
The Veterinary Anti-Parasitic Selamectin Is a Novel Inhibitor of the Mycobacterium tuberculosis DprE1 Enzyme

 ,
,  , , ,
, , ,  ,
,  , ,
, ,  and
and
Abstract
:1. Introduction
2. Results
2.1. Mycobacterium tuberculosis DprE1 Mutants Are More Susceptible to Selamectin
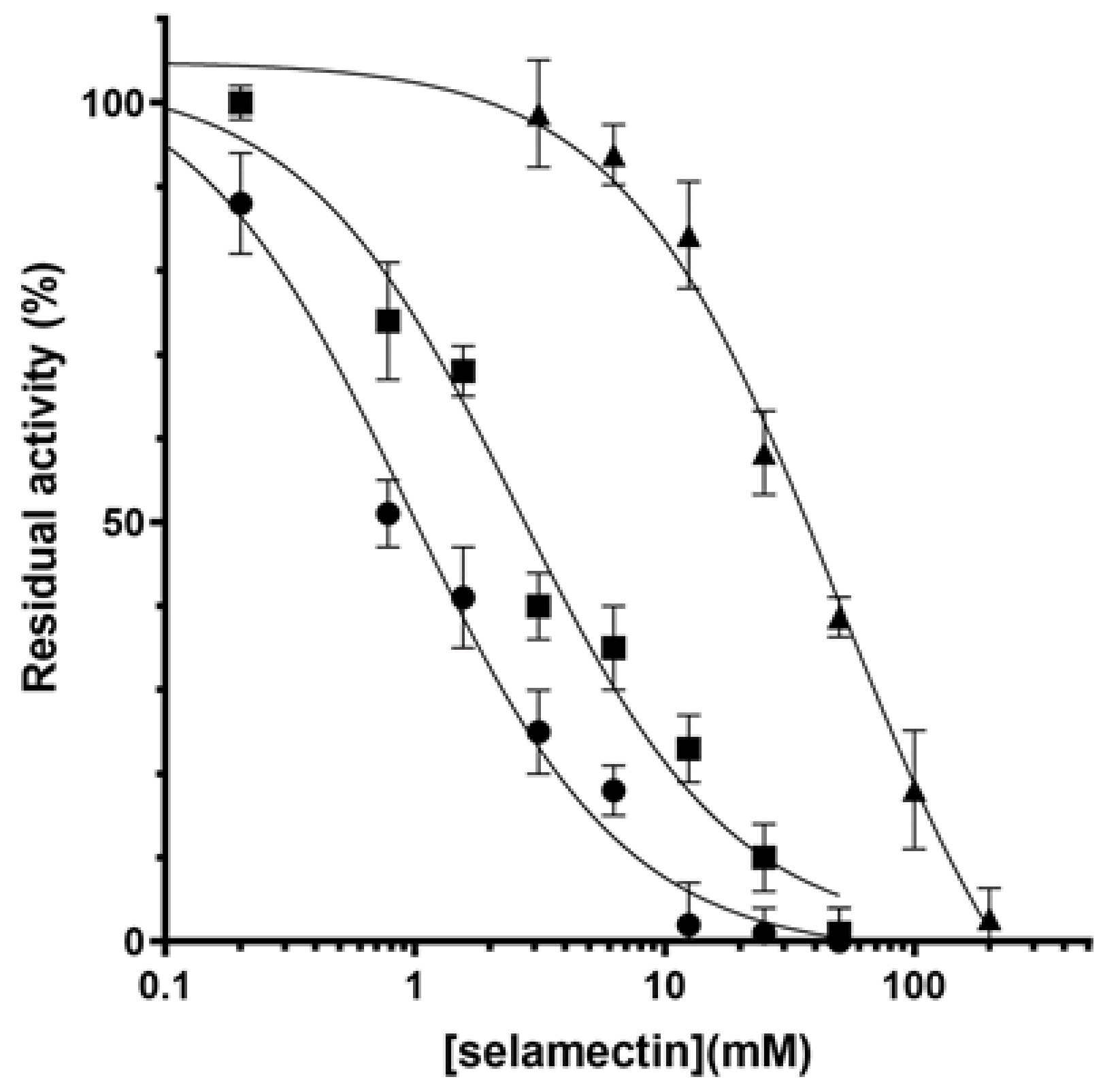
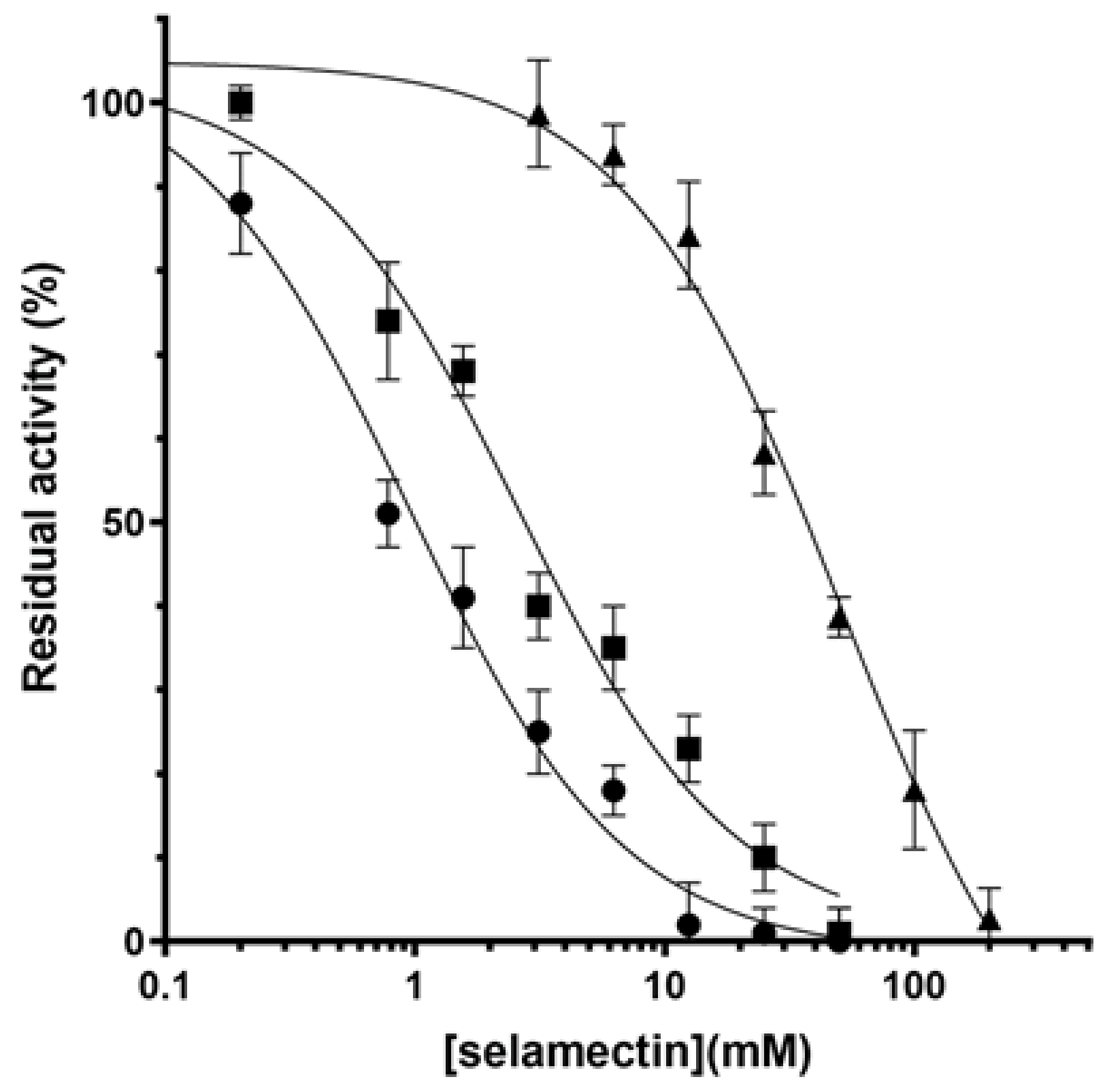
2.2. Avermectins Inhibit the DprE1 Enzyme In Vitro
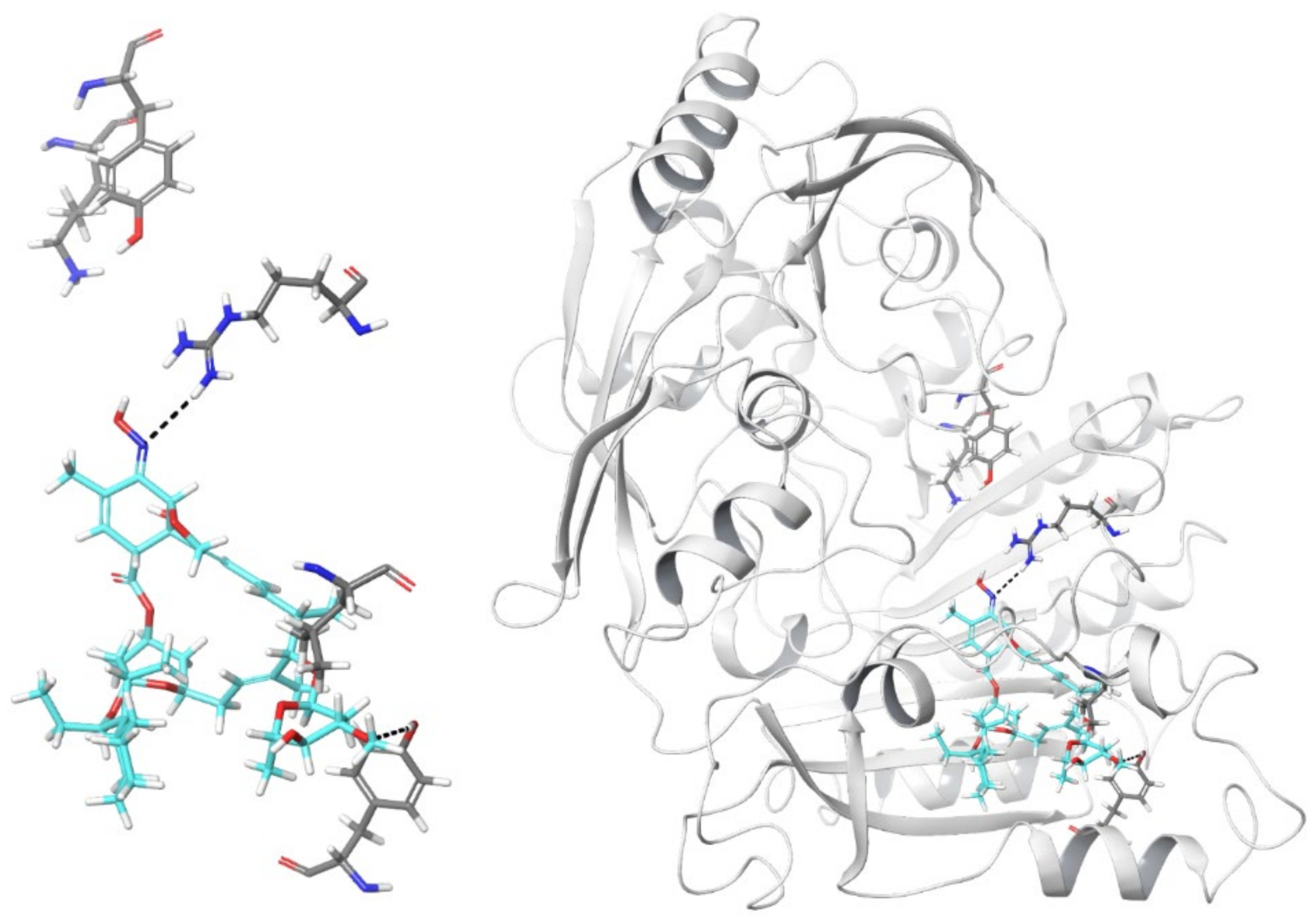
2.3. In Silico Model of Selamectin–DprE1 Binding
2.4. Position Leu282 Is Involved in the Binding of Selamectin to DprE1 but Lacks Molecular to Phenotypic Translation
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains, Reagents, and Culture Media
4.2. Drug Susceptibility Testing
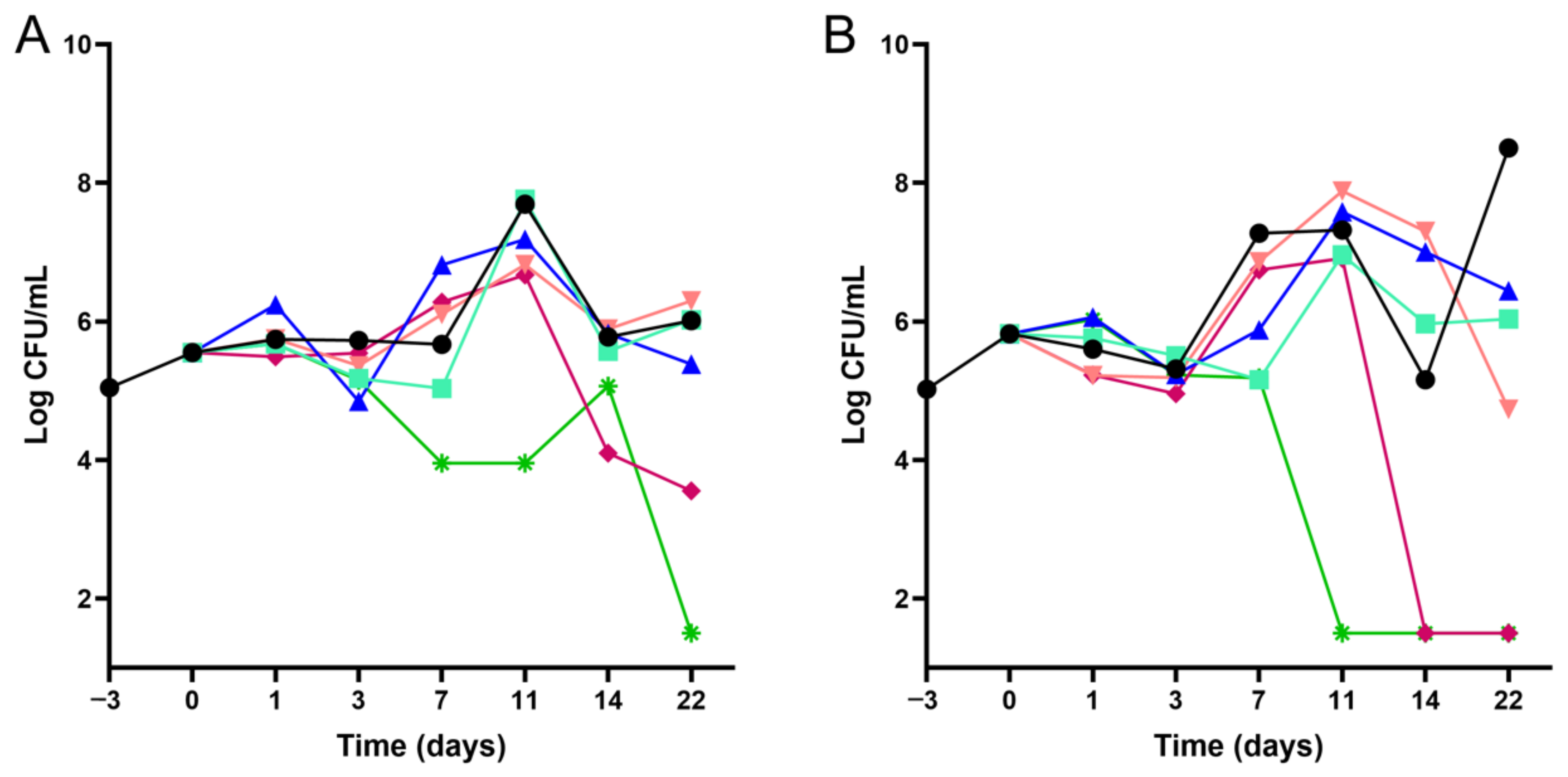
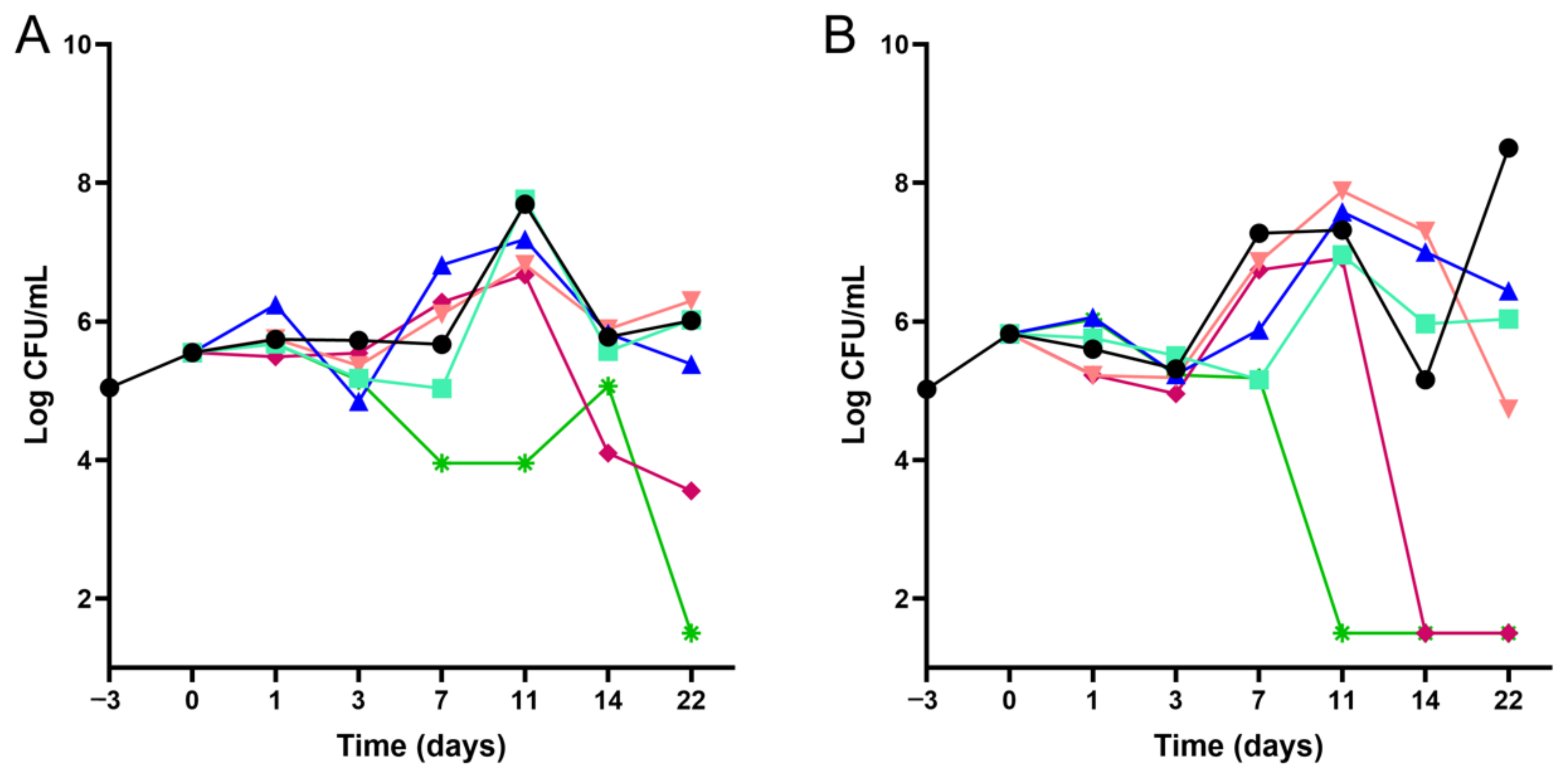
4.3. Time–Kill Kinetics Assays
4.4. Docking of Selamectin to DprE1 and Molecular Dynamics Simulations
4.5. DprE1 Enzymatic Studies
4.6. Construction of M. Smegmatis DprE1 Point Mutants
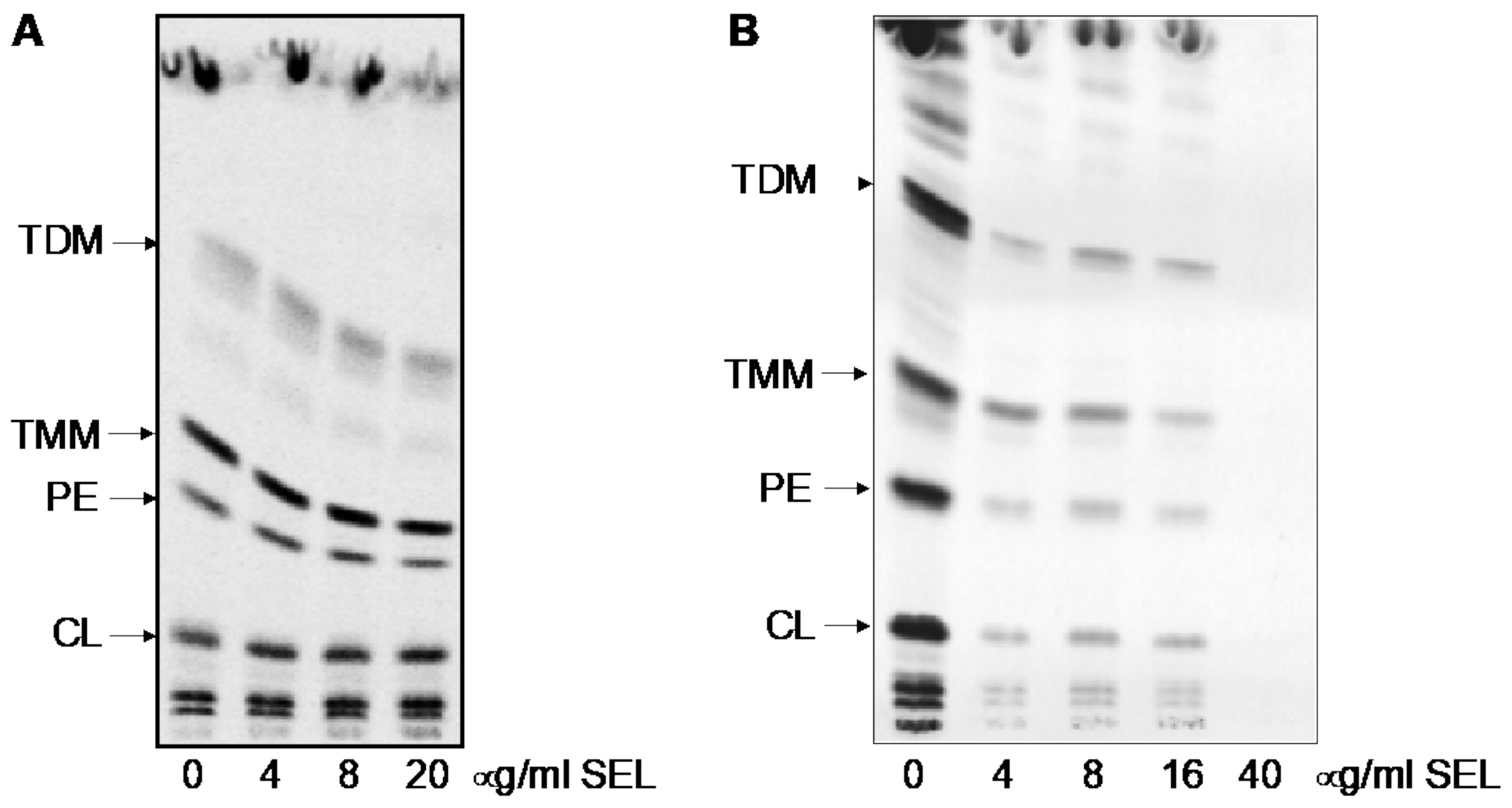
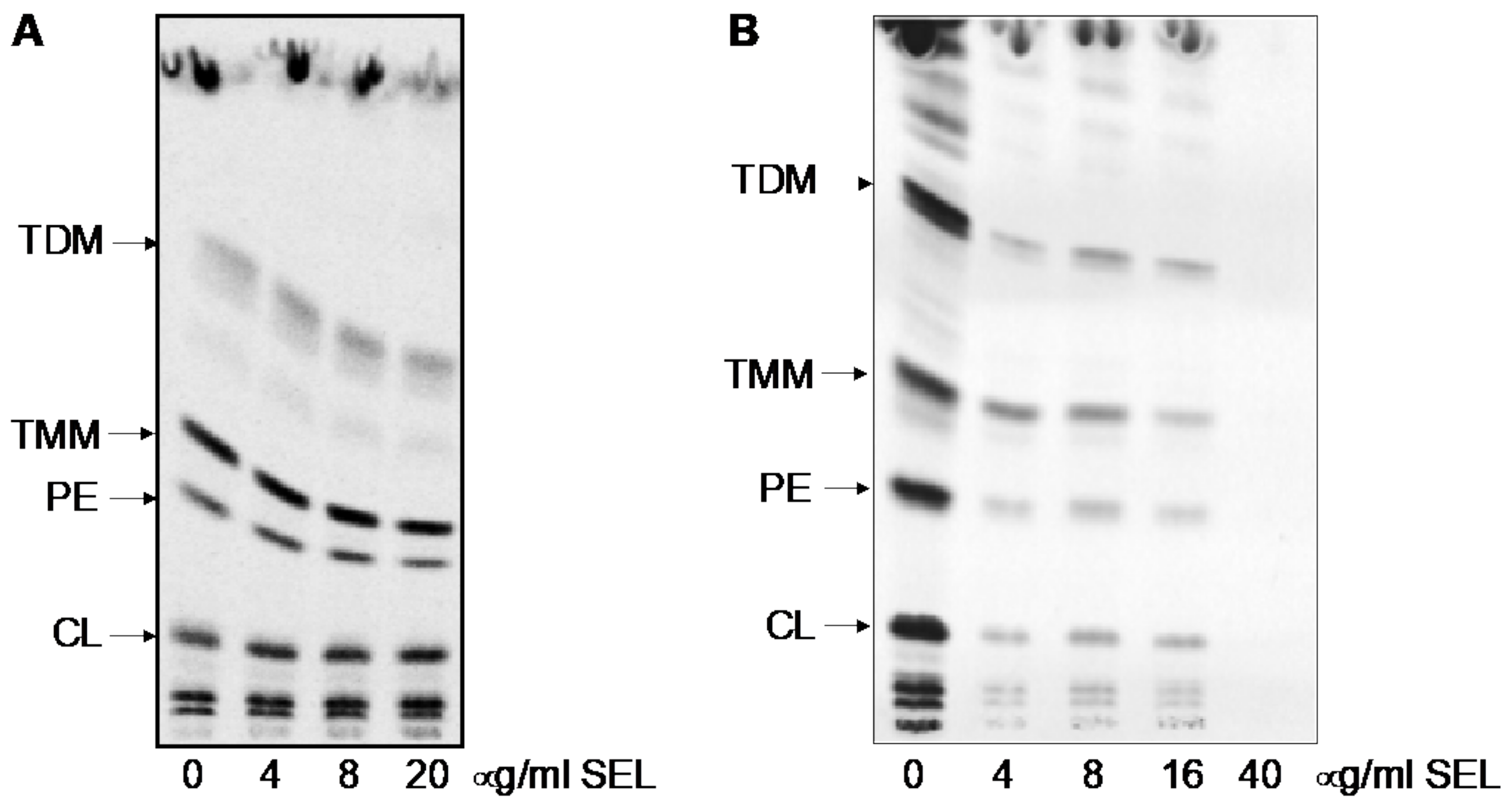
4.7. Analysis of Mycobacterial Lipids Using 14C Metabolic Labeling
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Global Tuberculosis Report. 2020. Available online: https://apps.who.int/iris/rest/bitstreams/1312164/retrieve (accessed on 5 July 2021).
- Conradie, F.; Diacon, A.H.; Ngubane, N.; Howell, P.; Everitt, D.; Crook, A.M.; Mendel, C.M.; Egizi, E.; Moreira, J.; Timm, J.; et al. Bedaquiline, Pretomanid and Linezolid for Treatment of Extensively Drug Resistant, Intolerant or Non-Responsive Multidrug Resistant Pulmonary Tuberculosis. N. Engl. J. Med. 2020, 382, 893–902. [Google Scholar] [CrossRef]
- Dorman, S.E.; Nahid, P.; Kurbatova, E.V.; Goldberg, S.V.; Bozeman, L.; Burman, W.J.; Chang, K.-C.; Chen, M.; Cotton, M.; Dooley, K.E.; et al. High-Dose Rifapentine with or without Moxifloxacin for Shortening Treatment of Pulmonary Tuberculosis: Study Protocol for TBTC Study 31/ACTG A5349 Phase 3 Clinical Trial. Contemp. Clin. Trials. 2020, 90, 105938. [Google Scholar] [CrossRef] [PubMed]
- Pipeline|Working Group for New TB Drugs. Available online: https://www.newtbdrugs.org/pipeline/clinical (accessed on 5 July 2021).
- World Health Organization; Global Tuberculosis Programme. WHO Treatment Guidelines for Drug-Resistant Tuberculosis: 2016 Update; WHO: Geneva, Switzerland, 2016; Available online: https://apps.who.int/iris/rest/bitstreams/1061087/retrieve (accessed on 5 July 2021).
- World Health Organization. Prioritization of Pathogens to Guide Discovery, Research and Development of New Antibiotics for Drug-Resistant Bacterial Infections Including Tuberculosis; WHO: Geneva, Switzerland, 2017; Available online: https://apps.who.int/iris/handle/10665/311820 (accessed on 5 July 2021).
- World Health Organization. 2020 Antibacterial Agents in Clinical and Preclinical Development: An Overview and Analysis; WHO: Geneva, Switzerland, 2021; Available online: https://apps.who.int/iris/rest/bitstreams/1341746/retrieve (accessed on 5 July 2021).
- Farha, M.A.; Brown, E.D. Drug Repurposing for Antimicrobial Discovery. Nat. Microbiol. 2019, 4, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Lim, L.E.; Vilchèze, C.; Ng, C.; Jacobs, W.R.; Ramón-García, S.; Thompson, C.J. Anthelmintic Avermectins Kill Mycobacterium tuberculosis, Including Multidrug-Resistant Clinical Strains. Antimicrob. Agents Chemother. 2013, 57, 1040–1046. [Google Scholar] [CrossRef] [Green Version]
- Scherr, N.; Pluschke, G.; Thompson, C.J.; Ramón-García, S. Selamectin Is the Avermectin with the Best Potential for Buruli Ulcer Treatment. PLoS Negl. Trop. Dis. 2015, 9, e0003996. [Google Scholar] [CrossRef]
- Muñoz-Muñoz, L.; Shoen, C.; Sweet, G.; Vitoria, A.; Bull, T.J.; Cynamon, M.; Thompson, C.J.; Ramón-García, S. Repurposing Avermectins and Milbemycins against Mycobacteroides Abscessus and Other Nontuberculous Mycobacteria. Antibiotics 2021, 10, 381. [Google Scholar] [CrossRef] [PubMed]
- Burg, R.W.; Miller, B.M.; Baker, E.E.; Birnbaum, J.; Currie, S.A.; Hartman, R.; Kong, Y.-L.; Monaghan, R.L.; Olson, G.; Putter, I.; et al. Avermectins, New Family of Potent Anthelmintic Agents: Producing Organism and Fermentation. Antimicrob. Agents Chemother. 1979, 15, 361–367. [Google Scholar] [CrossRef] [Green Version]
- Ōmura, S. A Splendid Gift from the Earth: The Origins and Impact of the Avermectins (Nobel Lecture). Angew. Chem. Int. Ed. 2016, 55, 10190–10209. [Google Scholar] [CrossRef]
- The Ivermectin Roadmappers; Billingsley, P.; Binka, F.; Chaccour, C.; Foy, B.; Gold, S.; Gonzalez-Silva, M.; Jacobson, J.; Jagoe, G.; Jones, C.; et al. A Roadmap for the Development of Ivermectin as a Complementary Malaria Vector Control Tool. Am. J. Trop. Med. Hyg. 2020, 102, 3–24. [Google Scholar] [CrossRef] [Green Version]
- Nolan, T.J.; Lok, J.B. Macrocyclic Lactones in the Treatment and Control of Parasitism in Small Companion Animals. Curr. Pharm. Biotechnol. 2012, 13, 1078–1094. [Google Scholar] [CrossRef]
- Revolution. Prescribing Information. Available online: https://www.zoetisus.com/_locale-assets/mcm-portal-assets/products/pdf/revolution-prescribing-information.pdf (accessed on 12 July 2021).
- Wolstenholme, A.J.; Rogers, A.T. Glutamate-gated chloride channels and the mode of action of the avermectin/milbemycin anthelmintics. Parasitology 2005, 131, S85–S95. [Google Scholar] [CrossRef] [PubMed]
- Mikusová, K.; Huang, H.; Yagi, T.; Holsters, M.; Vereecke, D.; D’Haeze, W.; Scherman, M.S.; Brennan, P.J.; McNeil, M.R.; Crick, D.C. Decaprenylphosphoryl Arabinofuranose, the Donor of the D-Arabinofuranosyl Residues of Mycobacterial Arabinan, Is Formed via a Two-Step Epimerization of Decaprenylphosphoryl Ribose. J. Bacteriol. 2005, 187, 8020–8025. [Google Scholar] [CrossRef] [Green Version]
- Abrahams, K.A.; Besra, G.S. Mycobacterial Cell Wall Biosynthesis: A Multifaceted Antibiotic Target. Parasitology 2018, 145, 116–133. [Google Scholar] [CrossRef] [Green Version]
- Kolly, G.S.; Boldrin, F.; Sala, C.; Dhar, N.; Hartkoorn, R.C.; Ventura, M.; Serafini, A.; McKinney, J.D.; Manganelli, R.; Cole, S.T. Assessing the essentiality of the decaprenyl-phospho-d-arabinofuranose pathway in Mycobacterium tuberculosis using conditional mutants. Mol. Microbiol. 2014, 92, 194–211. [Google Scholar] [CrossRef] [Green Version]
- Degiacomi, G.; Belardinelli, J.M.; Pasca, M.R.; De Rossi, E.; Riccardi, G.; Chiarelli, L.R. Promiscuous Targets for Antitubercular Drug Discovery: The Paradigm of DprE1 and MmpL3. Appl. Sci. 2020, 10, 623. [Google Scholar] [CrossRef] [Green Version]
- Makarov, V.; Manina, G.; Mikusova, K.; Möllmann, U.; Ryabova, O.; Saint-Joanis, B.; Dhar, N.; Pasca, M.R.; Buroni, S.; Lucarelli, A.P.; et al. Benzothiazinones Kill Mycobacterium tuberculosis by Blocking Arabinan Synthesis. Science 2009, 324, 801–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, G.; Orena, B.S.; Chiarelli, L.R.; Degiacomi, G.; Riabova, O.; Sammartino, J.C.; Makarov, V.; Riccardi, G.; Pasca, M.R. Rv0579 Is Involved in the Resistance to the TP053 Antitubercular Prodrug. Front. Microbiol. 2020, 11, 292. [Google Scholar] [CrossRef] [PubMed]
- Poce, G.; Bates, R.H.; Alfonso, S.; Cocozza, M.; Porretta, G.C.; Ballell, L.; Rullas, J.; Ortega, F.; De Logu, A.; Agus, E.; et al. Improved BM212 MmpL3 Inhibitor Analogue Shows Efficacy in Acute Murine Model of Tuberculosis Infection. PLoS ONE 2013, 8, e56980. [Google Scholar] [CrossRef]
- Makarov, V.; Neres, J.; Hartkoorn, R.C.; Ryabova, O.B.; Kazakova, E.; Šarkan, M.; Huszár, S.; Piton, J.; Kolly, G.S.; Vocat, A.; et al. The 8-Pyrrole-Benzothiazinones Are Noncovalent Inhibitors of DprE1 from Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2015, 59, 4446–4452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, G.; Chiarelli, L.R.; Esposito, M.; Makarov, V.; Bellinzoni, M.; Hartkoorn, R.C.; Degiacomi, G.; Boldrin, F.; Ekins, S.; de Jesus Lopes Ribeiro, A.L.; et al. Thiophenecarboxamide Derivatives Activated by EthA Kill Mycobacterium tuberculosis by Inhibiting the CTP Synthetase PyrG. Chem. Biol. 2015, 22, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Degiacomi, G.; Sammartino, J.C.; Sinigiani, V.; Marra, P.; Urbani, A.; Pasca, M.R. In Vitro Study of Bedaquiline Resistance in Mycobacterium tuberculosis Multi-Drug Resistant Clinical Isolates. Front. Microbiol. 2020, 11, 559469. [Google Scholar] [CrossRef]
- Neres, J.; Pojer, F.; Molteni, E.; Chiarelli, L.R.; Dhar, N.; Boy-Rottger, S.; Buroni, S.; Fullam, E.; Degiacomi, G.; Lucarelli, A.P.; et al. Structural Basis for Benzothiazinone-Mediated Killing of Mycobacterium tuberculosis. Sci. Transl. Med. 2012, 4, 150ra121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naik, M.; Humnabadkar, V.; Tantry, S.J.; Panda, M.; Narayan, A.; Guptha, S.; Panduga, V.; Manjrekar, P.; Jena, L.K.; Koushik, K.; et al. 4-aminoquinolone piperidine amides: Noncovalent inhibitors of DprE1 with long residence time and potent antimycobacterial activity. J. Med. Chem. 2014, 57, 5419–5434. [Google Scholar] [CrossRef] [PubMed]
- Neres, J.; Hartkoorn, R.C.; Chiarelli, L.R.; Gadupudi, R.; Pasca, M.R.; Mori, G.; Venturelli, A.; Savina, S.; Makarov, V.; Kolly, G.S.; et al. 2-Carboxyquinoxalines Kill Mycobacterium tuberculosis through Noncovalent Inhibition of DprE1. ACS Chem. Biol. 2015, 10, 705–714. [Google Scholar] [CrossRef]
- Karabanovich, G.; Dušek, J.; Savková, K.; Pavliš, O.; Pávková, I.; Korábečný, J.; Kučera, T.; Kočová Vlčková, H.; Huszár, S.; Konyariková, Z.; et al. Development of 3,5-Dinitrophenyl-Containing 1,2,4-Triazoles and Their Trifluoromethyl Analogues as Highly Efficient Antitubercular Agents Inhibiting Decaprenylphosphoryl-β- d -Ribofuranose 2′-Oxidase. J. Med. Chem. 2019, 62, 8115–8139. [Google Scholar] [CrossRef] [PubMed]
- Batiha, G.E.-S.; Alqahtani, A.; Ilesanmi, O.B.; Saati, A.A.; El-Mleeh, A.; Hetta, H.F.; Beshbishy, A.M. Avermectin Derivatives, Pharmacokinetics, Therapeutic and Toxic Dosages, Mechanism of Action, and Their Biological Effects. Pharmaceuticals 2020, 13, 196. [Google Scholar] [CrossRef]
- Stelitano, G.; Sammartino, J.C.; Chiarelli, L.R. Multitargeting Compounds: A Promising Strategy to Overcome Multi-Drug Resistant Tuberculosis. Molecules 2020, 25, 1239. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger LLC. Maestro; Schrödinger LLC: New York, NY, USA, 2019. [Google Scholar]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; ACM Press: New York, NY, USA, 2006. [Google Scholar]
- Kessel, J.C.V.; Hatfull, G.F. Efficient Point Mutagenesis in Mycobacteria Using Single-Stranded DNA Recombineering: Characterization of Antimycobacterial Drug Targets. Mol. Microbiol. 2008, 67, 1094–1107. [Google Scholar] [CrossRef]
- Kessel, J.C.V.; Hatfull, G.F. Recombineering in Mycobacterium tuberculosis. Nat. Methods 2007, 4, 147–152. [Google Scholar] [CrossRef]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A Simple Method for the Isolation and Purification of Total Lipides from Animal Tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| M. tuberculosis Strain | Strain Resistance Profile | MIC INH (µg/mL) | MIC SEL (µg/mL) | MIC BTZ043 (ng/mL) | Reference |
|---|---|---|---|---|---|
| H37Rv | 0.025 | 4 | 1 | ||
| H37Rv-pSODIT | 1 | ||||
| 53.3 (Rv2466c, W28S) | TP053 | 0.025 | 2–4 | nd | |
| 53.8 (Rv0579, L240V) | TP053 | 0.025 | 2–4 | nd | [23] |
| NTB1 (DprE1 C387S) | BTZ | 0.025 | 2 | >1000 | [22] |
| NTB9 (DprE1 C387G | BTZ | 0.025 | 2 | 1000 | [22] |
| H37Rv-pSODIT:dprE1 | BTZ | 0.025 | 4 | 1000 | |
| DR1 (mmpL3, V681I) | 1,5-diarylpyrroles | 0.025 | 2–4 | nd | [24] |
| Ty1 (Rv3405c, c190t) | Ty38c | 0.025 | 2–4 | nd | [25] |
| 88.1 (coaA, Q207R) | Thiophenecarboxamide derivatives | 0.025 | 2–4 | nd | [26] |
| 88.7 (pyrG, V186G) | Thiophenecarboxamide derivatives | 0.025 | 2–4 | nd | [26] |
| 81.10 (ethA, D1109-37) | Thiophenecarboxamide derivatives | 0.025 | 4–8 | nd | [26] |
| IC1 | STR, INH, RIF, EMB | >0.2 | 4–8 | nd | [27] |
| IC2 | STR, INH, RIF, EMB, PYR, ETH, CAP | >0.2 | 4–8 | nd | [27] |
| Avermectin | IC50 (µM) |
|---|---|
| Ivermectin | 13.2 ± 3.3 |
| Milbemycin oxime | 25.5 ± 5.6 |
| Moxidectin | 6.1 ± 0.9 |
| Selamectin | 2.6 ± 1.1 |
| BTZ-043 1 | 0.004 |
| DprE1 Variant | Km (µM) | kcat (min−1) | kcat/Km (min−1 µM−1) | IC50 SEL (µM) |
|---|---|---|---|---|
| Wild-type | 0.19 ± 0.20 | 5.1 ± 0.4 | 26.8 ± 1.1 | 2.6 ± 1.1 |
| C394A | 0.33 ± 0.22 | 2.8 ± 0.5 | 8.5 ± 0.6 | 3.9 ± 1.0 |
| C394G | nd | nd | nd | nd |
| C394S | 0.31 ± 0.15 | 1.6 ± 0.3 | 5.2 ± 0.6 | 12.1 ± 1.2 |
| L282F | 0.16 ± 0.12 | 4.5 ± 0.3 | 28.1 ± 1.4 | 1.0 ± 0.7 |
| L282V | 0.21 ± 0.15 | 4.3 ± 0.4 | 20.4 ± 1.1 | 50.6 ± 9.2 |
| M. smegmatis Strain | MIC BTZ-043 (ng/mL) | MIC SEL (µg/mL) |
|---|---|---|
| mc2155 | 8 | 4 |
| mc2155-pJV53H | 8 | 4 |
| mc2155-pJV53H DprE1 L282F | 8 | 4 |
| mc2155-pJV53H DprE1 L282V | 4 | 4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ezquerra-Aznárez, J.M.; Degiacomi, G.; Gašparovič, H.; Stelitano, G.; Sammartino, J.C.; Korduláková, J.; Governa, P.; Manetti, F.; Pasca, M.R.; Chiarelli, L.R.; et al. The Veterinary Anti-Parasitic Selamectin Is a Novel Inhibitor of the Mycobacterium tuberculosis DprE1 Enzyme. Int. J. Mol. Sci. 2022, 23, 771. https://doi.org/10.3390/ijms23020771
Ezquerra-Aznárez JM, Degiacomi G, Gašparovič H, Stelitano G, Sammartino JC, Korduláková J, Governa P, Manetti F, Pasca MR, Chiarelli LR, et al. The Veterinary Anti-Parasitic Selamectin Is a Novel Inhibitor of the Mycobacterium tuberculosis DprE1 Enzyme. International Journal of Molecular Sciences. 2022; 23(2):771. https://doi.org/10.3390/ijms23020771
Chicago/Turabian StyleEzquerra-Aznárez, José Manuel, Giulia Degiacomi, Henrich Gašparovič, Giovanni Stelitano, Josè Camilla Sammartino, Jana Korduláková, Paolo Governa, Fabrizio Manetti, Maria Rosalia Pasca, Laurent Roberto Chiarelli, and et al. 2022. "The Veterinary Anti-Parasitic Selamectin Is a Novel Inhibitor of the Mycobacterium tuberculosis DprE1 Enzyme" International Journal of Molecular Sciences 23, no. 2: 771. https://doi.org/10.3390/ijms23020771
APA StyleEzquerra-Aznárez, J. M., Degiacomi, G., Gašparovič, H., Stelitano, G., Sammartino, J. C., Korduláková, J., Governa, P., Manetti, F., Pasca, M. R., Chiarelli, L. R., & Ramón-García, S. (2022). The Veterinary Anti-Parasitic Selamectin Is a Novel Inhibitor of the Mycobacterium tuberculosis DprE1 Enzyme. International Journal of Molecular Sciences, 23(2), 771. https://doi.org/10.3390/ijms23020771