
COVID-19 in Joint Ageing and Osteoarthritis: Current Status and Perspectives



Abstract

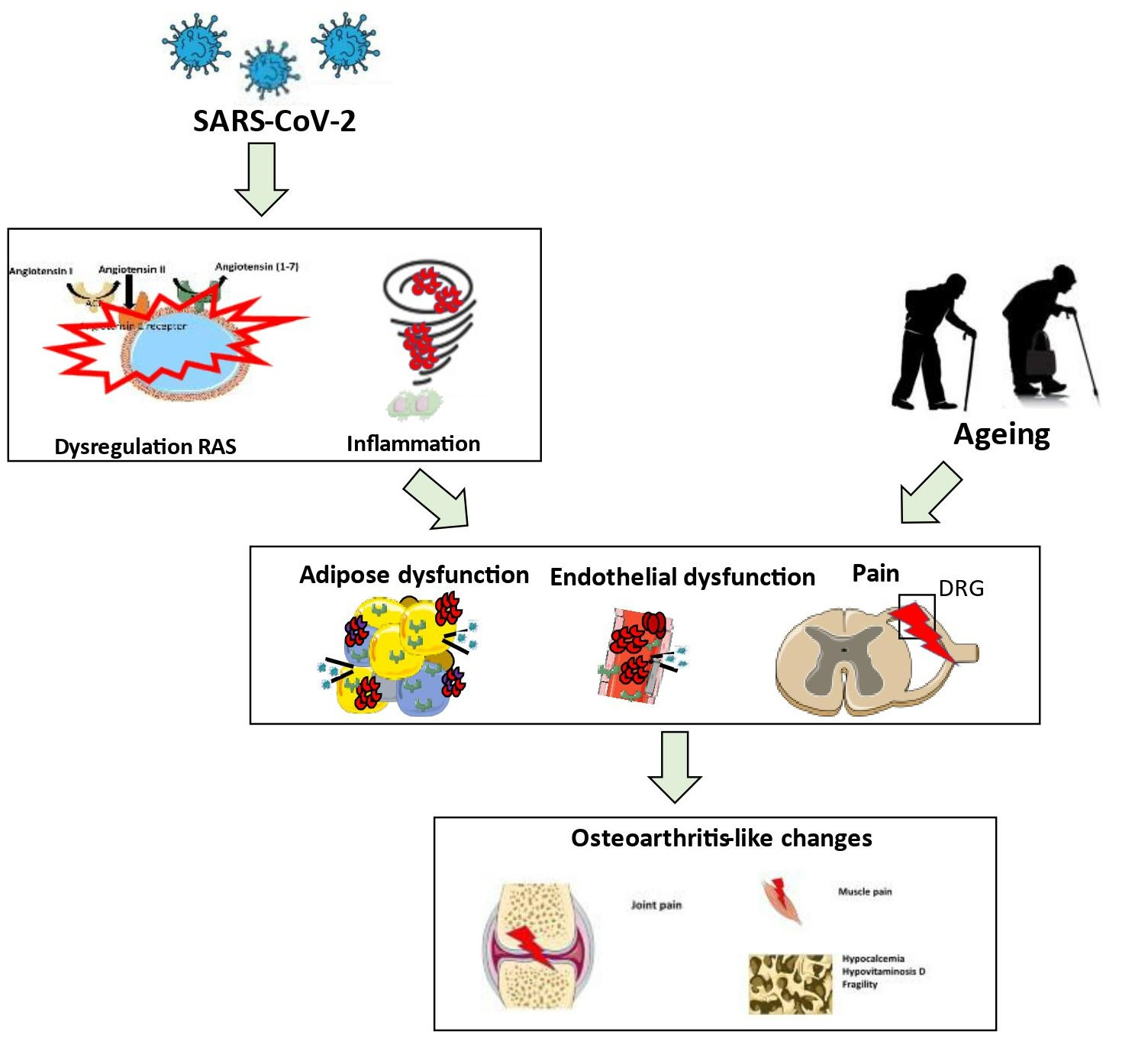{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. Acute COVID-19 Infection
1.2. Post-Acute and Chronic COVID-19
2. COVID-19 and Musculoskeletal Dysfunction
2.1. Osteoarticular Changes in COVID-19 Patients
2.2. Musculoskeletal Pain in COVID-19 Patients
2.3. Will COVID-19 Lead to Viral Arthritis?
3. Early Ageing Alterations in Endothelial and Adipose Tissue of COVID-19 Patients
3.1. Endothelial Dysfunction
3.2. Adipose Tissue Dysfunction
4. Neuronal Sensitization in COVID-19 Patients
5. Exploring Underlying Molecular Mechanisms
5.1. Renin-Angiotensin System
5.2. COVID-19 and Systemic Inflammation
6. New Approach in Treating Long-COVID: The Role of the Nicotinic Cholinergic Receptor
7. Conclusions and Hypothesis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| SARS-CoV-1 | Severe Acute Respiratory Syndrome Coronavirus-1 |
| MERS-CoV | Middle East Respiratory Syndrome Coronavirus |
| SARS-CoV-2 | Severe Acute Respiratory Syndrome Coronavirus-2 |
| COVID-19 | Coronavirus disease emerged in 2019 |
| WHO | World Health Organization |
| ACE2 | Angiotensin-Converting enzyme 2 |
| MRI | Magnetic Resonance Imaging |
| NSAID | Non-steroidal anti-inflammatory drug |
| NO | Nitric Oxide |
| OA | Osteoarthritis |
| iNOS | Inducible nitric oxide synthase |
| COX-2 | Cyclooxygenase 2 |
| PGE2 | Prostaglandin E2 |
| RAS | Renin-angiotensin system |
| IL-6 | Interleukin 6 |
| IL-1β | Interleukin 1 beta |
| TNFα | Tumor necrosis factor alfa |
| SASP | Senescence-associated secretory phenotype |
| CCL2/3/4 | C–C motif chemokine ligand 2/3/4 |
| CXCL2 | Chemokine (C-X-C motif) ligand 2 |
| Peptide hormone EREG | Protein encoded by EREG gene, epiregulin |
| DRG | Dorsal root ganglia |
| AT1R | Angiotensin II type 1 receptor |
| PI3K | Phosphoinositide 3-kinase |
| MAPKs | Mitogen-activated protein kinase |
| JNK | c-Jun N-terminal kinases |
| ARB | Angiotensin receptor blocker |
| AT1R blocker | Angiotensin II type I receptor blocker |
| AT2R | Angiotensin II type 2 receptor |
| ACEI | Angiotensin-converting enzyme inhibitor |
| TMPRSS2 | Transmembrane serine protease 2 |
| Bmal1 | Brain and muscle Arnt-like protein-1 |
| Angiotensin (1–7)/MAS axis | Angiotensin (1–7)/ mitochondrial assembly receptor axis |
| COPD | Chronic obstructive pulmonary disease |
| nAchR | Nicotinic acetylcholine receptor |
| CHRNA7 | Gene for the Cholinergic Receptor Nicotinic Alpha 7 Subunit |
| α7nAchR | Alfa7 nicotinic acetylcholine receptor |
| CHRFAM7A | human partial duplication of the CHRNA7 gene which encodes for the α7nAchR |
References
- Seyed Hosseini, E.; Riahi Kashani, N.; Nikzad, H.; Azadbakht, J.; Hassani Bafrani, H.; Haddad Kashani, H. The novel coronavirus Disease-2019 (COVID-19): Mechanism of action, detection and recent therapeutic strategies. Virology 2020, 551, 1–9. [Google Scholar] [CrossRef]
- Yesudhas, D.; Srivastava, A.; Gromiha, M.M. COVID-19 outbreak: History, mechanism, transmission, structural studies and therapeutics. Infection 2021, 49, 199–213. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. WHO Coronavirus Dashboard. Available online: https://covid19.who.int/ (accessed on 21 December 2021).
- Papageorgiou, A.C.; Mohsin, I. The SARS-CoV-2 Spike Glycoprotein as a Drug and Vaccine Target: Structural Insights into Its Complexes with ACE2 and Antibodies. Cells 2020, 9, 2343. [Google Scholar] [CrossRef] [PubMed]
- Raveendran, A.V.; Jayadevan, R.; Sashidharan, S. Long COVID: An overview. Diabetes Metab. Syndr. Clin. Res. Rev. 2021, 15, 869–875. [Google Scholar] [CrossRef]
- Angelo Carfì, M.; Roberto Bernabei, M.; Francesco Landi, M.D.P. Persistent Symptoms in Patients After Acute COVID-19. JAMA 2020, 324, 603–605. [Google Scholar] [CrossRef]
- Amenta, E.M.; Spallone, A.; Rodriguez-Barradas, M.C.; Sahly, H.M.E.; Atmar, R.L.; Kulkarni, P.A. Postacute COVID-19: An overview and approach to classification. Open Forum Infect. Dis. 2020, 7, ofaa509. [Google Scholar] [CrossRef]
- Tenforde, M.W.; Kim, S.S.; Lindsell, C.J.; Rose, E.B.; Shapiro, N.I.; Files, D.C.; Gibbs, K.W.; Erickson, H.L.; Steingrub, J.C.; Smithline, H.A.; et al. Symptom Duration and Risk Factors for Delayed Return to Usual Health Among Outpatients with COVID-19 in a Multistate Health Care Systems Network—United States, March–June 2020. MMWR Morb. Mortal Wkly. Rep. 2020, 69, 993–998. [Google Scholar] [CrossRef]
- Griffith, J.F. Musculoskeletal complications of severe acute respiratory syndrome. Semin. Musculoskelet. Radiol. 2011, 15, 554–560. [Google Scholar] [CrossRef] [PubMed]
- di Filippo, L.; Frara, S.; Giustina, A. The emerging osteo-metabolic phenotype of COVID-19: Clinical and pathophysiological aspects. Nat. Rev. Endocrinol. 2021, 17, 445–446. [Google Scholar] [CrossRef]
- di Filippo, L.; Doga, M.; Frara, S.; Giustina, A. Hypocalcemia in COVID-19: Prevalence, clinical significance and therapeutic implications. Rev. Endocr. Metab. Disord. 2021, 1–10. [Google Scholar] [CrossRef]
- Puig-Domingo, M.; Marazuela, M.; Yildiz, B.O.; Giustina, A. COVID-19 and endocrine and metabolic diseases. An updated statement from the European Society of Endocrinology. Endocrine 2021, 72, 301–316. [Google Scholar] [CrossRef]
- Bossoni, S.; Chiesa, L.; Giustina, A. Severe hypocalcemia in a thyroidectomized woman with COVID-19 infection. Endocrine 2020, 68, 253–254. [Google Scholar] [CrossRef] [PubMed]
- Carpagnano, G.E.; Di Lecce, V.; Quaranta, V.N.; Zito, A.; Buonamico, E.; Capozza, E.; Palumbo, A.; Di Gioia, G.; Valerio, V.N.; Resta, O. Vitamin D deficiency as a predictor of poor prognosis in patients with acute respiratory failure due to COVID-19. J. Endocrinol. Investig. 2021, 44, 765–771. [Google Scholar] [CrossRef]
- Kumar, R.; Rahthi, H.; Haq, A.; Wimalawansa, S.J.; Sharma, A. Putative roles of vitamin D in modulating immune response and immunopathology associated with COVID-19. Virus Res. 2020, 292, 198235. [Google Scholar] [CrossRef] [PubMed]
- Hadizadeh, F. Supplementation with vitamin D in the COVID-19 pandemic? Nutr. Rev. 2021, 79, 200–208. [Google Scholar] [CrossRef]
- Leung, T.W.; Wong, K.S.; Hui, A.C.; To, K.F.; Lai, S.T.; Ng, W.F.; Ng, H.K. Myopathic changes associated with severe acute respiratory syndrome: A postmortem case series. Arch. Neurol. 2005, 62, 1113–1117. [Google Scholar] [CrossRef]
- Tsai, L.-K.; Hsieh, S.-T.; Chang, Y.-C. Neurological manifestations in severe acute respiratory symptom. Acta Neurol. Taiwan. 2005, 14, 113–119. [Google Scholar] [PubMed]
- Vehar, S.; Boushra, M.; Ntiamoah, P.; Biehl, M. Post-acute sequelae of SARS-CoV-2 infection: Caring for the ‘long-haulers’. Cleve. Clin. J. Med. 2021, 88, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Aiyegbusi, O.L.; Hughes, S.E.; Turner, G.; Rivera, S.C.; McMullan, C.; Chandan, J.S.; Haroon, S.; Price, G.; Davies, E.H. Symptoms, complications and management of long COVID: A review. J. R. Soc. Med. 2021, 114, 428–442. [Google Scholar] [CrossRef]
- Tosato, M.; Carfì, A.; Martis, I.; Pais, C.; Ciciarello, F.; Rota, E.; Tritto, M.; Salerno, A.; Zazzara, M.B.; Martone, A.M.; et al. Prevalence and Predictors of Persistence of COVID-19 Symptoms in Older Adults: A Single-Center Study. J. Am. Med. Dir. Assoc. 2021, 22, 1840–1844. [Google Scholar] [CrossRef]
- Tuzun, S.; Keles, A.; Okutan, D.; Yildiran, T.; Palamar, D. Assessment of musculoskeletal pain, fatigue and grip strength in hospitalized patients with COVID-19. Eur. J. Phys. Rehabil. Med. 2021, 57, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Fernández-de-las-Peñas, C.; Rodríguez-Jiménez, J.; Fuensalida-Novo, S.; Palacios-Ceña, M.; Gómez-Mayordomo, V.; Florencio, L.L.; Hernández-Barrera, V.; Arendt-Nielsen, L. Myalgia as a symptom at hospital admission by severe acute respiratory syndrome coronavirus 2 infection is associated with persistent musculoskeletal pain as long-term post-COVID sequelae: A case-control study. Pain 2021, 162, 2832–2840. [Google Scholar] [CrossRef]
- Ghosn, J.; Piroth, L.; Epaulard, O.; Le Turnier, P.; Mentr, F.; Covid, F. Persistent COVID-19 symptoms are highly prevalent 6 months after hospitalization: Results from a large prospective cohort. Clin. Microbiol. Infect. 2021, 27, 1041. [Google Scholar] [CrossRef] [PubMed]
- Karaarslan, F.; Güneri, F.D.; Kardeş, S. Long COVID: Rheumatologic/musculoskeletal symptoms in hospitalized COVID-19 survivors at 3 and 6 months. Clin. Rheumatol. 2022, 41, 289–296. [Google Scholar] [CrossRef]
- Fernández-de-las-Peñas, C.; Navarro-Santana, M.; Plaza-Manzano, G.; Palacios-Ceña, D.; Arendt-Nielsen, L. Time Course Prevalence of Post-COVID Pain Symptoms of Musculoskeletal Origin in Patients Who Had Survived to SARS-CoV-2 Infection. Pain 2021. [Google Scholar] [CrossRef] [PubMed]
- Sykes, D.L.; Holdsworth, L.; Jawad, N.; Gunasekera, P.; Morice, A.H.; Crooks, M.G. Post-COVID-19 Symptom Burden: What is Long-COVID and How Should We Manage It? Lung 2021, 199, 113–119. [Google Scholar] [CrossRef]
- Gasparotto, M.; Framba, V.; Piovella, C.; Doria, A.; Iaccarino, L. Post-COVID-19 arthritis: A case report and literature review. Clin. Rheumatol. 2021, 40, 3357–3362. [Google Scholar] [CrossRef]
- Kocyigit, B.F.; Akyol, A. Reactive arthritis after COVID-19: A case-based review. Rheumatol. Int. 2021, 41, 2031–2039. [Google Scholar] [CrossRef]
- Ono, K.; Kishimoto, M.; Shimasaki, T.; Uchida, H.; Kurai, D.; Deshpande, G.A.; Komagata, Y.; Kaname, S. Reactive arthritis after COVID-19 infection. RMD Open 2020, 6, 2–5. [Google Scholar] [CrossRef] [PubMed]
- Grassi, M.; Giorgi, V.; Nebuloni, M.; Gismondo, M.R.; Salaffi, F.; Sarzi-Puttini, P.; Rimoldi, S.G.; Manzotti, A. SARS-CoV-2 in the knee joint: A cadaver study. Clin. Exp. Rheumatol. 2021. online ahead of print. [Google Scholar]
- Kuschner, Z.; Ortega, A.; Mukherji, P. A case of SARS-CoV-2-associated arthritis with detection of viral RNA in synovial fluid. J. Am. Coll. Emerg. Physicians Open 2021, 2, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Anyfanti, P.; Gavriilaki, E.; Douma, S.; Gkaliagkousi, E. Endothelial Dysfunction in Patients with Rheumatoid Arthritis: The Role of Hypertension. Curr. Hypertens. Rep. 2020, 22, 56. [Google Scholar] [CrossRef] [PubMed]
- Pantsulaia, I.; Ciszewski, W.M.; Niewiarowska, J. Senescent endothelial cells: Potential modulators of immunosenescence and ageing. Ageing Res. Rev. 2016, 29, 13–25. [Google Scholar] [CrossRef]
- Azzini, G.O.M.; Santos, G.S.; Visoni, S.B.C.; Azzini, V.O.M.; Santos, R.G.D.; Huber, S.C.; Lana, J.F. Metabolic syndrome and subchondral bone alterations: The rise of osteoarthritis—A review. J. Clin. Orthop. Trauma 2020, 11, S849–S855. [Google Scholar] [CrossRef]
- Le Clanche, S.; Bonnefont-Rousselot, D.; Sari-Ali, E.; Rannou, F.; Borderie, D. Inter-relations between osteoarthritis and metabolic syndrome: A common link? Biochimie 2016, 121, 238–252. [Google Scholar] [CrossRef]
- Zhuo, Q.; Yang, W.; Chen, J.; Wang, Y. Metabolic syndrome meets osteoarthritis. Nat. Rev. Rheumatol. 2012, 8, 729–737. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; He, C. Pro-inflammatory cytokines: The link between obesity and osteoarthritis. Cytokine Growth Factor Rev. 2018, 44, 38–50. [Google Scholar] [CrossRef]
- Goldring, S.R.; Goldring, M.B. The role of cytokines in cartilage matrix degeneration in osteoarthritis. Clin. Orthop. Relat. Res. 2004, 427, S27–S36. [Google Scholar] [CrossRef]
- Gheblawi, M.; Wang, K.; Viveiros, A.; Munan, M.; MacIntyre, E.; O’Neil, C.; Poglitsch, M.; Colombo, D.; Del Nonno, F.; Kassiri, Z.; et al. Angiotensin-Converting Enzyme 2: SARS-CoV-2 Receptor and Regulator of the Renin-Angiotensin System: Celebrating the 20th Anniversary of the Discovery of ACE2. Circ. Res. 2020, 126, 1456–1474. [Google Scholar] [CrossRef]
- Datta, P.K.; Liu, F.; Fischer, T.; Rappaport, J.; Qin, X. SARS-CoV-2 pandemic and research gaps: Understanding SARS-CoV-2 interaction with the ACE2 receptor and implications for therapy. Theranostics 2020, 10, 7448–7464. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Mezoh, G.; Crowther, N.J. Endothelial Dysfunction as a Primary Consequence of SARS-CoV-2 Infection. Adv. Exp. Med. Biol. 2021, 1321, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Amraei, R.; Rahimi, N. COVID-19, Renin-Angiotensin System and Endothelial Dysfunction. Cells 2020, 9, 1652. [Google Scholar] [CrossRef] [PubMed]
- Hang, W.; Chen, C.; Zhang, X.A.; Wang, D.W. Endothelial dysfunction in COVID-19 calls for immediate attention: The emerging roles of the endothelium in inflammation caused by SARS-CoV-2. Front. Med. 2021, 15, 638–643. [Google Scholar] [CrossRef]
- Spinelli, R.; Parrillo, L.; Longo, M.; Florese, P.; Desiderio, A.; Zatterale, F.; Miele, C.; Raciti, G.A.; Beguinot, F. Molecular basis of ageing in chronic metabolic diseases. J. Endocrinol. Investig. 2020, 43, 1373–1389. [Google Scholar] [CrossRef] [PubMed]
- Frasca, D.; Blomberg, B.B. Adipose tissue, immune aging, and cellular senescence. Semin. Immunopathol. 2020, 42, 573–587. [Google Scholar] [CrossRef]
- De Carvalho, F.G.; Justice, J.N.; de Freitas, E.C.; Kershaw, E.E.; Sparks, L.M. Adipose Tissue Quality in Aging: How Structural Skeletal Muscle Quality? Nutrients 2019, 11, 2553. [Google Scholar] [CrossRef] [PubMed]
- Copcu, H.E. Expert Opinion on Biological Therapy New normal: Two aspects of adipose tissue in COVID-19—Treat and threat? Expert Opin. Biol. Ther. 2020, 20, 1283–1292. [Google Scholar] [CrossRef]
- Slamkova, M.; Zorad, S.; Krskova, K. Alternative renin-angiotensin system pathways in adipose tissue and their role in the pathogenesis of obesity. Endocr. Regul. 2016, 50, 229–240. [Google Scholar] [CrossRef][Green Version]
- Heialy, S.A.; Hachim, M.Y.; Senok, A.; Gaudet, M.; Abou Tayoun, A.; Hamoudi, R.; Alsheikh-Ali, A.; Hamid, Q. Regulation of Angiotensin- Converting Enzyme 2 in Obesity: Implications for COVID-19. Front. Physiol. 2020, 11, 555039. [Google Scholar] [CrossRef]
- Al-benna, S. Association of high level gene expression of ACE2 in adipose tissue with mortality of COVID-19 in obese patients. Obes. Med. 2020, 19, 100283. [Google Scholar] [CrossRef] [PubMed]
- Ilias, I.; Zabuliene, L. Hyperglycemia and the novel COVID-19 infection: Possible pathophysiologic mechanisms. Med. Hypotheses 2020, 139, 109699. [Google Scholar] [CrossRef]
- Reiterer, M.; Rajan, M.; Gomez-Banoy, N.; Lau, J.D.; Gomez-Escobar, L.G.; Ma, L.; Gilani, A.; Alvarez-Mulett, S.; Sholle, E.T.; Chandar, V.; et al. Hyperglycemia in Acute COVID-19 is Characterized by Insulin resistance and adipose tissue infectivity by SARS-CoV-2. Cell Metab. 2021, 33, 2174–2188. [Google Scholar] [CrossRef]
- Maurya, R.; Sebastian, P.; Namdeo, M.; Devender, M. COVID-19 Severity in Obesity: Leptin and Inflammatory Cytokine Interplay in the Link Between High Morbidity and Mortality. Front. Immunol. 2021, 12, 649359. [Google Scholar] [CrossRef]
- Syx, D.; Tran, P.B.; Miller, R.E.; Malfait, A.M. Peripheral mechanisms contributing to osteoarthritis pain. Curr. Rheumatol. Rep. 2019, 20, 9. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, T.W.; Felson, D.T. Mechanisms of Osteoarthritis (OA) Pain. Curr. Osteoporos. Rep. 2018, 16, 611–616. [Google Scholar] [CrossRef] [PubMed]
- McFarland, A.J.; Yousuf, M.S.; Shiers, S.; Price, T.J. Neurobiology of SARS-CoV-2 interactions with the peripheral nervous system: Implications for COVID-19 and pain. Pain Rep. 2021, 6, e885. [Google Scholar] [CrossRef] [PubMed]
- Khatoon, F.; Prasad, K.; Kumar, V. COVID-19 associated nervous system manifestations. Sleep Med. 2021, S1389-9457(21)00387-7. [Google Scholar] [CrossRef]
- Roy, D.; Ghosh, R.; Dubey, S.; Dubey, M.J.; Benito-Leon, J.; Kanti Ray, B. Neurological and Neuropsychiatric Impacts of COVID-19 Pandemic. Can. J. Neurol. Sci. 2021, 48, 9–24. [Google Scholar] [CrossRef]
- Harapan, B.N.; Yoo, H.J. Neurological symptoms, manifestations, and complications associated with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease 19 (COVID-19). J. Neurol. 2021, 268, 3059–3071. [Google Scholar] [CrossRef] [PubMed]
- Shiers, S.; Ray, P.R.; Wangzhou, A.; Sankaranarayanan, I.; Tatsui, C.E.; Rhines, L.D.; Li, Y.; Uhelski, M.L.; Dougherty, P.M.; Price, T.J. ACE2 and SCARF expression in human dorsal root ganglion nociceptors: Implications for SARS-CoV-2 virus neurological effects. Pain 2020, 161, 2494–2501. [Google Scholar] [CrossRef] [PubMed]
- Cascella, M.; Del Gaudio, A.; Vittori, A.; Bimonte, S.; Del Prete, P.; Forte, C.A.; Cuomo, A.; De Blasio, E. COVID-pain: Acute and late-onset painful clinical manifestations in COVID-19—Molecular mechanisms and research perspectives. J. Pain Res. 2021, 14, 2403–2412. [Google Scholar] [CrossRef]
- Nemoto, W.; Nakagawasai, O.; Yaoita, F.; Kanno, S.; Yomogida, S.; Ishikawa, M.; Tadano, T.; Tan-No, K. Angiotensin II produces nociceptive behavior through spinal AT1 receptor-mediated p38 mitogen-activated protein kinase activation in mice. Mol. Pain 2013, 9, 38. [Google Scholar] [CrossRef] [PubMed]
- Forte, B.L.; Slosky, L.M.; Zhang, H.; Arnold, M.R.; Staatz, W.D.; Hay, M.; Largent-Milnes, T.M.; Vanderah, T.W. Angiotensin-(1-7)/Mas receptor as an antinociceptive agent in cancer-induced bone pain. Pain 2016, 157, 2709–2721. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Lu, X.; Li, M.; Zeng, J.; Zeng, J.; Shen, B.; Zeng, Y. Renin-angiotensin system in osteoarthritis: A new potential therapy. Int. Immunopharmacol. 2019, 75, 105796. [Google Scholar] [CrossRef]
- Tsukamoto, I.; Akagi, M.; Inoue, S.; Yamagishi, K.; Mori, S.; Asada, S. Expressions of local renin-angiotensin system components in chondrocytes. Eur. J. Histochem. 2014, 58, 132–138. [Google Scholar] [CrossRef]
- de Sá, G.A.; dos Santos, A.C.P.M.; Nogueira, J.M.; Dos Santos, D.M.; Amaral, F.A.; Jorge, E.C.; Caliari, M.V.; Queiroz-Junior, C.M.; Ferreira, A.J. Angiotensin II triggers knee joint lesions in experimental osteoarthritis. Bone 2021, 145, 115842. [Google Scholar] [CrossRef]
- Yamagishi, K.; Tsukamoto, I.; Nakamura, F.; Hashimoto, K.; Ohtani, K.; Akagi, M. Activation of the reninangiotensin system in mice aggravates mechanical loadinginduced knee osteoarthritis. Eur. J. Histochem. 2018, 62, 177–187. [Google Scholar] [CrossRef]
- Gul, R.; Kim, U.; Alfadda, A.A. Renin-angiotensin system at the interface of COVID-19 infection. Eur. J. Pharm. 2021, 890, 173656. [Google Scholar] [CrossRef]
- Bian, J.; Li, Z. Angiotensin-converting enzyme 2 (ACE2): SARS-CoV-2 receptor and RAS modulator. Acta Pharm. Sin. B 2021, 11, 1–12. [Google Scholar] [CrossRef]
- Zhuang, X.; Tsukuda, S.; Wrensch, F.; Wing, P.A.; Schilling, M.; Harris, J.M.; Borrmann, H.; Morgan, S.B.; Cane, J.L.; Mailly, L.; et al. The circadian clock component BMAL1 regulates SARS-CoV-2 entry and replication in lung epithelial cells. iScience 2021, 24, 103144. [Google Scholar] [CrossRef] [PubMed]
- Sultan, A.; Ali, R.; Sultan, T.; Ali, S.; Khan, N.J.; Parganiha, A. Circadian clock modulating small molecules repurposing as inhibitors of SARS-CoV-2 M pro for pharmacological interventions in COVID-19 pandemic. Chronobiol. Int. 2021, 38, 971–985. [Google Scholar] [CrossRef] [PubMed]
- Boechat, J.L.; Chora, I.; Morais, A.; Delgado, L. The immune response to SARS-CoV-2 and COVID-19 immunopathology—Current perspectives. Pulmonology 2021, 27, 423–437. [Google Scholar] [CrossRef]
- Ricci, D.; Etna, M.P.; Rizzo, F.; Sandini, S.; Severa, M.; Coccia, E.M. Innate immune response to SARS-CoV-2 infection: From cells to soluble mediators. Int. J. Mol. Sci. 2021, 22, 7017. [Google Scholar] [CrossRef]
- Chau, A.S.; Weber, A.G.; Maria, N.I.; Narain, S.; Liu, A.; Hajizadeh, N.; Malhotra, P.; Bloom, O.; Marder, G.; Kaplan, B. The Longitudinal Immune Response to Coronavirus Disease 2019: Chasing the Cytokine Storm. Arthritis Rheumatol. 2021, 73, 23–35. [Google Scholar] [CrossRef]
- Toor, S.M.; Saleh, R.; Sasidharan Nair, V.; Taha, R.Z.; Elkord, E. T-cell responses and therapies against SARS-CoV-2 infection. Immunology 2021, 162, 30–43. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Liu, S.; Liu, J.; Zhang, Z.; Wan, X.; Huang, B.; Chen, Y.; Zhang, Y. COVID-19: Immunopathogenesis and Immunotherapeutics. Signal Transduct. Target Ther. 2020, 5, 128. [Google Scholar] [CrossRef]
- Parasher, A. COVID-19: Current understanding of its Pathophysiology, Clinical presentation and Treatment. Postgrad. Med. J. 2021, 97, 312–320. [Google Scholar] [CrossRef]
- Khosroshahi, M.L.; Rokni, M.; Mokhtari, T.; Noorbakhsh, F. Immunology, immunopathogenesis and immunotherapeutics of COVID-19: An overview. Int. Immunopharmacol. 2021, 93, 107364. [Google Scholar] [CrossRef] [PubMed]
- Gavriatopoulou, M.; Ntanasis-Stathopoulos, I.; Korompoki, E.; Fotiou, D.; Migkou, M.; Tzanninis, I.G.; Psaltopoulou, T.; Kastritis, E.; Terpos, E.; Dimopoulos, M.A. Emerging treatment strategies for COVID-19 infection. Clin. Exp. Med. 2021, 21, 167–179. [Google Scholar] [CrossRef]
- Diallo, B.A.; Gay, L.; Coiffard, B.; Leone, M.; Mezouar, S.; Mege, J.L. Daytime variation in SARS-CoV-2 infection and cytokine production. Microb. Pathog. 2021, 158, 105067. [Google Scholar] [CrossRef]
- Sengupta, S.; Brooks, T.G.; Grant, G.R.; Fitzgerald, G.A. Accounting for Time: Circadian Rhythms in the Time of COVID-19. J. Biol. Rhythm. 2021, 36, 4–8. [Google Scholar] [CrossRef]
- Sengupta, S.; Ince, L.; Sartor, F.; Borrmann, H.; Zhuang, X.; Naik, A.; Curtis, A.; McKeating, J.A. Clocks, Viruses, and Immunity: Lessons for the COVID-19 Pandemic. J. Biol. Rhythm. 2021, 36, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Tamimi, F.; Abusamak, M.; Akkanti, B.; Chen, Z. The case for chronotherapy in COVID-19-induced acute respiratory distress syndrome. Br. J. Pharmacol. 2020, 177, 4845–4850. [Google Scholar] [CrossRef] [PubMed]
- Leung, J.M.; Yang, C.X.; Tam, A.; Shaipanich, T.; Hackett, T.L.; Singhera, G.K.; Dorscheid, D.R.; Sin, D.D. ACE-2 expression in the small airway epithelia of smokers and COPD patients: Implications for COVID-19. Eur. Respir. J. 2020, 55, 2000688. [Google Scholar] [CrossRef]
- Cai, G.; Bossé, Y.; Xiao, F.; Kheradmand, F.; Amos, C.I. Tobacco smoking increases the lung gene expression of ACE2, the Receptor of SARS-CoV-2. Am. J. Respir. Crit. Care Med. 2020, 201, 1557–1559. [Google Scholar] [CrossRef]
- Zhang, H.; Rostami, M.R.; Leopold, P.L.; Mezey, J.G.; O’Beirne, S.L.; Strulovici-Barel, Y.; Crystal, R.G. Expression of the SARS-CoV-2 ACE2 receptor in the human airway epithelium. Am. J. Respir. Crit. Care Med. 2020, 202, 219–229. [Google Scholar] [CrossRef]
- Jacobs, M.; van Eeckhoutte, H.P.; Wijnant, S.R.A.; Janssens, W.; Joos, G.F.; Brusselle, G.G.; Bracke, K.R. Increased expression of ACE2, the SARS-CoV-2 entry receptor, in alveolar and bronchial epithelium of smokers and COPD subjects. Eur. Respir. J. 2020, 56, 2002378. [Google Scholar] [CrossRef]
- Russo, P.; Bonassi, S.; Giacconi, R.; Malavolta, M.; Tomino, C.; Maggi, F. COVID-19 and smoking: Is nicotine the hidden link? Eur. Respir. J. 2020, 55, 2001116. [Google Scholar] [CrossRef]
- Leung, J.; Yang, C.; Sin, D. COVID-19 and nicotine as a mediator of ACE-2. Eur. Respir. J. 2020, 55, 2001261. [Google Scholar] [CrossRef] [PubMed]
- Di Maro, M.; Cataldi, M.; Santillo, M.; Chiurazzi, M.; Damiano, S.; De Conno, B.; Colantuoni, A.; Guida, B. The Cholinergic and ACE-2-Dependent Anti-Inflammatory Systems in the Lung: New Scenarios Emerging From COVID-19. Front. Physiol. 2021, 12, 653985. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, A.G.; Lin, M.; Wu, J.; Kennedy, T.P.; Daley, L.A.; Ashby, C.R., Jr.; Mantell, L.L. From nicotine to the cholinergic anti-inflammatory reflex–Can nicotine alleviate the dysregulated inflammation in COVID-19? J. Immunotoxicol. 2021, 18, 23–29. [Google Scholar] [CrossRef]
- Qin, Z.; Xiang, K.; Su, D.F.; Sun, Y.; Liu, X. Activation of the Cholinergic Anti-Inflammatory Pathway as a Novel Therapeutic Strategy for COVID-19. Front. Immunol. 2021, 11, 595342. [Google Scholar] [CrossRef] [PubMed]
- Farsalinos, K.; Eliopoulos, E.; Leonidas, D.D.; Papadopoulos, G.E.; Tzartos, S.; Poulas, K. Nicotinic cholinergic system and COVID-19: In silico identification of an interaction between SARS-CoV-2 and nicotinic receptors with potential therapeutic targeting implications. Int. J. Mol. Sci. 2020, 21, 5807. [Google Scholar] [CrossRef]
- Sinkus, M.L.; Graw, S.; Freedman, R.; Ross, R.G.; Lester, H.A.; Leonard, S. The human CHRNA7 and CHRFAM7A genes: A review of the genetics, regulation, and function. Neuropharmacology 2015, 96, 274–288. [Google Scholar] [CrossRef]
- Courties, A.; Boussier, J.; Hadjadj, J.; Yatim, N.; Barnabei, L.; Péré, H.; Veyer, D.; Kernéis, S.; Carlier, N.; Pène, F.; et al. Regulation of the acetylcholine/α7nAChR anti-inflammatory pathway in COVID-19 patients. Sci. Rep. 2021, 11, 11886. [Google Scholar] [CrossRef]
- Lupacchini, L.; Maggi, F.; Tomino, C.; De Dominicis, C.; Mollinari, C.; Fini, M.; Bonassi, S.; Merlo, D.; Russo, P. Nicotine Changes Airway Epithelial Phenotype and May Increase the SARS-CoV-2 Infection Severity. Molecules 2020, 26, 101. [Google Scholar] [CrossRef]
- Lauwers, M.; Courties, A.; Sellam, J.; Wen, C. The cholinergic system in joint health and osteoarthritis: A narrative-review. Osteoarthr. Cartil. 2021, 29, 643–653. [Google Scholar] [CrossRef] [PubMed]
- Courties, A.; Sellam, J.; Berenbaum, F. Role of the autonomic nervous system in osteoarthritis. Best Pract. Res. Clin. Rheumatol. 2017, 31, 661–675. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lauwers, M.; Au, M.; Yuan, S.; Wen, C. COVID-19 in Joint Ageing and Osteoarthritis: Current Status and Perspectives. Int. J. Mol. Sci. 2022, 23, 720. https://doi.org/10.3390/ijms23020720
Lauwers M, Au M, Yuan S, Wen C. COVID-19 in Joint Ageing and Osteoarthritis: Current Status and Perspectives. International Journal of Molecular Sciences. 2022; 23(2):720. https://doi.org/10.3390/ijms23020720
Chicago/Turabian StyleLauwers, Marianne, Manting Au, Shuofeng Yuan, and Chunyi Wen. 2022. "COVID-19 in Joint Ageing and Osteoarthritis: Current Status and Perspectives" International Journal of Molecular Sciences 23, no. 2: 720. https://doi.org/10.3390/ijms23020720
APA StyleLauwers, M., Au, M., Yuan, S., & Wen, C. (2022). COVID-19 in Joint Ageing and Osteoarthritis: Current Status and Perspectives. International Journal of Molecular Sciences, 23(2), 720. https://doi.org/10.3390/ijms23020720