
DNA Methylation and Non-Coding RNAs during Tissue-Injury Associated Pain

 , , , ,
, , , , 

Abstract
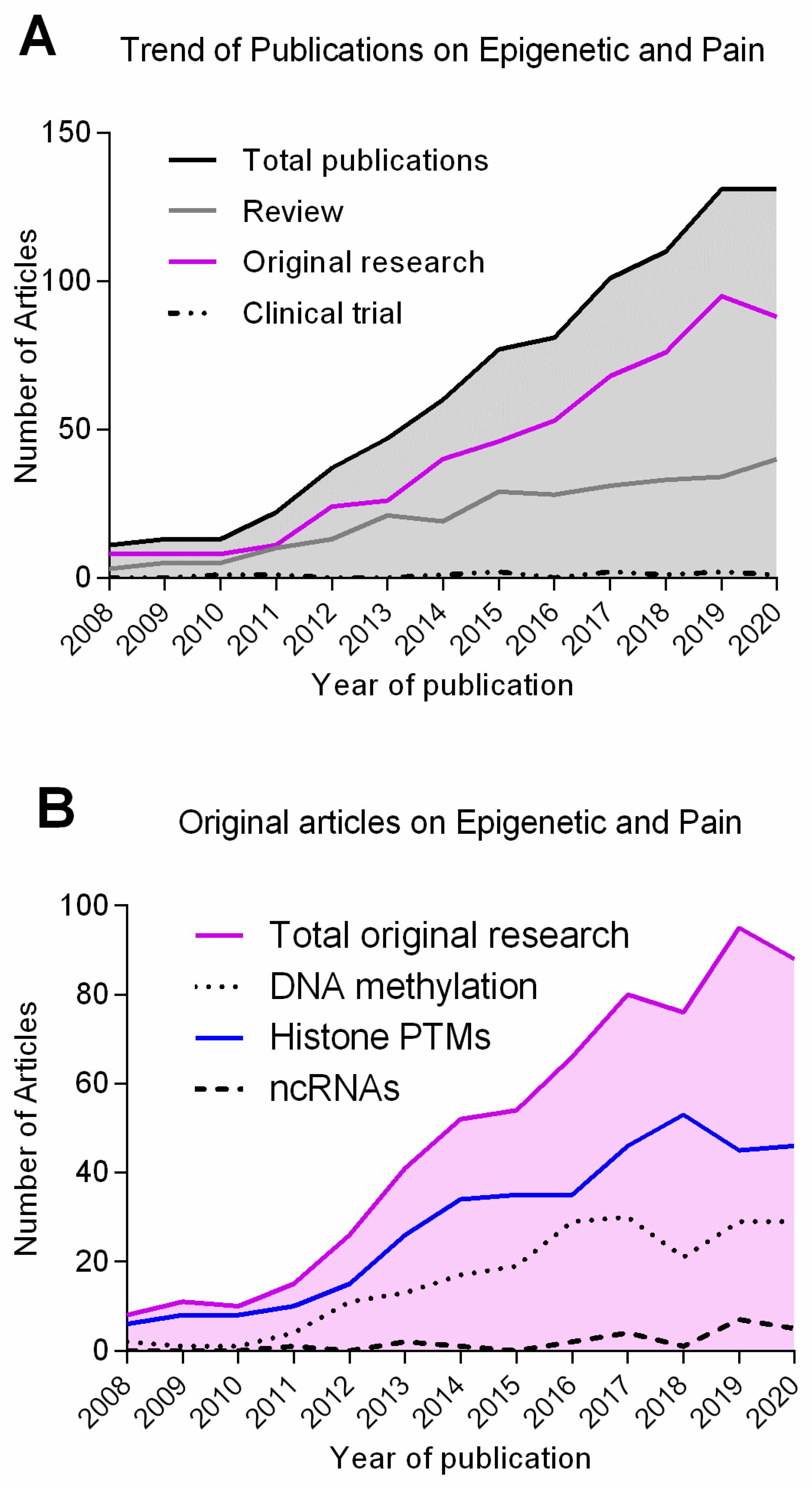
1. Introduction
1.1. Pain as a Disease
1.2. Transcriptional Changes Are Pivotal for the Persistence of Tissue Injury-Associated Pain
1.3. Epigenetic Mechanisms Regulate Gene Transcription in Adaptive and Maladaptive Responses
2. DNA Methylation and Pain
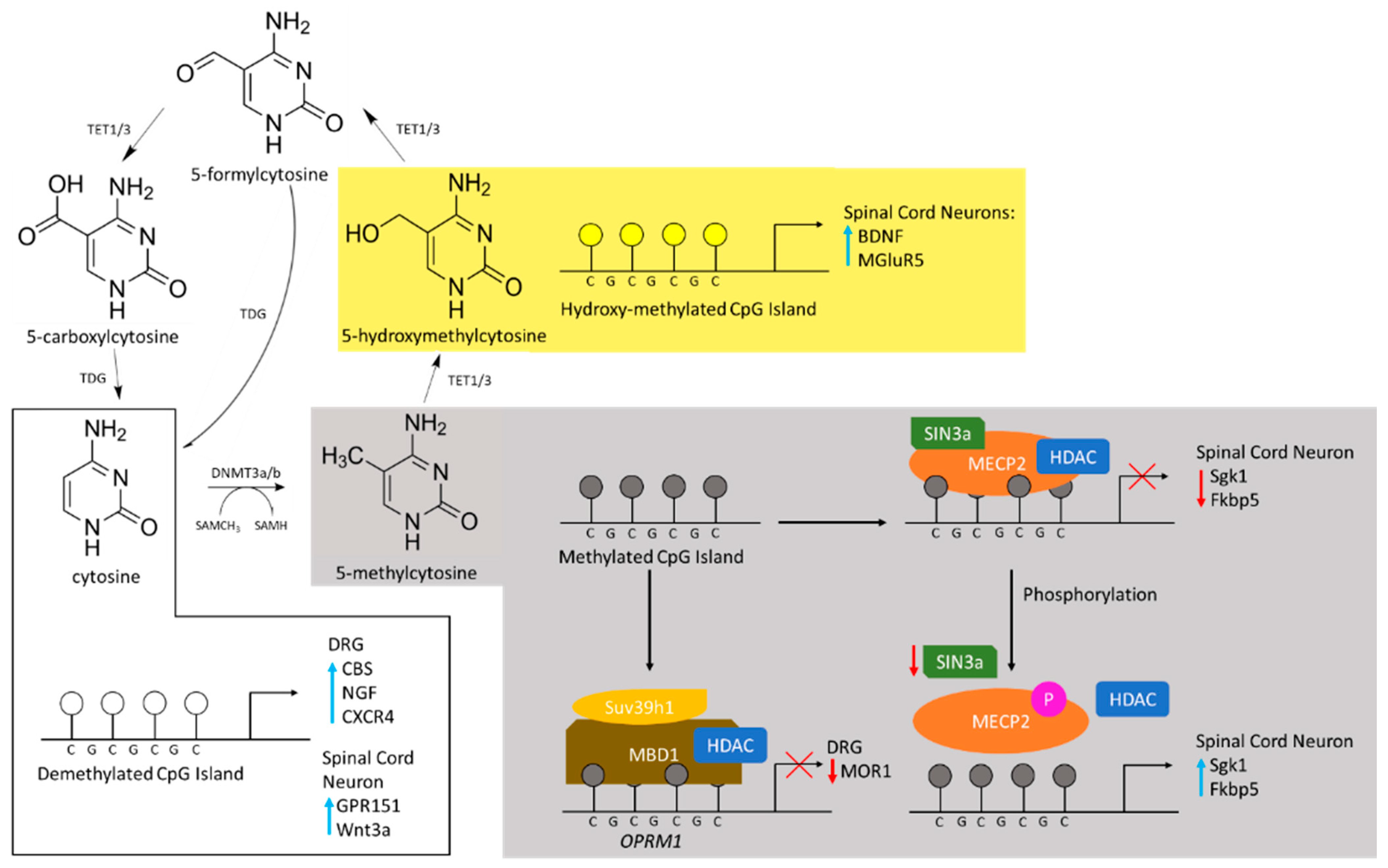
2.1. DNA Methylation Is Associated with Gene Silencing
2.2. DNA Methylation during Lasting Pain
2.3. Writers of DNA Methylation during Pain Consolidation
2.4. Readers of DNA Methylation Associated with Lasting Pain
2.5. Erasers of DNA Methylation during Inflammatory and Neuropathic Pain
2.6. DNA Methylation as a Therapeutic Target for Chronic Pain and Biomarker of Pain Progression
2.7. Future Directions to Study DNA Methylation during Pain Progression
3. Non-Coding RNAs and Pain
3.1. Types of Non-Coding RNAs, Their Function and Classification
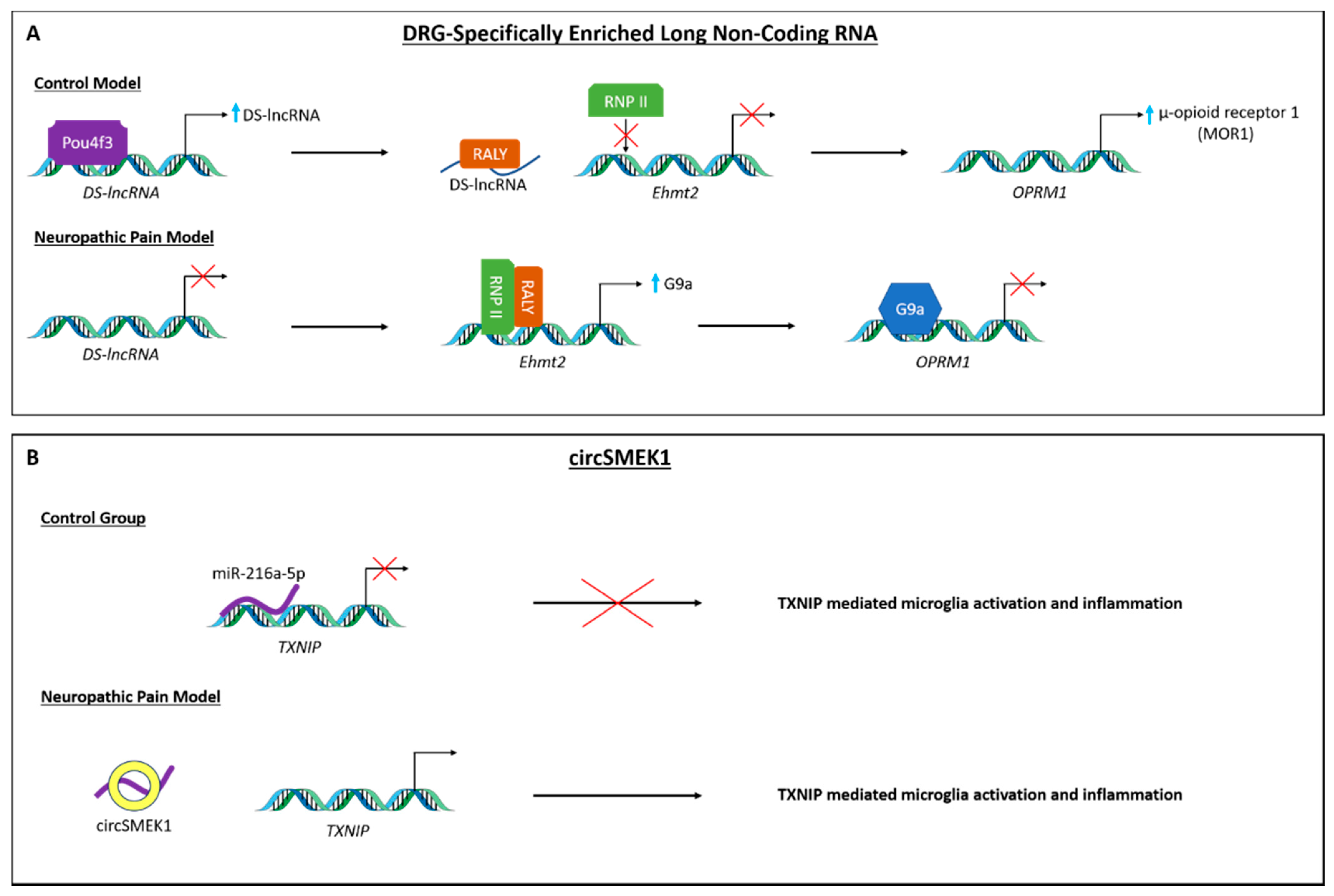
3.2. lncRNAs and circRNAs during Neuropathic Pain Processing
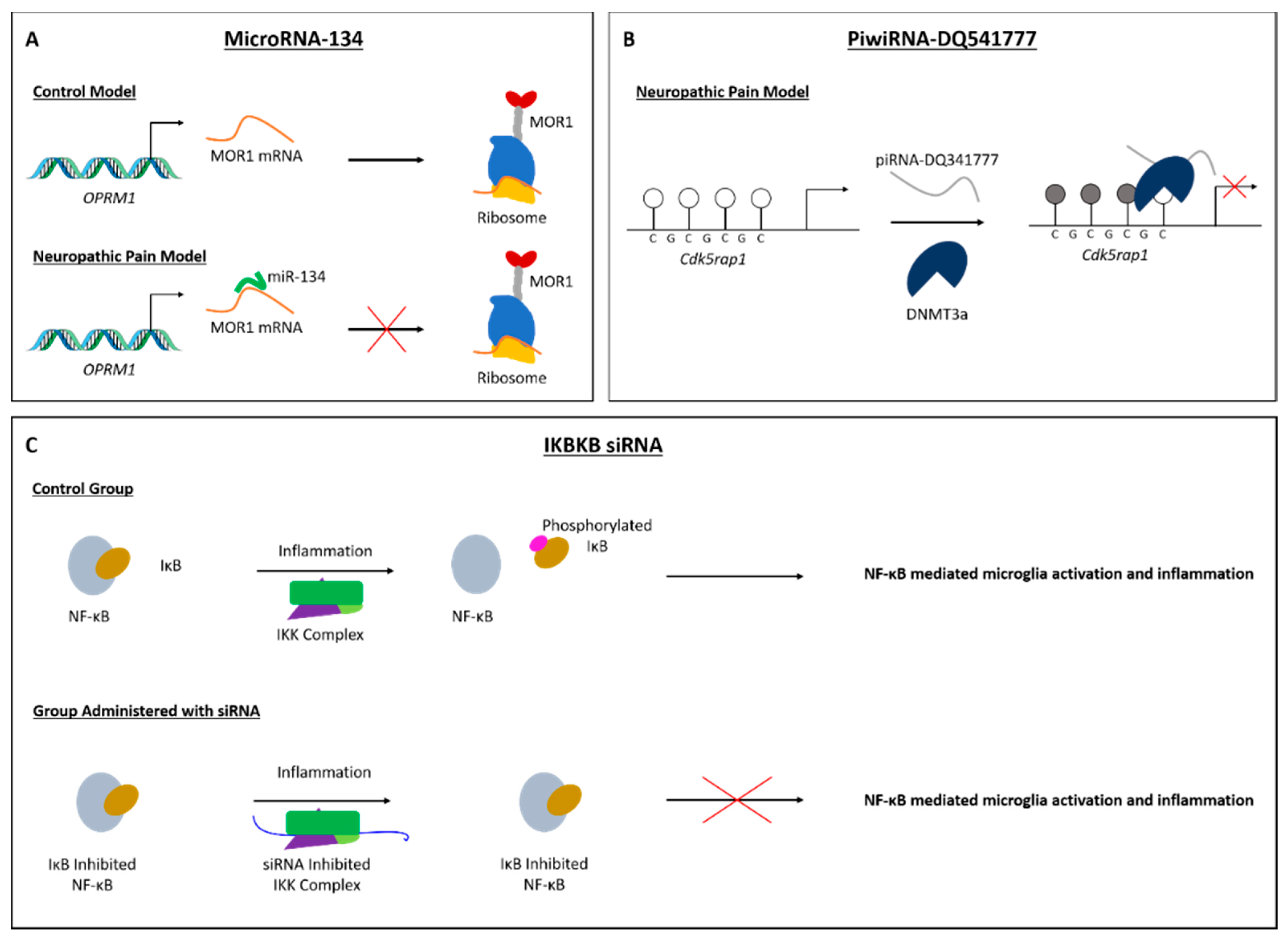
3.3. miRNAs in the Progression of Both Inflammatory and Neuropathic Pain
3.4. piRNA and siRNA in Neuropathic Pain
3.5. Could ncRNAs Be Potential Targets/Biomarkers for Pain Management?
3.6. Novel Approaches on ncRNAs for Pain Processing
4. Final Considerations and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Torres-Perez, J.V.; Irfan, J.; Febrianto, M.R.; Di Giovanni, S.; Nagy, I. Histone post-translational modifications as potential therapeutic targets for pain management. Trends Pharmacol. Sci. 2021, 42, 897–911. [Google Scholar] [CrossRef]
- Raja, S.N.; Carr, D.B.; Cohen, M.; Finnerup, N.B.; Flor, H.; Gibson, S.; Keefe, F.J.; Mogil, J.S.; Ringkamp, M.; Sluka, K.A.; et al. The revised International Association for the Study of Pain definition of pain: Concepts, challenges, and compromises. Pain 2020, 161, 1976–1982. [Google Scholar] [CrossRef]
- Raffaeli, W.; Arnaudo, E. Pain as a disease: An overview. J. Pain Res. 2017, 10, 2003–2008. [Google Scholar] [CrossRef]
- Costigan, M.; Scholz, J.; Woolf, C.J. Neuropathic pain: A maladaptive response of the nervous system to damage. Annu. Rev. Neurosci. 2009, 32, 1–32. [Google Scholar] [CrossRef]
- Finnerup, N.B.; Sindrup, S.H.; Jensen, T.S. The evidence for pharmacological treatment of neuropathic pain. Pain 2010, 150, 573–581. [Google Scholar] [CrossRef]
- Yam, M.F.; Loh, Y.C.; Tan, C.S.; Adam, S.K.; Manan, N.A.; Basir, R. General pathways of pain sensation and the major neurotransmitters involved in pain regulation. Int. J. Mol. Sci. 2018, 19, 2164. [Google Scholar] [CrossRef]
- Basbaum, A.I.; Bautista, D.M.; Scherrer, G.; Julius, D. Cellular and Molecular Mechanisms of Pain. Cell 2009, 139, 267–284. [Google Scholar] [CrossRef]
- Tsagareli, M.G. Pain and memory: Do they share similar mechanisms? World J. Neurosci. 2013, 3, 39–48. [Google Scholar] [CrossRef]
- Ji, R.R.; Kohno, T.; Moore, K.A.; Woolf, C.J. Central sensitization and LTP: Do pain and memory share similar mechanisms? Trends Neurosci. 2003, 26, 696–705. [Google Scholar] [CrossRef]
- Latremoliere, A.; Woolf, C.J. Central Sensitization: A Generator of Pain Hypersensitivity by Central Neural Plasticity. J. Pain 2009, 10, 895–926. [Google Scholar] [CrossRef]
- Pinho-Ribeiro, F.A.; Verri, W.A.; Chiu, I.M. Nociceptor Sensory Neuron–Immune Interactions in Pain and Inflammation. Trends Immunol. 2017, 38, 5–19. [Google Scholar] [CrossRef]
- Woolf, C.J. Central sensitization: Implications for the diagnosis and treatment of pain. Pain 2011, 152, S2–S15. [Google Scholar] [CrossRef]
- Godfrey, K.M.; Lillycrop, K.A.; Burdge, G.C.; Gluckman, P.D.; Hanson, M.A. Epigenetic mechanisms and the mismatch concept of the developmental origins of health and disease. Pediatr. Res. 2007, 61, 5R–10R. [Google Scholar] [CrossRef]
- Wu, C.T.; Morris, J.R. Genes, genetics, and epigenetics: A correspondence. Science 2001, 293, 1103–1105. [Google Scholar] [CrossRef]
- Bai, G.; Ren, K.; Dubner, R. Epigenetic regulation of persistent pain. Transl. Res. 2015, 165, 177–199. [Google Scholar] [CrossRef]
- Topham, L.; Gregoire, S.; Kang, H.; Salmon-Divon, M.; Lax, E.; Millecamps, M.; Szyf, M.; Stone, L. The Transition from Acute to Chronic Pain: Dynamic Epigenetic Reprogramming of the Mouse Prefrontal Cortex up to One Year Following Nerve Injury. Pain 2020, 161, 2394–2409. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, C.; Guo, Q.L.; Yan, J.Q.; Zhu, X.Y.; Huang, C.S.; Zou, W.Y. Intrathecal 5-azacytidine inhibits global DNA methylation and methyl- CpG-binding protein 2 expression and alleviates neuropathic pain in rats following chronic constriction injury. Brain Res. 2011, 1418, 64–69. [Google Scholar] [CrossRef]
- Géranton, S.M.; Morenilla-Palao, C.; Hunt, S.P. A role for transcriptional repressor methyl-CpG-binding protein 2 and plasticity-related gene serum- and glucocorticoid-inducible kinase 1 in the induction of inflammatory pain states. J. Neurosci. 2007, 27, 6163–6173. [Google Scholar] [CrossRef]
- Géranton, S.M.; Fratto, V.; Tochiki, K.K.; Hunt, S.P. Descending serotonergic controls regulate inflammation-induced mechanical sensitivity and methyl-CpG-binding protein 2 phosphorylation in the rat superficial dorsal horn. Mol. Pain 2008, 4, 35. [Google Scholar] [CrossRef]
- Tochiki, K.K.; Cunningham, J.; Hunt, S.P.; Géranton, S.M. The expression of spinal methyl-CpG-binding protein 2, DNA methyltransferases and histone deacetylases is modulated in persistent pain states. Mol. Pain 2012, 8, 14. [Google Scholar] [CrossRef]
- Zhang, R.; Huang, M.; Cao, Z.; Qi, J.; Qiu, Z.; Chiang, L.Y. MeCP2 plays an analgesic role in pain transmission through regulating CREB / miR-132 pathway. Mol. Pain 2015, 11, 19. [Google Scholar] [CrossRef]
- Sun, L.; Gu, X.; Pan, Z.; Guo, X.; Liu, J.; Atianjoh, F.E.; Wu, S.; Mo, K.; Xu, B.; Liang, L.; et al. Contribution of DNMT1 to Neuropathic Pain Genesis Partially through Epigenetically Repressing Kcna2 in Primary Afferent Neurons. J. Neurosci. 2019, 39, 6595–6607. [Google Scholar] [CrossRef]
- Zhao, J.Y.; Liang, L.; Gu, X.; Li, Z.; Wu, S.; Sun, L.; Atianjoh, F.E.; Feng, J.; Mo, K.; Jia, S.; et al. DNA methyltransferase DNMT3a contributes to neuropathic pain by repressing Kcna2 in primary afferent neurons. Nat. Commun. 2017, 8, 14712. [Google Scholar] [CrossRef]
- Liu, L.; Xu, D.; Wang, T.; Zhang, Y.; Yang, X.; Wang, X.; Tang, Y. Epigenetic reduction of miR-214-3p upregulates astrocytic colony-stimulating factor-1 and contributes to neuropathic pain induced by nerve injury. Pain 2020, 161, 96–108. [Google Scholar] [CrossRef]
- Wang, W.; Li, C.; Cai, Y.; Pan, Z.Z. Pain vulnerability and DNA methyltransferase 3a involved in the affective dimension of chronic pain. Mol. Pain 2017, 13, 1744806917726713. [Google Scholar] [CrossRef]
- Mo, K.; Wu, S.; Gu, X.; Xiong, M.; Cai, W.; Atianjoh, F.E.; Jobe, E.E.; Zhao, X.; Tu, W.F.; Tao, Y.X. MBD1 contributes to the genesis of acute pain and neuropathic pain by epigenetic silencing of Oprm1 and Kcna2 genes in primary sensory neurons. J. Neurosci. 2018, 38, 9883–9899. [Google Scholar] [CrossRef]
- Oliveira, A.M.M.; Litke, C.; Paldy, E.; Hagenston, A.M.; Lu, J.; Kuner, R.; Bading, H.; Mauceri, D. Epigenetic control of hypersensitivity in chronic inflammatory pain by the de novo DNA methyltransferase Dnmt3a2. Mol. Pain 2019, 15, 1744806919827469. [Google Scholar] [CrossRef]
- Li, F.; Xue, Z.Y.; Yuan, Y.; Huang, S.S.; Fan, Y.H.; Zhu, X.; Wei, L. Upregulation of CXCR4 through promoter demethylation contributes to inflammatory hyperalgesia in rats. CNS Neurosci. Ther. 2018, 24, 947–956. [Google Scholar] [CrossRef]
- Yuan, H.; Du, S.; Chen, L.; Xu, X.; Wang, Y.; Ji, F. Hypomethylation of nerve growth factor (NGF) promotes binding of C/EBPα and contributes to inflammatory hyperalgesia in rats. J. Neuroinflamm. 2020, 17, 34. [Google Scholar] [CrossRef]
- Jiang, B.C.; He, L.N.; Wu, X.B.; Shi, H.; Zhang, W.W.; Zhang, Z.J.; Cao, D.L.; Li, C.H.; Gu, J.; Gao, Y.J. Promoted interaction of C/EBPα with demethylated Cxcr3 gene promoter contributes to neuropathic pain in mice. J. Neurosci. 2017, 37, 685–700. [Google Scholar] [CrossRef]
- Jiang, B.C.; Zhang, W.W.; Yang, T.; Guo, C.Y.; Cao, D.L.; Zhang, Z.J.; Gao, Y.J. Demethylation of G-protein-coupled receptor 151 promoter facilitates the binding of Krüppel-like factor 5 and enhances neuropathic pain after nerve injury in mice. J. Neurosci. 2018, 38, 10535–10551. [Google Scholar] [CrossRef]
- Feng, W.; Teng, R.; Zhao, Y.; Gao, J.; Chu, H. Epigenetic modulation of Wnt signaling contributes to neuropathic pain in rats. Mol. Med. Rep. 2015, 12, 4727–4733. [Google Scholar] [CrossRef][Green Version]
- Hsieh, M.C.; Lai, C.Y.; Ho, Y.C.; Wang, H.H.; Cheng, J.K.; Chau, Y.P.; Peng, H.Y. Tet1-dependent epigenetic modification of BDNF expression in dorsal horn neurons mediates neuropathic pain in rats. Sci. Rep. 2016, 6, 37411. [Google Scholar] [CrossRef]
- Wu, Q.; Wei, G.; Ji, F.; Jia, S.; Wu, S.; Guo, X.; He, L.; Pan, Z.; Miao, X.; Mao, Q.; et al. TET1 Overexpression Mitigates Neuropathic Pain Through Rescuing the Expression of μ-Opioid Receptor and Kv1.2 in the Primary Sensory Neurons. Neurotherapeutics 2019, 16, 491–504. [Google Scholar] [CrossRef]
- Hsieh, M.C.; Ho, Y.C.; Lai, C.Y.; Chou, D.; Wang, H.H.; Chen, G.D.; Lin, T.B.; Peng, H.Y. Melatonin impedes Tet1-dependent mGluR5 promoter demethylation to relieve pain. J. Pineal Res. 2017, 63, e12436. [Google Scholar] [CrossRef]
- Pan, Z.; Xue, Z.Y.; Li, G.F.; Sun, M.L.; Zhang, M.; Hao, L.Y.; Tang, Q.Q.; Zhu, L.J.; Cao, J.L. DNA Hydroxymethylation by Ten-eleven Translocation Methylcytosine Dioxygenase 1 and 3 Regulates Nociceptive Sensitization in a Chronic Inflammatory Pain Model. Anesthesiology 2017, 127, 147–163. [Google Scholar] [CrossRef]
- Qi, F.; Zhou, Y.; Xiao, Y.; Tao, J.; Gu, J.; Jiang, X.; Xu, G.Y. Promoter demethylation of cystathionine-β-synthetase gene contributes to inflammatory pain in rats. Pain 2013, 154, 34–45. [Google Scholar] [CrossRef]
- Massart, R.; Dymov, S.; Millecamps, M.; Suderman, M.; Gregoire, S.; Koenigs, K.; Alvarado, S.; Tajerian, M.; Stone, L.S.; Szyf, M. Overlapping signatures of chronic pain in the DNA methylation landscape of prefrontal cortex and peripheral T cells. Sci. Rep. 2016, 6, 19615. [Google Scholar] [CrossRef]
- Gölzenleuchter, M.; Kanwar, R.; Zaibak, M.; Al Saiegh, F.; Hartung, T.; Klukas, J.; Smalley, R.L.; Cunningham, J.M.; Figueroa, M.E.; Schroth, G.P.; et al. Plasticity of DNA methylation in a nerve injury model of pain. Epigenetics 2015, 10, 200–212. [Google Scholar] [CrossRef]
- Pan, Z.; Zhu, L.J.; Li, Y.Q.; Hao, L.Y.; Yin, C.; Yang, J.X.; Guo, Y.; Zhang, S.; Hua, L.; Xue, Z.Y.; et al. Epigenetic modification of spinal miR-219 expression regulates chronic inflammation pain by targeting CaMKIIγ. J. Neurosci. 2014, 34, 9476–9483. [Google Scholar] [CrossRef]
- Tajerian, M.; Alvarado, S.; Millecamps, M.; Szyf, M.; Stone, L.S. The epigenetic signature of chronic pain in the mouse brain. Mol. Pain 2014, 10, O17. [Google Scholar] [CrossRef][Green Version]
- Tajerian, M.; Alvarado, S.; Millecamps, M.; Dashwood, T.; Anderson, K.M.; Haglund, L.; Ouellet, J.; Szyf, M.; Stone, L.S. DNA methylation of SPARC and chronic low back pain. Mol. Pain 2011, 7, 65. [Google Scholar] [CrossRef]
- Takenaka, S.; Sukenaga, N.; Ohmuraya, M.; Matsuki, Y.; Maeda, L.; Takao, Y.; Hirose, M.; Schaller, B. Association between neuropathic pain characteristics and DNA methylation of transient receptor potential ankyrin 1 in human peripheral blood. Medicine 2020, 99, e19325. [Google Scholar] [CrossRef]
- Gombert, S.; Rhein, M.; Winterpacht, A.; Münster, T.; Hillemacher, T.; Leffler, A.; Frieling, H. Transient receptor potential ankyrin 1 promoter methylation and peripheral pain sensitivity in Crohn’s disease. Clin. Epigenetics 2019, 12, 1. [Google Scholar] [CrossRef]
- Stenz, L.; Carré, J.L.; Luthi, F.; Vuistiner, P.; Burrus, C.; Paoloni-Giacobino, A.; Léger, B. Genome-Wide Epigenomic Analyses in Patients with Nociceptive and Neuropathic Chronic Pain Subtypes Reveals Alterations in Methylation of Genes Involved in the Neuro-Musculoskeletal System. J. Pain 2021. [Google Scholar] [CrossRef]
- Aroke, E.N.; Overstreet, D.S.; Penn, T.M.; Crossman, D.K.; Jackson, P.; Tollefsbol, T.O.; Quinn, T.L.; Yi, N.; Goodin, B.R. Identification of DNA methylation associated enrichment pathways in adults with non-specific chronic low back pain. Mol. Pain 2020, 16, 1744806920972889. [Google Scholar] [CrossRef]
- Bell, J.T.; Loomis, A.K.; Butcher, L.M.; Gao, F.; Zhang, B.; Hyde, C.L.; Sun, J.; Wu, H.; Ward, K.; Harris, J.; et al. Differential methylation of the TRPA1 promoter in pain sensitivity. Nat. Commun. 2014, 5, 2978. [Google Scholar] [CrossRef]
- Li, Z.; Li, A.; Yan, L.; Yang, T.; Xu, W.; Fan, P. Downregulation of long noncoding RNA DLEU1 attenuates hypersensitivity in chronic constriction injury-induced neuropathic pain in rats by targeting miR-133a-3p/SRPK1 axis. Mol. Med. 2020, 26, 104. [Google Scholar] [CrossRef]
- Pan, Z.; Du, S.; Wang, K.; Guo, X.; Mao, Q.; Feng, X.; Huang, L.; Wu, S.; Hou, B.; Chang, Y.J.; et al. Downregulation of a Dorsal Root Ganglion-Specifically Enriched Long Noncoding RNA is Required for Neuropathic Pain by Negatively Regulating RALY-Triggered Ehmt2 Expression. Adv. Sci. 2021, 8, 2004515. [Google Scholar] [CrossRef]
- Ma, X.; Wang, H.; Song, T.; Wang, W.; Zhang, Z. lncRNA MALAT1 contributes to neuropathic pain development through regulating miR-129-5p/HMGB1 axis in a rat model of chronic constriction injury. Int. J. Neurosci. 2020, 130, 1215–1224. [Google Scholar] [CrossRef]
- Xian, S.; Ding, R.; Li, M.; Chen, F. LncRNA NEAT1/miR-128-3p/AQP4 axis regulating spinal cord injury-induced neuropathic pain progression. J. Neuroimmunol. 2021, 351, 577457. [Google Scholar] [CrossRef]
- Li, Y.; Yin, C.; Liu, B.; Nie, H.; Wang, J.; Zeng, D.; Chen, R.; He, X.; Fang, J.; Du, J.; et al. Transcriptome profiling of long noncoding RNAs and mRNAs in spinal cord of a rat model of paclitaxel-induced peripheral neuropathy identifies potential mechanisms mediating neuroinflammation and pain. J. Neuroinflamm. 2021, 18, 48. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, T.; Li, X.; Wen, C.C.; Yan, X.T.; Peng, C.; Xiao, Y. Circ_0005075 targeting miR-151a-3p promotes neuropathic pain in CCI rats via inducing NOTCH2 expression. Gene 2021, 767, 145079. [Google Scholar] [CrossRef]
- Xin, Y.; Song, X.; Ge, Q. Circular RNA SMEK1 promotes neuropathic pain in rats through targeting microRNA-216a-5p to mediate Thioredoxin Interacting Protein (TXNIP) expression. Bioengineered 2021, 12, 5540–5551. [Google Scholar] [CrossRef]
- Xiong, W.; Liu, F.; Wang, J.; Wang, Z. Circular RNA expression profiling in dorsal root ganglion of rats with peripheral nerve injury-induced neuropathic pain. Res. Sq. 2020, 1–13. [Google Scholar] [CrossRef]
- Chen, H.W.; Zhang, X.X.; Peng, Z.D.; Xing, Z.M.; Zhang, Y.W.; Li, Y.L. The circular RNA circSlc7a11 promotes bone cancer pain pathogenesis in rats by modulating LLC-WRC 256 cell proliferation and apoptosis. Mol. Cell. Biochem. 2021, 476, 1751–1763. [Google Scholar] [CrossRef]
- Wei, M.; Li, L.; Zhang, Y.; Zhang, M.; Su, Z. Downregulated circular RNA zRANB1 mediates Wnt5a/β-Catenin signaling to promote neuropathic pain via miR-24-3p/LPAR3 axis in CCI rat models. Gene 2020, 761, 145038. [Google Scholar] [CrossRef]
- Zhou, J.; Xiong, Q.; Chen, H.; Yang, C.; Fan, Y. Identification of the spinal expression profile of non-coding rnas involved in neuropathic pain following spared nerve injury by sequence analysis. Front. Mol. Neurosci. 2017, 10, 91. [Google Scholar] [CrossRef]
- He, J.; Wang, H.; Huang, J.; Zhang, L.; Li, D.; He, W.; Xiong, Q.; Qin, Z. Diabetic neuropathic pain induced by streptozotocin alters the expression profile of non-coding RNAs in the spinal cord of mice as determined by sequencing analysis. Exp. Ther. Med. 2021, 22, 775. [Google Scholar] [CrossRef]
- Dai, D.; Wang, J.; Jiang, Y.; Yuan, L.; Lu, Y.; Zhang, A.; Zou, D.; Chen, X. Small RNA sequencing reveals microRNAs related to neuropathic pain in rats. Braz. J. Med. Biol. Res. 2019, 52, e8380. [Google Scholar] [CrossRef]
- Bai, G.; Ambalavanar, R.; Wei, D.; Dessem, D. Downregulation of selective microRNAs in trigeminal ganglion neurons following inflammatory muscle pain. Mol. Pain 2007, 3, 15. [Google Scholar] [CrossRef]
- Favereaux, A.; Thoumine, O.; Bouali-Benazzouz, R.; Roques, V.; Papon, M.A.; Salam, S.A.; Drutel, G.; Léger, C.; Calas, A.; Nagy, F.; et al. Bidirectional integrative regulation of Cav1.2 calcium channel by microRNA miR-103: Role in pain. EMBO J. 2011, 30, 3830–3841. [Google Scholar] [CrossRef]
- Braun, A.; Evdokimov, D.; Frank, J.; Sommer, C.; Üçeyler, N. MiR103a-3p and miR107 are related to adaptive coping in a cluster of fibromyalgia patients. PLoS ONE 2020, 15, e0239286. [Google Scholar] [CrossRef]
- Zhang, L.; Wu, R.; Xu, M.J.; Sha, J.; Xu, G.Y.; Wu, J.; Zhang, P.A. MiRNA-107 contributes to inflammatory pain by down-regulating GLT-1 expression in rat spinal dorsal horn. Eur. J. Pain 2021, 25, 1254–1263. [Google Scholar] [CrossRef]
- Elramah, S.; López-González, M.J.; Bastide, M.; Dixmérias, F.; Roca-Lapirot, O.; Wielanek-Bachelet, A.C.; Vital, A.; Leste-Lasserre, T.; Brochard, A.; Landry, M.; et al. Spinal miRNA-124 regulates synaptopodin and nociception in an animal model of bone cancer pain. Sci. Rep. 2017, 7, 10949. [Google Scholar] [CrossRef]
- Bali, K.K.; Gandla, J.; Rangel, D.R.; Castaldi, L.; Mouritzen, P.; Agarwal, N.; Schmelz, M.; Heppenstall, P.; Kuner, R. A genome-wide screen reveals microRNAs in peripheral sensory neurons driving painful diabetic neuropathy. Pain 2021, 162, 1334–1351. [Google Scholar] [CrossRef]
- Kynast, K.L.; Russe, O.Q.; Möser, C.V.; Geisslinger, G.; Niederberger, E. Modulation of central nervous system-specific microRNA-124a alters the inflammatory response in the formalin test in mice. Pain 2013, 154, 368–376. [Google Scholar] [CrossRef]
- Li, X.; Wang, D.; Zhou, J.; Yan, Y.; Chen, L. Evaluation of circulating microRNA expression in patients with trigeminal neuralgia: An observational study. Medicine 2020, 99, e22972. [Google Scholar] [CrossRef]
- Ni, J.; Gao, Y.; Gong, S.; Guo, S.; Hisamitsu, T.; Jiang, X. Regulation of μ-opioid type 1 receptors by microRNA134 in dorsal root ganglion neurons following peripheral inflammation. Eur. J. Pain 2013, 17, 313–323. [Google Scholar] [CrossRef]
- Liu, M.; Cheng, X.; Yan, H.; Chen, J.; Liu, C.; Chen, Z. MiR-135-5p Alleviates Bone Cancer Pain by Regulating Astrocyte-Mediated Neuroinflammation in Spinal Cord through JAK2/STAT3 Signaling Pathway. Mol. Neurobiol. 2021, 58, 4802–4815. [Google Scholar] [CrossRef]
- Zhang, J.; Rong, L.; Shao, J.; Zhang, Y.; Liu, Y.; Zhao, S.; Li, L.; Yu, W.; Zhang, M.; Ren, X.; et al. Epigenetic restoration of voltage-gated potassium channel Kv1.2 alleviates nerve injury-induced neuropathic pain. J. Neurochem. 2021, 156, 367–378. [Google Scholar] [CrossRef]
- Li, J.; Zhu, Y.; Ma, Z.; Liu, Y.; Sun, Z.; Wu, Y. miR-140 ameliorates neuropathic pain in CCI rats by targeting S1PR1. J. Recept. Signal Transduct. 2021, 41, 401–407. [Google Scholar] [CrossRef]
- Guo, J.B.; Chen, B.L.; Song, G.; Zheng, Y.L.; Zhu, Y.; Yang, Z.; Su, X.; Wang, Y.; Cao, Q.; Chen, P.J.; et al. Comparative Transcriptome Profiling Reveals Changes of microRNAs Response to Exercise in Rats with Neuropathic Pain. Neural Plast. 2021, 2021, 5597139. [Google Scholar] [CrossRef]
- Poh, K.W.; Yeo, J.F.; Ong, W.Y. MicroRNA changes in the mouse prefrontal cortex after inflammatory pain. Eur. J. Pain 2011, 15, 801.e1–801.e12. [Google Scholar] [CrossRef]
- Tao, Z.; Zhou, Y.; Zeng, B.; Yang, X.; Su, M. MicroRNA-183 attenuates osteoarthritic pain by inhibiting the TGFα-mediated CCL2/CCR2 signalling axis. Bone Jt. Res. 2021, 10, 548–557. [Google Scholar] [CrossRef]
- Sakai, A.; Suzuki, H. Nerve injury-induced upregulation of miR-21 in the primary sensory neurons contributes to neuropathic pain in rats. Biochem. Biophys. Res. Commun. 2013, 435, 176–181. [Google Scholar] [CrossRef]
- Wang, W.; Li, R. MiR-216a-5p alleviates chronic constriction injury-induced neuropathic pain in rats by targeting KDM3A and inactivating Wnt/β-catenin signaling pathway. Neurosci. Res. 2021, 170, 255–264. [Google Scholar] [CrossRef]
- Wu, X.; Wang, X.; Yin, Y.; Zhu, L.; Zhang, F.; Yang, J. Investigation of the role of miR-221 in diabetic peripheral neuropathy and related molecular mechanisms. Adv. Clin. Exp. Med. 2021, 30, 623–632. [Google Scholar] [CrossRef]
- Zhu, J.; Yang, J.; Xu, J. MiR-223 Inhibits the Polarization and Recruitment of Macrophages via NLRP3/IL-1 β Pathway to Meliorate Neuropathic Pain. Pain Res. Manag. 2021, 2021, 6674028. [Google Scholar] [CrossRef]
- Tan, M.; Shen, L.; Hou, Y. Epigenetic modification of BDNF mediates neuropathic pain via miR-30a-3p/EP300 axis in CCI rats. Biosci. Rep. 2020, 40, BSR20194442. [Google Scholar] [CrossRef]
- Xu, H.; Zhang, J.; Shi, X.; Li, X.; Zheng, C. NF-κB inducible miR-30b-5p aggravates joint pain and loss of articular cartilage via targeting SIRT1-FoxO3a-mediated NLRP3 inflammasome. Aging 2021, 13, 20774–20792. [Google Scholar] [CrossRef]
- Tramullas, M.; Francés, R.; De La Fuente, R.; Velategui, S.; Carcelén, M.; García, R.; Llorca, J.; Hurlé, M.A. MicroRNA-30c-5p modulates neuropathic pain in rodents. Sci. Transl. Med. 2018, 10, eaao6299. [Google Scholar] [CrossRef]
- Zhu, M.; Wang, L.; Zhu, J.; Xu, H.; Wei, K.; Chen, Q.; Wu, X.; Miao, X.; Lu, Z. MicroRNA-330 Directs Downregulation of the GABABR2 in the Pathogenesis of Pancreatic Cancer Pain. J. Mol. Neurosci. 2020, 70, 1541–1551. [Google Scholar] [CrossRef]
- Gandla, J.; Lomada, S.K.; Lu, J.; Kuner, R.; Bali, K.K. MiR-34c-5p functions as pronociceptive microRNA in cancer pain by targeting Cav2.3 containing calcium channels. Pain 2017, 158, 1765–1779. [Google Scholar] [CrossRef]
- Li, H.; Tao, R.; Wang, J.; Xia, L. Upregulation of miR-375 level ameliorates morphine analgesic tolerance in mouse dorsal root ganglia by inhibiting the JAK2/STAT3 pathway. J. Pain Res. 2017, 10, 1279–1287. [Google Scholar] [CrossRef]
- Xu, M.; Wu, R.; Zhang, L.; Zhu, H.Y.; Xu, G.Y.; Qian, W.; Zhang, P.A. Decreased mir-485-5p contributes to inflammatory pain through post-transcriptional upregulation of asic1 in rat dorsal root ganglion. J. Pain Res. 2020, 13, 3013–3022. [Google Scholar] [CrossRef]
- Wu, Y.; Gu, Y.; Shi, B. miR-590-3p Alleviates diabetic peripheral neuropathic pain by targeting RAP1A and suppressing infiltration by the T cells. Acta Biochim. Pol. 2020, 67, 587–593. [Google Scholar] [CrossRef]
- Sakai, A.; Saitow, F.; Miyake, N.; Miyake, K.; Shimada, T.; Suzuki, H. MiR-7a alleviates the maintenance of neuropathic pain through regulation of neuronal excitability. Brain 2013, 136, 2738–2750. [Google Scholar] [CrossRef]
- Chen, H.P.; Zhou, W.; Kang, L.M.; Yan, H.; Zhang, L.; Xu, B.H.; Cai, W.H. Intrathecal miR-96 inhibits nav1.3 expression and alleviates neuropathic pain in rat following chronic construction injury. Neurochem. Res. 2014, 39, 76–83. [Google Scholar] [CrossRef]
- Zhang, C.; Sha, H.; Peng, Y.; Wang, Y.; Liu, C.; Zhou, X. PiRNA-DQ541777 Contributes to Neuropathic Pain via Targeting Cdk5rap1. J. Neurosci. 2019, 39, 9028–9039. [Google Scholar] [CrossRef]
- Wu, W.; Ji, X.; Zhao, Y. Emerging Roles of Long Non-coding RNAs in Chronic Neuropathic Pain. Front. Neurosci. 2019, 13, 1097. [Google Scholar] [CrossRef]
- Li, Z.; Li, X.; Chen, X.; Li, S.; Ho, I.H.T.; Liu, X.; Chan, M.T.V.; Wu, W.K.K. Emerging roles of long non-coding RNAs in neuropathic pain. Cell Prolif. 2019, 52, e12528. [Google Scholar] [CrossRef]
- Wu, S.; Bono, J.; Tao, Y.X. Long noncoding RNA (lncRNA): A target in neuropathic pain. Expert Opin. Ther. Targets 2019, 23, 15–20. [Google Scholar] [CrossRef]
- Xu, D.; Ma, X.; Sun, C.; Han, J.; Zhou, C.; Chan, M.T.V.; Wu, W.K.K. Emerging roles of circular RNAs in neuropathic pain. Cell Prolif. 2021, 54, e13139. [Google Scholar] [CrossRef]
- de la Peña, J.B.I.; Song, J.J.; Campbell, Z.T. RNA control in pain: Blame it on the messenger. Wiley Interdiscip. Rev. RNA 2019, 10, e1546. [Google Scholar]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef]
- Auclair, G.; Weber, M. Mechanisms of DNA methylation and demethylation in mammals. Biochimie 2012, 94, 2202–2211. [Google Scholar] [CrossRef]
- Fatemi, M.; Pao, M.M.; Jeong, S.; Gal-Yam, E.N.; Egger, G.; Weisenberger, D.J.; Jones, P.A. Footprinting of mammalian promoters: Use of a CpG DNA methyltransferase revealing nucleosome positions at a single molecule level. Nucleic Acids Res. 2005, 33, e176. [Google Scholar] [CrossRef]
- Russo, G.; Landi, R.; Pezone, A.; Morano, A.; Zuchegna, C.; Romano, A.; Muller, M.T.; Gottesman, M.E.; Porcellini, A.; Avvedimento, E.V. DNA damage and Repair Modify DNA methylation and Chromatin Domain of the Targeted Locus: Mechanism of allele methylation polymorphism. Sci. Rep. 2016, 6, 33222. [Google Scholar] [CrossRef]
- Barman, P.; Reddy, D.; Bhaumik, S.R. Mechanisms of antisense transcription initiation with implications in gene expression, genomic integrity and disease pathogenesis. Non-Coding RNA 2019, 5, 11. [Google Scholar] [CrossRef]
- Hermann, A.; Goyal, R.; Jeltsch, A. The Dnmt1 DNA-(cytosine-C5)-methyltransferase methylates DNA processively with high preference for hemimethylated target sites. J. Biol. Chem. 2004, 279, 48350–48359. [Google Scholar] [CrossRef]
- Clouaire, T.; Stancheva, I. Methyl-CpG binding proteins: Specialized transcriptional repressors or structural components of chromatin? Cell. Mol. Life Sci. 2008, 65, 1509–1522. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517–534. [Google Scholar] [CrossRef]
- Zhou, X.; Zhuang, Z.; Wang, W.; He, L.; Wu, H.; Cao, Y.; Pan, F.; Zhao, J.; Hu, Z.; Sekhar, C.; et al. OGG1 is essential in oxidative stress induced DNA demethylation. Cell. Signal. 2016, 28, 1163–1171. [Google Scholar] [CrossRef]
- Bayraktar, G.; Kreutz, M.R. The role of activity-dependent DNA demethylation in the adult brain and in neurological disorders. Front. Mol. Neurosci. 2018, 11, 169. [Google Scholar] [CrossRef]
- Liu, L.; van Groen, T.; Kadish, I.; Tollefsbol, T.O. DNA methylation impacts on learning and memory in aging. Neurobiol. Aging 2009, 30, 549–560. [Google Scholar] [CrossRef]
- Fehrenbacher, J.C.; Guo, C.; Kelley, M.R.; Vasko, M.R. DNA damage mediates changes in neuronal sensitivity induced by the inflammatory mediators, MCP-1 and LPS, and can be reversed by enhancing the DNA repair function of APE1. Neuroscience 2017, 366, 23–35. [Google Scholar] [CrossRef]
- Garriga, J.; Laumet, G.; Chen, S.R.; Zhang, Y.; Madzo, J.; Issa, J.P.J.; Pan, H.L.; Jelinek, J. Nerve injury-induced chronic pain is associated with persistent DNA methylation reprogramming in dorsal root ganglion. J. Neurosci. 2018, 38, 6090–6101. [Google Scholar] [CrossRef]
- Downs, J.; Géranton, S.M.; Bebbington, A.; Jacoby, P.; Bahi-Buisson, N.; Ravine, D.; Leonard, H. Linking MECP2 and pain sensitivity: The example of Rett syndrome. Am. J. Med. Genet. Part A 2010, 152A, 1197–1205. [Google Scholar] [CrossRef]
- Banerjee, A.; Castro, J.; Sur, M. Rett syndrome: Genes, synapses, circuits, and therapeutics. Front. Psychiatry 2012, 3, 34. [Google Scholar] [CrossRef]
- Jiang, Y.; Langley, B.; Lubin, F.D.; Renthal, W.; Wood, M.A.; Yasui, D.H.; Kumar, A.; Nestler, E.J.; Akbarian, S.; Beckel-Mitchener, A.C. Epigenetics in the nervous system. J. Neurosci. 2008, 28, 11753–11759. [Google Scholar] [CrossRef]
- Chahrour, M.; Sung, Y.J.; Shaw, C.; Zhou, X.; Wong, S.T.C.; Qin, J.; Zoghbi, H.Y. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 2008, 320, 1224–1229. [Google Scholar] [CrossRef]
- Murgatroyd, C.; Patchev, A.V.; Wu, Y.; Micale, V.; Bockmühl, Y.; Fischer, D.; Holsboer, F.; Wotjak, C.T.; Almeida, O.F.X.; Spengler, D. Dynamic DNA methylation programs persistent adverse effects of early-life stress. Nat. Neurosci. 2009, 12, 1559–1566. [Google Scholar] [CrossRef]
- Decosterd, I.; Woolf, C.J. Spared nerve injury: An animal model of persistent peripheral neuropathic pain. Pain 2000, 87, 149–158. [Google Scholar] [CrossRef]
- Tajerian, M.; Alvarado, S.; Millecamps, M.; Vachon, P.; Crosby, C.; Bushnell, M.C.; Szyf, M.; Stone, L.S. Peripheral Nerve Injury Is Associated with Chronic, Reversible Changes in Global DNA Methylation in the Mouse Prefrontal Cortex. PLoS ONE 2013, 8, e55259. [Google Scholar] [CrossRef]
- Rudenko, A.; Dawlaty, M.M.; Seo, J.; Cheng, A.W.; Meng, J.; Le, T.; Faull, K.F.; Jaenisch, R.; Tsai, L.H. Tet1 is critical for neuronal activity-regulated gene expression and memory extinction. Neuron 2013, 79, 1109–1122. [Google Scholar] [CrossRef]
- Grégoire, S.; Millecamps, M.; Naso, L.; Do Carmo, S.; Cuello, A.C.; Szyf, M.; Stone, L.S. Therapeutic benefits of the methyl donor S-adenosylmethionine on nerve injury-induced mechanical hypersensitivity and cognitive impairment in mice. Pain 2017, 158, 802–810. [Google Scholar] [CrossRef]
- Diatchenko, L.; Nackley, A.G.; Slade, G.D.; Bhalang, K.; Belfer, I.; Max, M.B.; Goldman, D.; Maixner, W. Catechol-O-methyltransferase gene polymorphisms are associated with multiple pain-evoking stimuli. Pain 2006, 125, 216–224. [Google Scholar] [CrossRef]
- Kandlur, A.; Satyamoorthy, K.; Gangadharan, G. Oxidative Stress in Cognitive and Epigenetic Aging: A Retrospective Glance. Front. Mol. Neurosci. 2020, 13, 41. [Google Scholar] [CrossRef]
- DNA Methylation and Perioperative Pain Treatment—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/study/NCT02938455?term=epigenetic&cond=pain&draw=2&rank=9 (accessed on 2 July 2020).
- Li, Y.; Tollefsbol, T.O. DNA methylation detection: Bisulfite genomic sequencing analysis. Methods Mol. Biol. 2011, 791, 11–21. [Google Scholar] [CrossRef]
- Li, S.; Tollefsbol, T.O. DNA methylation methods: Global DNA methylation and methylomic analyses. Methods 2021, 187, 28–43. [Google Scholar] [CrossRef]
- Yong, W.S.; Hsu, F.M.; Chen, P.Y. Profiling genome-wide DNA methylation. Epigenetics Chromatin 2016, 9, 26. [Google Scholar] [CrossRef]
- Wreczycka, K.; Gosdschan, A.; Yusuf, D.; Grüning, B.; Assenov, Y.; Akalin, A. Strategies for analyzing bisulfite sequencing data. J. Biotechnol. 2017, 261, 105–115. [Google Scholar] [CrossRef]
- Sun, Z.; Dai, N.; Borgaro, J.G.; Quimby, A.; Sun, D.; Corrêa, I.R.; Zheng, Y.; Zhu, Z.; Guan, S. A Sensitive approach to map genome-wide 5-Hydroxymethylcytosine and 5-Formylcytosine at single-base resolution. Mol. Cell 2015, 57, 750–761. [Google Scholar] [CrossRef][Green Version]
- Yu, M.; Hon, G.C.; Szulwach, K.E.; Song, C.X.; Zhang, L.; Kim, A.; Li, X.; Dai, Q.; Shen, Y.; Park, B.; et al. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell 2012, 149, 1368–1380. [Google Scholar] [CrossRef]
- Jelinek, J.; Madzo, J. DREAM: A simple method for DNA methylation profiling by high-throughput sequencing. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2016; Volume 1465. [Google Scholar]
- Nair, S.S.; Coolen, M.W.; Stirzaker, C.; Song, J.Z.; Statham, A.L.; Strbenac, D.; Robinson, M.D.; Clark, S.J. Comparison of methyl-DNA immunoprecipitation (MeDIP) and methyl-CpG binding domain (MBD) protein capture for genome-wide DNA methylation analysis reveal CpG sequence coverage bias. Epigenetics 2011, 6, 34–44. [Google Scholar] [CrossRef]
- Stevens, M.; Cheng, J.B.; Li, D.; Xie, M.; Hong, C.; Maire, C.L.; Ligon, K.L.; Hirst, M.; Marra, M.A.; Costello, J.F.; et al. Estimating absolute methylation levels at single-CpG resolution from methylation enrichment and restriction enzyme sequencing methods. Genome Res. 2013, 23, 1541–1553. [Google Scholar] [CrossRef]
- Kirschner, S.A.; Hunewald, O.; Mériaux, S.B.; Brunnhoefer, R.; Muller, C.P.; Turner, J.D. Focussing reduced representation CpG sequencing through judicious restriction enzyme choice. Genomics 2016, 107, 109–119. [Google Scholar] [CrossRef]
- Zhao, M.T.; Whyte, J.J.; Hopkins, G.M.; Kirk, M.D.; Prather, R.S. Methylated DNA immunoprecipitation and high-throughput sequencing (MeDIP-seq) using low amounts of genomic DNA. Cell. Reprogram. 2014, 16, 175–184. [Google Scholar] [CrossRef]
- Harris, R.A.; Wang, T.; Coarfa, C.; Nagarajan, R.P.; Hong, C.; Downey, S.L.; Johnson, B.E.; Fouse, S.D.; Delaney, A.; Zhao, Y.; et al. Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat. Biotechnol. 2010, 28, 1097–1105. [Google Scholar] [CrossRef]
- Kangaspeska, S.; Stride, B.; Métivier, R.; Polycarpou-Schwarz, M.; Ibberson, D.; Carmouche, R.P.; Benes, V.; Gannon, F.; Reid, G. Transient cyclical methylation of promoter DNA. Nature 2008, 452, 112–115. [Google Scholar] [CrossRef]
- Wang, P.; Chen, H.; Tian, J.; Dai, Z.; Zou, X. Electrochemical evaluation of DNA methylation level based on the stoichiometric relationship between purine and pyrimidine bases. Biosens. Bioelectron. 2013, 45, 34–39. [Google Scholar] [CrossRef]
- Yotani, T.; Yamada, Y.; Arai, E.; Tian, Y.; Gotoh, M.; Komiyama, M.; Fujimoto, H.; Sakamoto, M.; Kanai, Y. Novel method for DNA methylation analysis using high-performance liquid chromatography and its clinical application. Cancer Sci. 2018, 109, 1690–1700. [Google Scholar] [CrossRef]
- Pan, G.; Jiang, L.; Tang, J.; Guo, F. A novel computational method for detecting DNA methylation sites with DNA sequence information and physicochemical properties. Int. J. Mol. Sci. 2018, 19, 511. [Google Scholar] [CrossRef]
- Palazzo, A.F.; Lee, E.S. Non-coding RNA: What is functional and what is junk? Front. Genet. 2015, 6, 2. [Google Scholar] [CrossRef]
- Kutter, C.; Svoboda, P. miRNA, siRNA, piRNA: Knowns of the unknown. RNA Biol. 2008, 5, 181–188. [Google Scholar] [CrossRef]
- Yu, C.Y.; Kuo, H.C. The emerging roles and functions of circular RNAs and their generation. J. Biomed. Sci. 2019, 26, 1–12. [Google Scholar] [CrossRef]
- Sanchez Calle, A.; Kawamura, Y.; Yamamoto, Y.; Takeshita, F.; Ochiya, T. Emerging roles of long non-coding RNA in cancer. Cancer Sci. 2018, 109, 2093–2100. [Google Scholar] [CrossRef]
- Ng, S.Y.; Lin, L.; Soh, B.S.; Stanton, L.W. Long noncoding RNAs in development and disease of the central nervous system. Trends Genet. 2013, 29, 461–468. [Google Scholar] [CrossRef]
- Hombach, S.; Kretz, M. Non-coding RNAs: Classification, biology and functioning. Adv. Exp. Med. Biol. 2016, 937, 3–17. [Google Scholar] [CrossRef]
- Cavalcante, G.C.; Magalhães, L.; Ribeiro-Dos-santos, Â.; Vidal, A.F. Mitochondrial epigenetics: Non-coding RNAs as a novel layer of complexity. Int. J. Mol. Sci. 2020, 21, 1838. [Google Scholar] [CrossRef]
- van den Ameele, J.; Fuge, J.; Pitceathly, R.D.S.; Berry, S.; McIntyre, Z.; Hanna, M.G.; Lee, M.; Chinnery, P.F. Chronic pain is common in mitochondrial disease. Neuromuscul. Disord. 2020, 30, 413–419. [Google Scholar] [CrossRef]
- van Tilburg, M.A.L.; Parisien, M.; Boles, R.G.; Drury, G.L.; Smith-Voudouris, J.; Verma, V.; Khoury, S.; Chabot-Doré, A.J.; Nackley, A.G.; Smith, S.B.; et al. A genetic polymorphism that is associated with mitochondrial energy metabolism increases risk of fibromyalgia. Pain 2020, 161, 2860–2871. [Google Scholar] [CrossRef]
- Zhao, J.; Lee, M.C.; Momin, A.; Cendan, C.M.; Shepherd, S.T.; Baker, M.D.; Asante, C.; Bee, L.; Bethry, A.; Perkins, J.R.; et al. Small RNAs control sodium channel expression, nociceptor excitability, and pain thresholds. J. Neurosci. 2010, 30, 10860–10871. [Google Scholar] [CrossRef]
- Yu, B.; Zhou, S.; Wang, Y.; Ding, G.; Ding, F.; Gu, X. Profile of microRNAs following rat sciatic nerve injury by deep sequencing: Implication for mechanisms of nerve regeneration. PLoS ONE 2011, 6, e24612. [Google Scholar] [CrossRef]
- Tang, S.; Jing, H.; Song, F.; Huang, H.; Li, W.; Xie, G.; Zhou, J. MicroRNAs in the Spinal Microglia Serve Critical Roles in Neuropathic Pain. Mol. Neurobiol. 2021, 58, 132–142. [Google Scholar] [CrossRef]
- Bali, K.K.; Selvaraj, D.; Satagopam, V.P.; Lu, J.; Schneider, R.; Kuner, R. Genome-wide identification and functional analyses of microRNA signatures associated with cancer pain. EMBO Mol. Med. 2013, 5, 1740–1758. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, H. TRPA1 involved in miR-141-5p-alleviated neuropathic pain induced by oxaliplatin. Neuroreport 2021, 32, 284–290. [Google Scholar] [CrossRef]
- Peng, L.; Wu, B.; Shi, L.; Zou, L.; Li, L.; Yang, R.; Xu, X.; Li, G.; Liu, S.; Zhang, C.; et al. Long Non-coding RNA Uc.48+ Small Interfering RNA Alleviates Neuroinflammatory Hyperalgesia in Gp120-Treated Rats via the P2Y12 Receptor. Front. Neurosci. 2021, 15, 663962. [Google Scholar] [CrossRef]
- Lee, S.; Shin, H.J.; Noh, C.; Kim, S.I.; Ko, Y.K.; Lee, S.Y.; Lim, C.; Hong, B.; Yang, S.Y.; Kim, D.W.; et al. Ikbkb sirna-encapsulated poly (Lactic-co-glycolic acid) nanoparticles diminish neuropathic pain by inhibiting microglial activation. Int. J. Mol. Sci. 2021, 22, 5657. [Google Scholar] [CrossRef]
- Hanna, J.; Hossain, G.S.; Kocerha, J. The potential for microRNA therapeutics and clinical research. Front. Genet. 2019, 10, 478. [Google Scholar] [CrossRef]
- Kuebart, A.; Wollborn, V.; Huhn, R.; Hermanns, H.; Werdehausen, R.; Brandenburger, T. Intraneural application of microRNA-1 mimetic nucleotides does not resolve neuropathic pain after chronic constriction injury in rats. J. Pain Res. 2020, 13, 2907–2914. [Google Scholar] [CrossRef]
- Dayer, C.F.; Luthi, F.; Le Carré, J.; Vuistiner, P.; Terrier, P.; Benaim, C.; Giacobino, J.P.; Léger, B. Differences in the miRNA signatures of chronic musculoskeletal pain patients from neuropathic or nociceptive origins. PLoS ONE 2019, 14, e0219311. [Google Scholar] [CrossRef]
- Al-Rawaf, H.A.; Gabr, S.A.; Alghadir, A.H. Vitamin D Deficiency and Molecular Changes in Circulating MicroRNAs in Older Adults with Lower Back Pain. Pain Res. Manag. 2021, 2021, 6662651. [Google Scholar] [CrossRef]
- MicroRNAs as Biomarkers of Pain Intensity in Patients with Chronic Fatigue Syndrome (CFS)—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/record/NCT03892954?term=miRNA&cond=pain&draw=4&rank=1&view=record (accessed on 2 July 2020).
- Biomarker Signatures of the Sleep-Pain Enigma—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03850743?term=miRNA&cond=pain&draw=4 (accessed on 2 July 2020).
- Bussotti, G.; Notredame, C.; Enright, A.J. Detecting and comparing non-coding RNAs in the high-throughput era. Int. J. Mol. Sci. 2013, 14, 15423–15458. [Google Scholar] [CrossRef]
- Schena, M.; Shalon, D.; Davis, R.W.; Brown, P.O. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 1995, 270, 467–470. [Google Scholar] [CrossRef]
- Shi, H.; Zhou, Y.; Jia, E.; Pan, M.; Bai, Y.; Ge, Q. Bias in RNA-seq Library Preparation: Current Challenges and Solutions. Biomed Res. Int. 2021, 2021, 6647597. [Google Scholar] [CrossRef]
- Landgraf, P.; Rusu, M.; Sheridan, R.; Sewer, A.; Iovino, N.; Aravin, A.; Pfeffer, S.; Rice, A.; Kamphorst, A.O.; Landthaler, M.; et al. A Mammalian microRNA Expression Atlas Based on Small RNA Library Sequencing. Cell 2007, 129, 1401–1414. [Google Scholar] [CrossRef]
- Wright, C.; Rajpurohit, A.; Burke, E.E.; Williams, C.; Collado-Torres, L.; Kimos, M.; Brandon, N.J.; Cross, A.J.; Jaffe, A.E.; Weinberger, D.R.; et al. Comprehensive assessment of multiple biases in small RNA sequencing reveals significant differences in the performance of widely used methods. BMC Genom. 2019, 20, 513. [Google Scholar] [CrossRef]
- Hagemann-Jensen, M.; Abdullayev, I.; Sandberg, R.; Faridani, O.R. Small-seq for single-cell small-RNA sequencing. Nat. Protoc. 2018, 13, 2407–2424. [Google Scholar] [CrossRef]
- Wang, J.; Zhao, Y.; Zhou, X.; Hiebert, S.W.; Liu, Q.; Shyr, Y. Nascent RNA sequencing analysis provides insights into enhancer-mediated gene regulation. BMC Genom. 2018, 19, 633. [Google Scholar] [CrossRef]
- Angelova, M.T.; Dimitrova, D.G.; Dinges, N.; Lence, T.; Worpenberg, L.; Carré, C.; Roignant, J.Y. The emerging field of epitranscriptomics in neurodevelopmental and neuronal disorders. Front. Bioeng. Biotechnol. 2018, 6, 46. [Google Scholar] [CrossRef]
- Thrupp, N.; Sala Frigerio, C.; Wolfs, L.; Skene, N.G.; Fattorelli, N.; Poovathingal, S.; Fourne, Y.; Matthews, P.M.; Theys, T.; Mancuso, R.; et al. Single-Nucleus RNA-Seq Is Not Suitable for Detection of Microglial Activation Genes in Humans. Cell Rep. 2020, 32, 108189. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Epigenetic Mechanism | Epigenetic Mark/Enzyme | Animal Model | Pain Model | Site | Behaviour Assessed | Targets (Genes or Enzymes) | References |
|---|---|---|---|---|---|---|---|
| DNA Methylation | (p)MeCP2 | Rat | CCI | SC neurons | Thermal, mechanical | - | [17] |
| CFA injection in ankle joint | Mechanical | SGK1, FKBP5, Sin3a | [18] | ||||
| CFA | Thermal, mechanical | Zif268, Fos | [19] | ||||
| CFA injection in ankle joint, SNI | - | - | [20] | ||||
| Mouse | SNI | SC neurons, DRG | Thermal, mechanical | miR-132, CREB | [21] | ||
| DNMT1 | Mouse | SNL | DRG | Thermal, mechanical, cold | Kcna2 | [22] | |
| DNMT3a | Rat, mouse | SNL, CCI, CFA | DRG | Thermal, mechanical, cold | Kcna2 | [23] | |
| Rat | SNL | SC neurons | Thermal, mechanical | CSF1 | [24] | ||
| Mouse | Partial SNL | Amygdala | Thermal, mechanical, stress | - | [25] | ||
| MBD1, DNMT3a | Mouse | SNL, CFA | DRG | Thermal, mechanical, cold, capsaicin | Oprm1, Kcna2 | [26] | |
| DNMT3a2 | Mouse | CFA | SC neurons | Thermal, mechanical | Ptgs2 | [27] | |
| DNMT3b | Rat | CFA | DRG | - | CXCR4, NFkB | [28] | |
| Thermal, mechanical | miR-29B | [29] | |||||
| Mouse | SNL | SC neurons | Thermal, mechanical | CXCR3, C/EBPα | [30] | ||
| GPR151 | [31] | ||||||
| Wnt3a | Rat | CCI | SC neurons | Thermal, mechanical | Wnt3a | [32] | |
| Tet1 | Rat | SNL | SC neurons | Mechanical | BDNF | [33] | |
| DRG | Thermal, mechanical | Oprm1, Kcna2 | [34] | ||||
| CFA | SC neurons | Thermal, mechanical | mGluR5 | [35] | |||
| Tet1, Tet3, 5hmC | Mouse | CFA | SC neurons | Thermal, mechanical | Stat3 | [36] | |
| Promoter de-/hyper-methylation | Rat | CFA | DRG | Mechanical | CBS, MBD4, Gadd45α | [37] | |
| CCI | SC | - | Wnt3a | [32] | |||
| SNI | PFC, T cells | Mechanical | Pak1, Pax6, Clip3, Srp54a, Xxpo4 and others | [38] | |||
| SNL | DRG | - | AFT3, A2m, NGF, SOCS3, SOX11, STAT3 and others | [39] | |||
| Mouse | CFA | SC neurons | Thermal, mechanical | miR-219 | [40] | ||
| SNI | PFC, | Mechanical | Syt2 | [41] | |||
| Mouse and Human | Aging, low back pain | IVD | - | SPARC | [42] | ||
| Promoter de-/hyper-methylation | Human | - | Blood tissue | Preoperative and chronic pain | TRPA1 | [43] | |
| Crohn’s disease | [44] | ||||||
| Chronic nociceptive pain vs. chronic neuropathic pain | RAB10, BMP1, LRRC59, PNPLA6, P3H3 and others | [45] | |||||
| Chronic lower back pain | CELSR1, KIF11, MINK1, NAV1 and others | [46] | |||||
| Blood tissue and multiple brain regions | Discordant heat pain sensitivity | TRPA1, ST6GALNAC3, MICAL2 and others | [47] | ||||
| lncRNA * | DLEU1 | Rat | CCI | SC | Thermal, mechanical | miR-133a-3p/SRPK1 | [48] |
| DS-lncRNA | Mouse | CCI, SNL | DRG | Thermal, mechanical | - | [49] | |
| MALAT1 | Rat | CCI | SC | Thermal, mechanical | miR-129-5p/HMGB1 | [50] | |
| NEAT1 | Rat | SCI | SC | Thermal, mechanical | miR-128-3p/AQP4 | [51] | |
| 145 up- and 267 down-regulated lncRNAs | Rat | Paclitaxel-induced peripheral neuropathy | SC | - | Multiple targets | [52] | |
| circRNA * | Circ_0005075 | Rat | CCI | SC | Thermal, mechanical | miR-151a-3p/NOTCH2 | [53] |
| circSMEK1 | Rat | CCI | SC microglia | Thermal, mechanical | miR-216a-5p | [54] | |
| 374 different circRNA | Rat | CCI | DRG | - | Dopaminergic synapse, renin secretion, MAPK pathway and neurogenesis | [55] | |
| circSlc7a11 and 7 others | Rat | Model of bone cancer pain | SC | - | Altered cell proliferation and apoptosis | [56] | |
| zRANB1 | Rat | CCI | SC | Thermal, mechanical | miR-24-3p/LPAR3, Wnt5a/β-catenin pathway | [57] | |
| 134 lncRNA, 12 miRNA and 188 circRNA | Rat | SNI | SC | - | Multiple pathways | [58] | |
| 9 lncRNA, 148 miRNA and 135 circRNA | Mouse | Model of diabetic neuropathic pain | SC | Mechanical | Multiple pathways | [59] | |
| miRNA * | 72 different miRNA | Rat | SNI | DRG | Thermal, mechanical | 17,316 target genes | [60] |
| miR-10a, −29a, −98, −99a, −124a, −134, -183 | Rat | CFA injection to masseter muscle | Trigeminal ganglia | - | - | [61] | |
| miR-103 | Rat | SNL | SC neurons | Thermal, mechanical | Cacna1c, Cacna2d1 Cacnb1 | [62] | |
| miR-103a-3p, -107 | Human | Fibromyalgia | White blood cells | Coping strategy | Multiple genetic targets (in silico) | [63] | |
| miR-107 | Rat | CFA | SC | Mechanical | Slc1a2 (glutamate transporter 1, GL-1) | [64] | |
| miR-124 | Mouse | Model of cancer pain | SC | - | Synpo | [65] | |
| miR-124-1, -33, -380 | Mouse | Model of diabetic peripheral neuropathy | DRG neurons | Mechanical | Kcnab2, Serpinb6a, Emb, Pacsin1 and others. | [66] | |
| miR-124a | Mouse | Formalin Injection | SC neurons | Formalin | MeCP2 | [67] | |
| miR-132-3p, -146b-5p, -155-5p, -384 | Human | Trigeminal neuralgia | Serum | - | - | [68] | |
| miR-134 | Rat | CFA | DRG | Thermal, mechanical | Oprm1 | [69] | |
| miR-135-5p | Mouse | Model of bone cancer pain | SC astrocytes | Mechanical, spontaneous flinching | JAK2/STAT3 signalling | [70] | |
| miR-137 | Rat | CCI | DRG neurons, SC | Thermal, mechanical | Kcna2 (Kv1.2) | [71] | |
| miR-140 | Rat | CCI | DRG | Thermal, mechanical | S1PR1 | [72] | |
| miR-145-5p, -341, -300-5p, -653-5p | Rat | CCI vs. CCI-exercise | DRG | Thermal, mechanical | Multiple targets | [73] | |
| miR-155, miR-223 | Mouse | Facial carrageenan injection | Prefrontal cortex | Mechanical | C/EBPβ, GCSF | [74] | |
| miR-183 | Mouse | Model of osteoarthritis | DRG | Weight distribution between paws | TGFα, CCL2/CCR2 signal | [75] | |
| Human | Patients with osteoarthritis pain | Joint fluid (serum) | - | - | |||
| miR-21 | Rat | SNL, CCI | DRG | Thermal, mechanical | - | [76] | |
| miR-214-3p | Rat | SNL | SC neurons | Thermal, mechanical | CSF1 | [24] | |
| miR-216a-5p | Rat | CCI | SC | Thermal, mechanical | IL-6, TNF-α, IL-1β, KDM3A and Wnt/β-catenin pathway | [77] | |
| miR-219 | Mouse | CFA, formalin injection | SC neurons | Thermal, mechanical, formalin | CaMKIIγ | [40] | |
| miR-221 | Rat | Model of diabetic peripheral neuropathy | Serum exosomes from blood samples | Thermal, mechanical | SOCS3 | [78] | |
| miR-223 | Mouse | CCI | SC | Thermal, mechanical | NLRP3/IL-1β pathway | [79] | |
| miR-30a-3p | Rat | CCI | SC | Thermal, mechanical | EP300, bdnf | [80] | |
| miR-30b-5p | Rat | Model of osteoarthritis | Cartilage | - | NF-κB | [81] | |
| Human | Patients | Cancerous tissues | |||||
| miR-30c-5p | Mouse, rat | SNI | CSF, DRG, plasma, SC | Mechanical | TGF-β1 | [82] | |
| Human | Ischemic neuropathic pain | CSF, plasma | Severity of neuropathic pain | - | |||
| miR-330 | Mouse | Model of pancreatic cancer pain | SC | Mechanical (abdominal) | Gababr2 | [83] | |
| miR-34c-5p | Mouse | Model of bone metastatic pain | DRG | - | Cav2.3, P2rx6, Oprd1, Oprm1 | [84] | |
| miR-375 | Mouse | Morphine analgesic tolerance | DRG | Thermal | Janus kinase 2 (JAK2) | [85] | |
| miR-485-5p | Rat | CFA | DRG | Mechanical | Asic1 | [86] | |
| miR-590-3p | Mouse | Model of diabetic peripheral neuropathy | DRG | Thermal | RAP1A | [87] | |
| miR-7a | Rat | SNL, CCI, CFA | DRG | Thermal, mechanical | Scn2b | [88] | |
| miR-96 | Rat | CCI | DRG | Thermal, mechanical | Nav1.3 | [89] | |
| piRNA | piRNA-DQ541777 | Mouse | CCI, CFA | SC neurons | Thermal, mechanical | Cdk5rap1 | [90] |
| Techniques | Mechanism of Action | Strength | Weakness | Resolution | Cost | References |
|---|---|---|---|---|---|---|
| Sodium bisulphite sequencing: Conversion of unmethylated cytosine residues to uracil then to thymine. Gold standard technique. | ||||||
| RRBS | Use of restrictions enzymes to enrich CpG sites, stabilising methylated sites. |
|
| Single base | Moderate (~£200–£500) | [45,123] |
| T-WGBS | Gold standard assaying technique. Genome wide analysis of methylated sites. |
|
| Single base | High (~£700–£3000) | [124] |
| TAB-seq | Oxidation of TET proteins combined with 5mC to localise 5hmC. |
|
| Single base | High (~£1000) | [125,126] |
| Differential enzymatic cleavage of DNA: Enzymatic restriction of DNA resulting in methylated CpG fragments. | ||||||
| DREAM | Enzymatic digestion of DNA through utilisation of restriction endonucleases. |
|
| High | Moderate | [126,127] |
| Affinity capture of methylated DNA: Use of methyl- CpG-binding domain (MBD) proteins or MeDIP to bind to methylated DNA. | ||||||
| MBDCap-Seq | Methy-CpG binding domain based (MBD) protein captures DNA methylation, identifying highly differentiated regions. |
|
| 150 bp | Moderate (~£100) | [123] |
| MeDIP | Methylated DNA immunoprecipitation uses antibodies specific to 5mC to precipitate methylated DNA. |
|
| 100 bp | Moderate (~£100) | [128] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Irfan, J.; Febrianto, M.R.; Sharma, A.; Rose, T.; Mahmudzade, Y.; Di Giovanni, S.; Nagy, I.; Torres-Perez, J.V. DNA Methylation and Non-Coding RNAs during Tissue-Injury Associated Pain. Int. J. Mol. Sci. 2022, 23, 752. https://doi.org/10.3390/ijms23020752
Irfan J, Febrianto MR, Sharma A, Rose T, Mahmudzade Y, Di Giovanni S, Nagy I, Torres-Perez JV. DNA Methylation and Non-Coding RNAs during Tissue-Injury Associated Pain. International Journal of Molecular Sciences. 2022; 23(2):752. https://doi.org/10.3390/ijms23020752
Chicago/Turabian StyleIrfan, Jahanzaib, Muhammad Rizki Febrianto, Anju Sharma, Thomas Rose, Yasamin Mahmudzade, Simone Di Giovanni, Istvan Nagy, and Jose Vicente Torres-Perez. 2022. "DNA Methylation and Non-Coding RNAs during Tissue-Injury Associated Pain" International Journal of Molecular Sciences 23, no. 2: 752. https://doi.org/10.3390/ijms23020752
APA StyleIrfan, J., Febrianto, M. R., Sharma, A., Rose, T., Mahmudzade, Y., Di Giovanni, S., Nagy, I., & Torres-Perez, J. V. (2022). DNA Methylation and Non-Coding RNAs during Tissue-Injury Associated Pain. International Journal of Molecular Sciences, 23(2), 752. https://doi.org/10.3390/ijms23020752