance clustering/Ward’s method for linkage | hierarchical clustering | [100] | 2017 |
| Uveal melanoma | UVM | 450K | Yes | most variable 1.0% of probes | 3859 | no | PAM clustering/Euclidean distance | consensus clustering | [101] | 2017 |
| Pancreatic adenocarcinoma | PAAD | 450K | Yes | cancer specific hypermethylation (beta value > 0.25) | 31,956 | yes | binary distance clustering/Ward’s method for linkage | consensus clustering | [102] | 2017 |
| Sarcoma | SARC | 450K | Yes | most variable 1.0% of probes | not provided | no | PAM clustering/Euclidean distance | consensus clustering | [103] | 2017 |
| Thymoma | THYM | 450K | Not reported | most variable 1.0% of probes | not provided | no | PAM clustering/Euclidean distance | consensus clustering | [104] | 2018 |
| Testicular cancer | TGTC | 450K | Yes | most variable (standard deviation >= 0.26) | 9614 | yes | Euclidean distance clustering/Ward’s method for linkage | hierarchical clustering | [105] | 2018 |
| Malignant pleural mesothelioma | MPM | 450K | Not reported | most variable 1.0% of probes | not provided | no | PAM clustering/Euclidean distance | consensus clustering | [106] | 2018 |
| Oesophageal carcinoma | ESCA | 450K | Yes | cancer specific hypermethylation (beta value > 0.25) | not provided | yes | binary distance clustering/Ward’s method for linkage | consensus clustering | [107] | 2017 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paweł, K.; Maria Małgorzata, S. CpG Island Methylator Phenotype—A Hope for the Future or a Road to Nowhere? Int. J. Mol. Sci. 2022, 23, 830. https://doi.org/10.3390/ijms23020830
Paweł K, Maria Małgorzata S. CpG Island Methylator Phenotype—A Hope for the Future or a Road to Nowhere? International Journal of Molecular Sciences. 2022; 23(2):830. https://doi.org/10.3390/ijms23020830
Chicago/Turabian StylePaweł, Karpiński, and Sąsiadek Maria Małgorzata. 2022. "CpG Island Methylator Phenotype—A Hope for the Future or a Road to Nowhere?" International Journal of Molecular Sciences 23, no. 2: 830. https://doi.org/10.3390/ijms23020830
APA StylePaweł, K., & Maria Małgorzata, S. (2022). CpG Island Methylator Phenotype—A Hope for the Future or a Road to Nowhere? International Journal of Molecular Sciences, 23(2), 830. https://doi.org/10.3390/ijms23020830