
Innate Immune System Response to Burn Damage—Focus on Cytokine Alteration

 , ,
, ,  , , , , ,
, , , , ,  and
and
Abstract
:1. Introduction
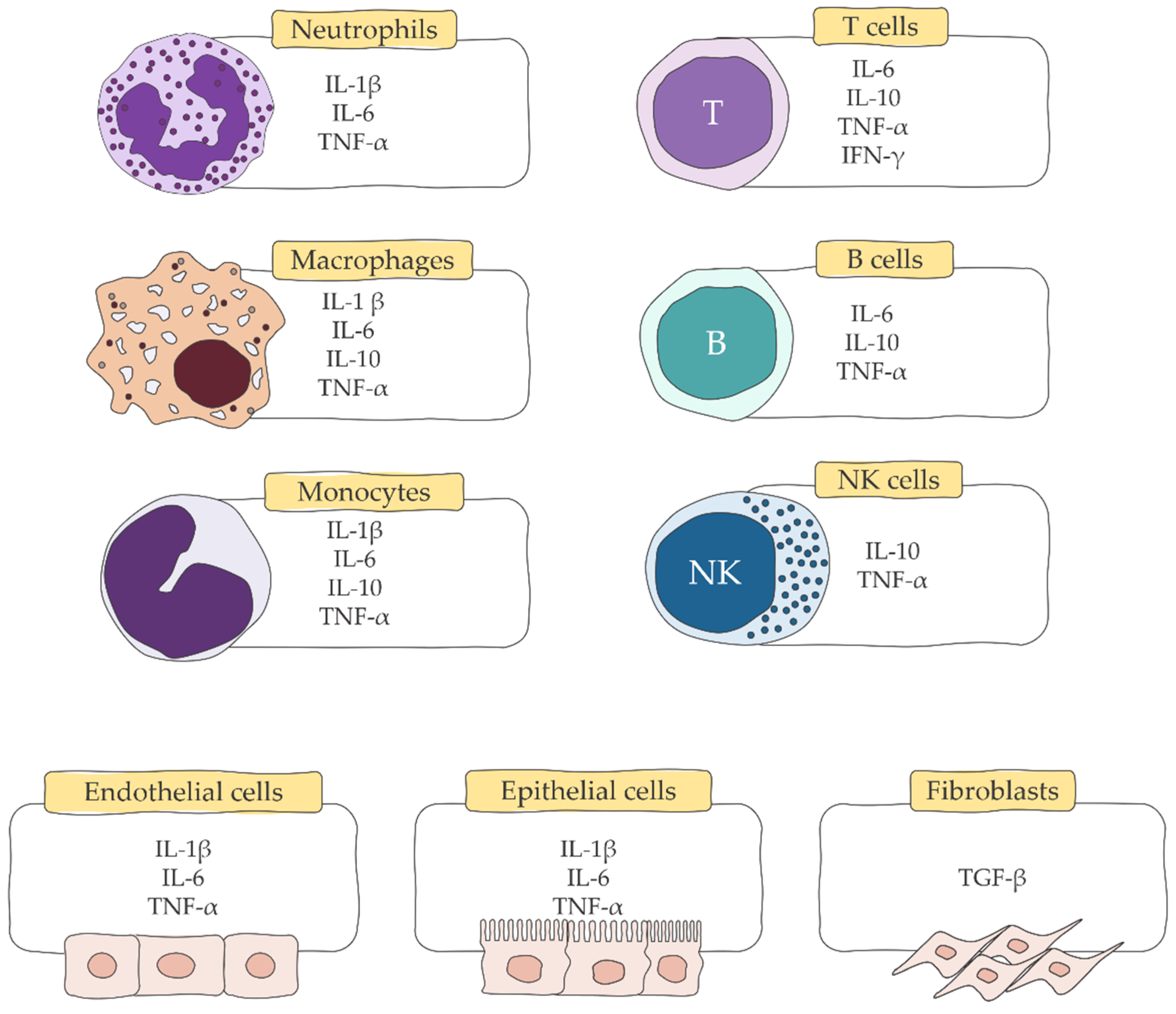
2. Host Immune Response to Burns
2.1. Mast Cells
2.2. Neutrophils
2.3. Dendritic Cells
2.4. Monocytes and Macrophages
2.5. Inflammasomes
2.6. NK and NKT Cells
2.7. Complement System
3. Infections
3.1. In the Wound
3.2. Pneumonias
3.3. In Blood and Urine
3.4. In the Vascular System
4. Cytokines
4.1. Proinflammatory Responses
4.1.1. IL-1
4.1.2. IL-6
4.1.3. TNF-α
4.1.4. IFN-γ
4.2. Anti-Inflammatory Responses
4.2.1. IL-10
4.2.2. TGF-β
5. Antioxidant and Trace Element Supplementation
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schaefer, T.J.; Tannan, S.C. Thermal Burns. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2021. Available online: https://www.ncbi.nlm.nih.gov/books/NBK430773/ (accessed on 4 November 2021).
- Boldeanu, L.; Boldeanu, M.V.; Bogdan, M.; Meca, A.D.; Coman, C.G.; Buca, B.R.; Tartau, C.G.; Tartau, L.M. Immunological Approaches and Therapy in Burns (Review). Exp. Ther. Med. 2020, 20, 2361–2367. [Google Scholar] [CrossRef] [PubMed]
- Rani, M.; Schwacha, M.G. Aging and the Pathogenic Response to Burn. Aging Dis 2012, 3, 171–180. [Google Scholar] [PubMed]
- Hettiaratchy, S.; Dziewulski, P. ABC of Burns: Pathophysiology and Types of Burns. BMJ 2004, 328, 1427–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norbury, W.; Herndon, D.N.; Tanksley, J.; Jeschke, M.G.; Finnerty, C.C. Infection in Burns. Surg. Infect. (Larchmt.) 2016, 17, 250–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.J.; Mahendraraj, K.; Houng, A.; Marano, M.; Petrone, S.; Lee, R.; Chamberlain, R.S. Pediatric Burns: A Single Institution Retrospective Review of Incidence, Etiology, and Outcomes in 2273 Burn Patients (1995-2013). J. Burn. Care Res. 2016, 37, e579–e585. [Google Scholar] [CrossRef]
- Williams, F.N.; Herndon, D.N.; Hawkins, H.K.; Lee, J.O.; Cox, R.A.; Kulp, G.A.; Finnerty, C.C.; Chinkes, D.L.; Jeschke, M.G. The Leading Causes of Death after Burn Injury in a Single Pediatric Burn Center. Crit. Care 2009, 13, R183. [Google Scholar] [CrossRef] [Green Version]
- Joshi, A.P.; Saad, M.; Mohan, M. A Review on Burn and Burn Models in Animals. J. Basic Pharmacol. Toxicol. 2017, 1, 1–8. [Google Scholar]
- Xiao, W.; Mindrinos, M.N.; Seok, J.; Cuschieri, J.; Cuenca, A.G.; Gao, H.; Hayden, D.L.; Hennessy, L.; Moore, E.E.; Minei, J.P.; et al. A Genomic Storm in Critically Injured Humans. J. Exp. Med. 2011, 208, 2581–2590. [Google Scholar] [CrossRef] [PubMed]
- Hampson, P.; Dinsdale, R.J.; Wearn, C.M.; Bamford, A.L.; Bishop, J.R.B.; Hazeldine, J.; Moiemen, N.S.; Harrison, P.; Lord, J.M. Neutrophil Dysfunction, Immature Granulocytes, and Cell-Free DNA are Early Biomarkers of Sepsis in Burn-Injured Patients: A Prospective Observational Cohort Study. Ann. Surg. 2017, 265, 1241–1249. [Google Scholar] [CrossRef]
- Jeschke, M.G.; Patsouris, D.; Stanojcic, M.; Abdullahi, A.; Rehou, S.; Pinto, R.; Chen, P.; Burnett, M.; Amini-Nik, S. Pathophysiologic Response to Burns in the Elderly. EBioMedicine 2015, 2, 1536–1548. [Google Scholar] [CrossRef] [Green Version]
- Al-Tarrah, K.; Hewison, M.; Moiemen, N.; Lord, J.M. Vitamin D Status and Its Influence on Outcomes Following Major Burn Injury and Critical Illness. Burns Trauma 2018, 6, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, D.M. The diagnosis of the depth of burning. Br. J. Surg. 1953, 40, 588–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strudwick, X.L.; Cowin, A.J. The Role of the Inflammatory Response in Burn Injury; IntechOpen: London, UK, 2017; ISBN 978-1-78923-131-1. [Google Scholar]
- Rendon, J.L.; Choudhry, M.A. Th17 Cells: Critical Mediators of Host Responses to Burn Injury and Sepsis. J. Leukoc. Biol. 2012, 92, 529–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- American Burn Association, National Burn Repository. American Burn Association National Burn Repository 2016 Report; Report of data from 2006–2015; American Burn Association, National Burn Repository: Chicago, IL, USA, 2016. [Google Scholar]
- Blears, E.; Sommerhalder, C.; Toliver-Kinsky, T.; Finnerty, C.C.; Herndon, D.N. Current Problems in Burn Immunology. Curr. Probl. Surg. 2020, 57, 100779. [Google Scholar] [CrossRef] [PubMed]
- Niedźwiedzka-Rystwej, P.; Ratajczak, W.; Tokarz-Deptuła, B.; Deptuła, W. Charakterystyka i rola inflamasomów. Post Biol. Kom. 2016, 43, 237–254. [Google Scholar]
- Tiwari, V.K. Burn Wound: How It Differs from Other Wounds? Indian J. Plast. Surg. 2012, 45, 364–373. [Google Scholar] [CrossRef]
- Vinish, M.; Cui, W.; Stafford, E.; Bae, L.; Hawkins, H.; Cox, R.; Toliver-Kinsky, T. Dendritic Cells Modulate Burn Wound Healing by Enhancing Early Proliferation. Wound Repair Regen. 2016, 24, 6–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiu, F.; Jeschke, M.G. Perturbed Mononuclear Phagocyte System in Severely Burned and Septic Patients. Shock 2013, 40, 81–88. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Cai, W.; Yang, X.; Jia, Y.; Zheng, Z.; Wang, H.; Li, J.; Li, Y.; Gao, J.; Fan, L.; et al. ROS-Mediated NLRP3 Inflammasome Activity Is Essential for Burn-Induced Acute Lung Injury. Mediat. Inflamm. 2015, 2015, e720457. [Google Scholar] [CrossRef] [Green Version]
- Niedzwiedzka, P.; Deptula, W. Rola komorek tucznych w odpowiedzi immunologicznej. Medycyna Weterynaryjna 2008, 64, 1291–1294. [Google Scholar]
- Barrett, L.W.; Fear, V.S.; Waithman, J.C.; Wood, F.M.; Fear, M.W. Understanding Acute Burn Injury as a Chronic Disease. Burns Trauma 2019, 7, s41038-019-0163-2. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Xu, T.; Ma, S.; Wen, H. Expression and Activity Levels of Chymase in Mast Cells of Burn Wound Tissues Increase during the Healing Process in a Hamster Model. Exp. Ther. Med. 2015, 9, 2190–2194. [Google Scholar] [CrossRef] [Green Version]
- Souza, H.R.; de Azevedo, L.R.; Possebon, L.; de Souza Costa, S.; Iyomasa-Pilon, M.M.; Oliani, S.M.; Girol, A.P. Heterogeneity of Mast Cells and Expression of Annexin A1 Protein in a Second Degree Burn Model with Silver Sulfadiazine Treatment. PLoS One 2017, 12, e0173417. [Google Scholar] [CrossRef] [Green Version]
- Lateef, Z.; Stuart, G.; Jones, N.; Mercer, A.; Fleming, S.; Wise, L. The Cutaneous Inflammatory Response to Thermal Burn Injury in a Murine Model. Int. J. Mol. Sci. 2019, 20, 538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devine, R.A.; Diltz, Z.; Hall, M.W.; Thakkar, R.K. The Systemic Immune Response to Pediatric Thermal Injury. Int. J. Burns Trauma 2018, 8, 6–16. [Google Scholar]
- Calum, H.; Moser, C.; Jensen, P.Ø.; Christophersen, L.; Maling, D.S.; Van Gennip, M.; Bjarnsholt, T.; Hougen, H.P.; Givskov, M.; Jacobsen, G.K.; et al. Thermal Injury Induces Impaired Function in Polymorphonuclear Neutrophil Granulocytes and Reduced Control of Burn Wound Infection. Clin. Exp. Immunol. 2009, 156, 102–110. [Google Scholar] [CrossRef] [Green Version]
- Hazeldine, J.; McGee, K.C.; Al-Tarrah, K.; Hassouna, T.; Patel, K.; Imran, R.; Bishop, J.R.B.; Bamford, A.; Barnes, D.; Wilson, Y.; et al. Multicentre, Longitudinal, Observational Cohort Study to Examine the Relationship between Neutrophil Function and Sepsis in Adults and Children with Severe Thermal Injuries: A Protocol for the Scientific Investigation of the Biological Pathways Following Thermal Injury-2 (SIFTI-2) Study. BMJ Open 2021, 11, e052035. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, L.; Guo, Z.; Li, L.; Shao, Y.; Song, M.; Sun, B. Investigation and Assessment of Neutrophil Dysfunction Early after Severe Burn Injury. Burns 2021, 47, 1851–1862. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.N.; Moore, M.; Dimisko, L.; Alexander, A.; Ibrahim, A.; Hassell, B.A.; Warren, H.S.; Tompkins, R.G.; Fagan, S.P.; Irimia, D. Spontaneous Neutrophil Migration Patterns during Sepsis after Major Burns. PLoS One 2014, 9, e114509. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.-C.; Wu, S.-Y.S.; Su, W.-Y.; Lin, Y.-C.; Lee, Y.-H.; Wu, W.-H.; Chen, C.-H.; Wen, Z.-H. Anti-Inflammatory and Burn Injury Wound Healing Properties of the Shell of Haliotis Diversicolor. BMC Complementary Altern. Med. 2016, 16, 487. [Google Scholar] [CrossRef] [Green Version]
- Gürbüz, V.; Corak, A.; Yeğen, B.C.; Kurtel, H.; Alican, I. Oxidative Organ Damage in a Rat Model of Thermal Injury: The Effect of Cyclosporin A. Burns 1997, 23, 37–42. [Google Scholar] [CrossRef]
- Parihar, A.; Parihar, M.S.; Milner, S.; Bhat, S. Oxidative Stress and Anti-Oxidative Mobilization in Burn Injury. Burns 2008, 34, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Kartchner, L.B.; Gode, C.J.; Dunn, J.L.M.; Glenn, L.I.; Duncan, D.N.; Wolfgang, M.C.; Cairns, B.A.; Maile, R. One-Hit Wonder: Late after Burn Injury, Granulocytes Can Clear One Bacterial Infection but Cannot Control a Subsequent Infection. Burns 2019, 45, 627–640. [Google Scholar] [CrossRef]
- Valvis, S.M.; Waithman, J.; Wood, F.M.; Fear, M.W.; Fear, V.S. The Immune Response to Skin Trauma Is Dependent on the Etiology of Injury in a Mouse Model of Burn and Excision. J. Invest. Dermatol. 2015, 135, 2119–2128. [Google Scholar] [CrossRef] [Green Version]
- van den Berg, L.M.; de Jong, M.A.W.P.; de Witte, L.; Ulrich, M.M.W.; Geijtenbeek, T.B.H. Burn Injury Suppresses Human Dermal Dendritic Cell and Langerhans Cell Function. Cell Immunol. 2011, 268, 29–36. [Google Scholar] [CrossRef]
- Kurmis, R.; Greenwood, J.; Aromataris, E. Trace Element Supplementation Following Severe Burn Injury: A Systematic Review and Meta-Analysis. J. Burn Care Res. 2016, 37, 143–159. [Google Scholar] [CrossRef] [PubMed]
- Merah-Mourah, F.; Cohen, S.O.; Charron, D.; Mooney, N.; Haziot, A. Identification of Novel Human Monocyte Subsets and Evidence for Phenotypic Groups Defined by Interindividual Variations of Expression of Adhesion Molecules. Sci. Rep. 2020, 10, 4397. [Google Scholar] [CrossRef] [PubMed]
- Galbraith, N.J.; Gardner, S.A.; Walker, S.P.; Trainor, P.; Carter, J.V.; Bishop, C.; Sarojini, H.; O’Brien, S.J.; Bhatnagar, A.; Polk, H.C.; et al. The Role and Function of IκKα/β in Monocyte Impairment. Sci. Rep. 2020, 10, 12222. [Google Scholar] [CrossRef]
- Rehou, S.; Shahrokhi, S.; Natanson, R.; Stanojcic, M.; Jeschke, M.G. Antioxidant and Trace Element Supplementation Reduce the Inflammatory Response in Critically Ill Burn Patients. J. Burn Care Res. 2018, 39, 1–9. [Google Scholar] [CrossRef]
- Strudwick, X.L.; Adams, D.H.; Pyne, N.T.; Samuel, M.S.; Murray, R.Z.; Cowin, A.J. Systemic Delivery of Anti-Integrin AL Antibodies Reduces Early Macrophage Recruitment, Inflammation, and Scar Formation in Murine Burn Wounds. Adv. Wound Care 2020, 9, 637–648. [Google Scholar] [CrossRef]
- Williams, K.N.; Szilagyi, A.; He, L.-K.; Conrad, P.; Halerz, M.; Gamelli, R.L.; Shankar, R.; Muthumalaiappan, K. Dendritic Cell Depletion in Burn Patients Is Regulated by MafB Expression. J. Burn Care Res. 2012, 33, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.-M.; Yu, Y.; Chai, J.-K.; Hu, S.; Sheng, Z.-Y.; Yao, Y.-M. Low HLA-DR Expression on CD14+ Monocytes of Burn Victims with Sepsis, and the Effect of Carbachol in Vitro. Burns 2008, 34, 1158–1162. [Google Scholar] [CrossRef] [PubMed]
- Venet, F.; Tissot, S.; Debard, A.-L.; Faudot, C.; Crampé, C.; Pachot, A.; Ayala, A.; Monneret, G. Decreased Monocyte Human Leukocyte Antigen-DR Expression after Severe Burn Injury: Correlation with Severity and Secondary Septic Shock. Crit. Care Med. 2007, 35, 1910–1917. [Google Scholar] [CrossRef]
- Kaczmarczyk, M.; Niedźwiedzka, P.; Deptuła, W. Characteristics of Dendritic Cells. Adv. Clin. Exp. Med. 2006, 15, 871. [Google Scholar]
- Schwacha, M.G. Macrophages and Post-Burn Immune Dysfunction. Burns 2003, 29, 1–14. [Google Scholar] [CrossRef]
- Grbic, J.T.; Mannick, J.A.; Gough, D.B.; Rodrick, M.L. The Role of Prostaglandin E2 in Immune Suppression Following Injury. Ann. Surg. 1991, 214, 253–263. [Google Scholar] [CrossRef]
- Horgan, A.F.; O’Riordain, D.S.; Chin, D.H.; Mannick, J.A.; Rodrick, M.L. The Role of Cyclic Adenosine Monophosphate in the Suppression of Cellular Immunity after Thermal Injury. Arch. Surg. 1994, 129, 1284–1289. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.W. Free Radicals and Lipid Peroxidation Mediated Injury in Burn Trauma: The Role of Antioxidant Therapy. Toxicology 2003, 189, 75–88. [Google Scholar] [CrossRef]
- Reddell, L.; Cotton, B.A. Antioxidants and Micronutrient Supplementation in Trauma Patients. Curr. Opin. Clin. Nutr. Metab. Care 2012, 15, 181–187. [Google Scholar] [CrossRef]
- Guo, H.; Callaway, J.B.; Ting, J.P.-Y. Inflammasomes: Mechanism of Action, Role in Disease, and Therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [Green Version]
- Schwacha, M.G.; Samy, T.S.; Catania, R.A.; Chaudry, I.H. Thermal Injury Alters Macrophage Responses to Prostaglandin E2: Contribution to the Enhancement of Inducible Nitric Oxide Synthase Activity. J. Leukoc. Biol. 1998, 64, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Stanojcic, M.; Chen, P.; Harrison, R.A.; Wang, V.; Antonyshyn, J.; Zúñiga-Pflücker, J.C.; Jeschke, M.G. Leukocyte Infiltration and Activation of the NLRP3 Inflammasome in White Adipose Tissue Following Thermal Injury. Crit. Care Med. 2014, 42, 1357–1364. [Google Scholar] [CrossRef] [Green Version]
- Vinaik, R.; Abdullahi, A.; Barayan, D.; Jeschke, M.G. NLRP3 Inflammasome Activity Is Required for Wound Healing after Burns. Transl. Res. 2020, 217, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Cuddihy, J.; Wu, G.; Ho, L.; Kudo, H.; Dannhorn, A.; Mandalia, S.; Collins, D.; Weir, J.; Spencer, A.; Vizcaychipi, M.; et al. Lactate Dehydrogenase Activity Staining Demonstrates Time-Dependent Immune Cell Infiltration in Human Ex-Vivo Burn-Injured Skin. Sci. Rep. 2021, 11, 21249. [Google Scholar] [CrossRef]
- Xiao, M.; Li, L.; Li, C.; Liu, L.; Yu, Y.; Ma, L. 3,4-Methylenedioxy-β-Nitrostyrene Ameliorates Experimental Burn Wound Progression by Inhibiting the NLRP3 Inflammasome Activation. Plast. Reconstr. Surg. 2016, 137, 566e. [Google Scholar] [CrossRef] [PubMed]
- Farag, S.S.; Fehniger, T.A.; Ruggeri, L.; Velardi, A.; Caligiuri, M.A. Natural Killer Cell Receptors: New Biology and Insights into the Graft-versus-Leukemia Effect. Blood 2002, 100, 1935–1947. [Google Scholar] [CrossRef]
- Tosi, M.F. Innate Immune Responses to Infection. J. Allergy Clin. Immunol. 2005, 116, 241–249. [Google Scholar] [CrossRef]
- Klimpel, G.R.; Herndon, D.N.; Fons, M.; Albrecht, T.; Asuncion, M.T.; Chin, R.; Stein, M.D. Defective NK Cell Activity Following Thermal Injury. Clin. Exp. Immunol. 1986, 66, 384–392. [Google Scholar]
- Blazar, B.A.; Rodrick, M.L.; O’Mahony, J.B.; Wood, J.J.; Bessey, P.Q.; Wilmore, D.W.; Mannick, J.A. Suppression of Natural Killer-Cell Function in Humans Following Thermal and Traumatic Injury. J. Clin. Immunol. 1986, 6, 26–36. [Google Scholar] [CrossRef]
- Dibirdik, I.; Durak, N.; Kişlaoǧlu, E.; Kutluay, T.; Aytemiz, C. Effects of Prophylactic Intravenous Immunoglobulin-G Therapy on Humoral and Cellular Immune Components and Their Functions in Burned Patients. Burns 1995, 21, 130–135. [Google Scholar] [CrossRef]
- Bender, B.S.; Winchurch, R.A.; Thupari, J.N.; Proust, J.J.; Adler, W.H.; Munster, A.M. Depressed Natural Killer Cell Function in Thermally Injured Adults: Successful in Vivo and in Vitro Immunomodulation and the Role of Endotoxin. Clin. Exp. Immunol. 1988, 71, 120–125. [Google Scholar]
- Zhan, J.; Li, G. A study on the effects of CD3AK cells on the improvement of cellular immune function in burned patients. Zhonghua Shao Shang Za Zhi = Zhonghua Shaoshang Zazhi = Chin. J. Burn. 2001, 17, 159–162. [Google Scholar]
- Korkmaz, H.I.; Krijnen, P.A.J.; Ulrich, M.M.W.; de Jong, E.; van Zuijlen, P.P.M.; Niessen, H.W.M. The Role of Complement in the Acute Phase Response after Burns. Burns 2017, 43, 1390–1399. [Google Scholar] [CrossRef]
- Mokline, A.; Garsallah, L.; Rahmani, I.; Jerbi, K.; Oueslati, H.; Tlaili, S.; Hammouda, R.; Gasri, B.; Messadi, A.A. Procalcitonin: A Diagnostic and Prognostic Biomarker of Sepsis in Burned Patients. Ann. Burns Fire Disasters 2015, 28, 116–120. [Google Scholar]
- Sinha, A.; Sharma, M.K.; Tripathi, K.; Duggal, N.; Tiwari, V.K. Evaluation of Serum Levels of Procalcitonin and C-Reactive Protein as Prognostic Indicators in Burns. Indian J. Plast. Surg. 2021, 54, 308–313. [Google Scholar] [CrossRef]
- Zbyrak, V.; Reverón, S.L.; Smoke, S.; Mehta, A.; Marano, M.A.; Lee, R. Antibiotic Usage After Procalcitonin-Guided Therapy Algorithm Implementation In A Burn Intensive Care Unit. Ann. Burns Fire Disasters 2020, 33, 317–321. [Google Scholar] [PubMed]
- Møller-Kristensen, M.; Hamblin, M.R.; Thiel, S.; Jensenius, J.C.; Takahashi, K. Burn Injury Reveals Altered Phenotype in Mannan-Binding Lectin-Deficient Mice. J. Investig. Dermatol. 2007, 127, 1524–1531. [Google Scholar] [CrossRef] [Green Version]
- Brownson, E.G.; Gibran, N.S. Evaluation of the Burn Wound: Management Decisions. In Total Burn Care; Elsevier: Amsterdam, The Netherlands, 2018; pp. 87–92. [Google Scholar]
- Church, D.; Elsayed, S.; Reid, O.; Winston, B.; Lindsay, R. Burn Wound Infections. Clin. Microbiol. Rev. 2006, 19, 403–434. [Google Scholar] [CrossRef] [Green Version]
- Cambiaso-Daniel, J.; Gallagher, J.J.; Norbury, W.B.; Finnerty, C.C.; Herndon, D.N.; Culnan, D.M. Treatment of Infection in Burn Patients. In Total Burn Care; Elsevier: Amsterdam, The Netherlands, 2018; pp. 93–113.e4. [Google Scholar] [CrossRef]
- Essayagh, T.; El Hamzaoui, S. Epidemiology of burn wound infection in Rabat, Morocco: Three-year review. Med. Sante Trop. 2014, 24, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Latifi, N.A.; Karimi, H. Correlation of Occurrence of Infection in Burn Patients. Ann. Burns Fire Disasters 2017, 30, 172–176. [Google Scholar] [PubMed]
- Sewunet, T.; Demissie, Y.; Mihret, A.; Abebe, T. Bacterial Profile and Antimicrobial Susceptibility Pattern of Isolates among Burn Patients at Yekatit 12 Hospital Burn Center, Addis Ababa, Ethiopia. Ethiop. J. Health Sci. 2013, 23, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Patil, N.K.; Luan, L.; Bohannon, J.K.; Hernandez, A.; Guo, Y.; Sherwood, E.R. Frontline Science: Anti-PD-L1 Protects against Infection with Common Bacterial Pathogens after Burn Injury. J. Leukoc. Biol. 2018, 103, 23–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanford, J.A.; Gallo, R.L. Functions of the Skin Microbiota in Health and Disease. Semin Immunol. 2013, 25, 370–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toliver-Kinsky, T.; Kobayashi, M.; Suzuki, F.; Sherwood, R.E. The systemic inflammatory response syndrome. In Total Burn Care; Elsevier: Amsterdam, The Netherlands, 2018; pp. 205–220. [Google Scholar]
- Plichta, J.K.; Gao, X.; Lin, H.; Dong, Q.; Toh, E.; Nelson, D.E.; Gamelli, R.L.; Grice, E.A.; Radek, K.A. Cutaneous Burn Injury Promotes Shifts in the Bacterial Microbiome in Autologous Donor Skin: Implications for Skin Grafting Outcomes. Shock 2017, 48, 441–448. [Google Scholar] [CrossRef]
- Corcione, S.; Lupia, T.; De Rosa, F.G. Microbiome in the Setting of Burn Patients: Implications for Infections and Clinical Outcomes. Burns Trauma 2020, 8, tkaa033. [Google Scholar] [CrossRef]
- Chan, C.-H.; Yang, S.-F.; Yeh, H.-W.; Yeh, Y.-T.; Wang, Y.-H.; Teng, Y.-H.; Yeh, C.-B. Risk of Pneumonia in Patients with Burn Injury: A Population-Based Cohort Study. Clin. Epidemiol. 2018, 10, 1083–1091. [Google Scholar] [CrossRef] [Green Version]
- Liodaki, E.; Kalousis, K.; Mauss, K.L.; Kisch, T.; Mailaender, P.; Stang, F. Epidemiology of Pneumonia in a Burn Care Unit: The Influence of Inhalation Trauma on Pneumonia and of Pneumonia on Burn Mortality. Ann. Burns Fire Disasters 2015, 28, 128–133. [Google Scholar] [PubMed]
- Woodson, L.C.; Sherwood, E.R.; Kinsky, M.P.; Talon, M.; Martinello, C.; Woodson, S.M. Anesthesia for burned patients. In Total Burn Care; Elsevier: Amsterdam, The Netherlands, 2018; pp. 131–157. [Google Scholar]
- de La Cal, M.A.; Cerdá, E.; García-Hierro, P.; Lorente, L.; Sánchez-Concheiro, M.; Díaz, C.; van Saene, H.K. Pneumonia in Patients with Severe Burns : A Classification According to the Concept of the Carrier State. Chest 2001, 119, 1160–1165. [Google Scholar] [CrossRef]
- Greenhalgh, D.G.; Saffle, J.R.; Holmes, J.H.; Gamelli, R.L.; Palmieri, T.L.; Horton, J.W.; Tompkins, R.G.; Traber, D.L.; Mozingo, D.W.; Deitch, E.A.; et al. American Burn Association Consensus Conference to Define Sepsis and Infection in Burns. J. Burn. Care Res. 2007, 28, 776–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, P.-H.; Kao, C.-C.; How, C.-K.; Yang, Y.-S.; Chen, M.-C.; Hung-Tsang Yen, D.; Lee, Y.-T. Initial White Blood Cell Count and Revised Baux Score Predict Subsequent Bloodstream Infection in Burn Patients: A Retrospective Analysis of Severe Burn Patients from the Formosa Color Dust Explosion of 2015. J. Formos Med. Assoc. 2021, 120, 1719–1728. [Google Scholar] [CrossRef] [PubMed]
- Wisplinghoff, H.; Bischoff, T.; Tallent, S.M.; Seifert, H.; Wenzel, R.P.; Edmond, M.B. Nosocomial Bloodstream Infections in US Hospitals: Analysis of 24,179 Cases from a Prospective Nationwide Surveillance Study. Clin. Infect. Dis. 2004, 39, 309–317. [Google Scholar] [CrossRef] [Green Version]
- Ballard, J.; Edelman, L.; Saffle, J.; Sheridan, R.; Kagan, R.; Bracco, D.; Cancio, L.; Cairns, B.; Baker, R.; Fillari, P.; et al. Positive Fungal Cultures in Burn Patients: A Multicenter Review. J. Burn. Care Res. 2008, 29, 213–221. [Google Scholar] [CrossRef]
- O’Neill, J.A.; Pruitt, B.A.; Foley, F.D.; Moncrief, J.A. Suppurative Thrombophlebitis--a Lethal Complication of Intravenous Therapy. J. Trauma 1968, 8, 256–267. [Google Scholar] [CrossRef]
- Samsoondar, W.; Freeman, J.B.; Coultish, I.; Oxley, C. Colonization of Intravascular Catheters in the Intensive Care Unit. Am. J. Surg. 1985, 149, 730–732. [Google Scholar] [CrossRef]
- Land, W.G. The Role of Damage-Associated Molecular Patterns in Human Diseases: Part I - Promoting Inflammation and Immunity. Sultan. Qaboos Univ. Med. J. 2015, 15, e9–e21. [Google Scholar] [PubMed]
- Kany, S.; Vollrath, J.T.; Relja, B. Cytokines in Inflammatory Disease. Int. J. Mol. Sci. 2019, 20, 6008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, O.; Akira, S. Pattern Recognition Receptors and Inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [Green Version]
- Curfs, J.H.; Meis, J.F.; Hoogkamp-Korstanje, J.A. A Primer on Cytokines: Sources, Receptors, Effects, and Inducers. Clin. Microbiol. Rev. 1997, 10, 742–780. [Google Scholar] [CrossRef]
- Rani, M.; Nicholson, S.E.; Zhang, Q.; Schwacha, M.G. Damage-Associated Molecular Patterns (DAMPs) Released after Burn Are Associated with Inflammation and Monocyte Activation. Burns 2017, 43, 297–303. [Google Scholar] [CrossRef] [Green Version]
- Kaneko, N.; Kurata, M.; Yamamoto, T.; Morikawa, S.; Masumoto, J. The Role of Interleukin-1 in General Pathology. Inflamm. Regen. 2019, 39, 12. [Google Scholar] [CrossRef] [Green Version]
- Roh, J.S.; Sohn, D.H. Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune. Netw. 2018, 18, e27. [Google Scholar] [CrossRef]
- Bidani, A.; Wang, C.Z.; Heming, T.A. Early Effects of Smoke Inhalation on Alveolar Macrophage Functions. Burns 1996, 22, 101–106. [Google Scholar] [CrossRef]
- Gery, I.; Gershon, R.K.; Waksman, B.H. Potentiation of the T-Lymphocyte Response to Mitogens. I. The Responding Cell. J. Exp. Med. 1972, 136, 128–142. [Google Scholar] [CrossRef] [Green Version]
- Rosenstreich, D.L.; Vogel, S.N.; Jacques, A.R.; Wahl, L.M.; Oppenheim, J.J. Macrophage Sensitivity to Endotoxin: Genetic Control by a Single Codominant Gene. J. Immunol. 1978, 121, 1664–1670. [Google Scholar]
- Cannon, J.G.; Friedberg, J.S.; Gelfand, J.A.; Tompkins, R.G.; Burke, J.F.; Dinarello, C.A. Circulating Interleukin-1 Beta and Tumor Necrosis Factor-Alpha Concentrations after Burn Injury in Humans. Crit. Care Med. 1992, 20, 1414–1419. [Google Scholar] [CrossRef]
- Finnerty, C.C.; Herndon, D.N.; Przkora, R.; Pereira, C.T.; Oliveira, H.M.; Queiroz, D.M.M.; Rocha, A.M.C.; Jeschke, M.G. Cytokine Expression Profile over Time in Severely Burned Pediatric Patients. Shock 2006, 26, 13–19. [Google Scholar] [CrossRef]
- Kupper, T.S.; Ballard, D.W.; Chua, A.O.; McGuire, J.S.; Flood, P.M.; Horowitz, M.C.; Langdon, R.; Lightfoot, L.; Gubler, U. Human Keratinocytes Contain MRNA Indistinguishable from Monocyte Interleukin 1 Alpha and Beta MRNA. Keratinocyte Epidermal Cell-Derived Thymocyte-Activating Factor Is Identical to Interleukin 1. J. Exp. Med. 1986, 164, 2095–2100. [Google Scholar] [CrossRef] [Green Version]
- Salgado, R.M.; Alcántara, L.; Mendoza-Rodríguez, C.A.; Cerbón, M.; Hidalgo-González, C.; Mercadillo, P.; Moreno, L.M.; Álvarez-Jiménez, R.; Krötzsch, E. Post-Burn Hypertrophic Scars Are Characterized by High Levels of IL-1β MRNA and Protein and TNF-α Type i Receptors. Burns 2012, 38, 668–676. [Google Scholar] [CrossRef]
- Ueyama, M.; Maruyama, I.; Osame, M.; Sawada, Y. Marked Increase in Plasma Interleukin-6 in Burn Patients. J. Lab Clin. Med. 1992, 120, 693–698. [Google Scholar]
- Pileri, D.; Accardo Palombo, A.; D’Amelio, L.; D’Arpa, N.; Amato, G.; Masellis, A.; Cataldo, V.; Mogavero, R.; Napoli, B.; Lombardo, C.; et al. Concentrations of Cytokines Il-6 and Il-10 in Plasma of Burn Patients: Their Relationship to Sepsis and Outcome. Ann. Burns Fire Disasters 2008, 21, 182–185. [Google Scholar]
- Gauglitz, G.G.; Finnerty, C.C.; Herndon, D.N.; Mlcak, R.P.; Jeschke, M.G. Are Serum Cytokines Early Predictors for the Outcome of Burn Patients with Inhalation Injuries Who Do Not Survive? Critical. Care 2008, 12, R81. [Google Scholar] [CrossRef] [Green Version]
- Qiao, Z.; Wang, W.; Yin, L.; Luo, P.; Greven, J.; Horst, K.; Hildebrand, F. Using IL-6 Concentrations in the First 24 h Following Trauma to Predict Immunological Complications and Mortality in Trauma Patients: A Meta-Analysis. Eur. J. Trauma Emerg. Surg. 2018, 44, 679–687. [Google Scholar] [CrossRef]
- Popko, K.; Gorska, E.; Stelmaszczyk-Emmel, A.; Plywaczewski, R.; Stoklosa, A.; Gorecka, D.; Pyrzak, B.; Demkow, U. Proinflammatory Cytokines IL-6 and TNF-α and the Development of Inflammation in Obese Subjects. Eur. J. Med. Res. 2010, 15, 120–122. [Google Scholar] [CrossRef]
- Old, L.J. Tumor Necrosis Factor (TNF). Science 1985, 230, 630–632. [Google Scholar] [CrossRef]
- Spooner, C.E.; Markowitz, N.P.; Saravolatz, L.D. The Role of Tumor Necrosis Factor in Sepsis. Clin. Immunol. Immunopathol. 1992, 62, S11–S17. [Google Scholar] [CrossRef] [Green Version]
- Stanojcic, M.; Chen, P.; Xiu, F.; Jeschke, M.G. Impaired Immune Response in Elderly Burn Patients: New Insights Into the Immune-Senescence Phenotype. Ann. Surg. 2016, 264, 195–202. [Google Scholar] [CrossRef]
- Torre-Amione, G.; Bozkurt, B.; Deswal, A.; Mann, D.L. An Overview of Tumor Necrosis Factor Alpha and the Failing Human Heart. Curr. Opin. Cardiol. 1999, 14, 206–210. [Google Scholar] [CrossRef]
- Marchi, L.F.; Sesti-Costa, R.; Ignacchiti, M.D.C.; Chedraoui-Silva, S.; Mantovani, B. In Vitro Activation of Mouse Neutrophils by Recombinant Human Interferon-Gamma: Increased Phagocytosis and Release of Reactive Oxygen Species and pro-Inflammatory Cytokines. Int. Immunopharmacol. 2014, 18, 228–235. [Google Scholar] [CrossRef] [Green Version]
- O’Sullivan, S.T.; Lederer, J.A.; Horgan, A.F.; Chin, D.H.; Mannick, J.A.; Rodrick, M.L. Major Injury Leads to Predominance of the T Helper-2 Lymphocyte Phenotype and Diminished Interleukin-12 Production Associated with Decreased Resistance to Infection. Ann. Surg. 1995, 222, 482–490; discussion 490–492. [Google Scholar] [CrossRef]
- Wolf, S.E.; Woodside, K.J.; Ramirez, R.J.; Kobayashi, M.; Suzuki, F.; Herndon, D.N. Insulin-like Growth Factor-I/Insulin-like Growth Factor Binding Protein-3 Alters Lymphocyte Responsiveness Following Severe Burn. J. Surg. Res. 2004, 117, 255–261. [Google Scholar] [CrossRef]
- Katsikis, P.D.; Chu, C.Q.; Brennan, F.M.; Maini, R.N.; Feldmann, M. Immunoregulatory Role of Interleukin 10 in Rheumatoid Arthritis. J. Exp. Med. 1994, 179, 1517–1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dehne, M.G.; Sablotzki, A.; Hoffmann, A.; Mühling, J.; Dietrich, F.E.; Hempelmann, G. Alterations of Acute Phase Reaction and Cytokine Production in Patients Following Severe Burn Injury. Burns 2002, 28, 535–542. [Google Scholar] [CrossRef]
- Sherry, R.M.; Cue, J.I.; Goddard, J.K.; Parramore, J.B.; DiPiro, J.T. Interleukin-10 Is Associated with the Development of Sepsis in Trauma Patients. J. Trauma Inj. Infect. Crit. Care 1996, 40, 613–617; discussion 616–617. [Google Scholar] [CrossRef]
- Lyons, A.; Kelly, J.L.; Rodrick, M.L.; Mannick, J.A.; Lederer, J.A. Major Injury Induces Increased Production of Interleukin-10 by Cells of the Immune System with a Negative Impact on Resistance to Infection. Ann. Surg. 1997, 226, 450–458; discussion 458–460. [Google Scholar] [CrossRef]
- Klass, B.R.; Grobbelaar, A.O.; Rolfe, K.J. Transforming Growth Factor Β1 Signalling, Wound Healing and Repair: A Multifunctional Cytokine with Clinical Implications for Wound Repair, a Delicate Balance. Postgrad. Med. J. 2009, 85, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.B.; Sporn, M.B. Differential Expression of the TGF-Beta Isoforms in Embryogenesis Suggests Specific Roles in Developing and Adult Tissues. Mol. Reprod. Dev. 1992, 32, 91–98. [Google Scholar] [CrossRef]
- Cowin, A.J.; Holmes, T.M.; Brosnan, P.; Ferguson, M.W. Expression of TGF-Beta and Its Receptors in Murine Fetal and Adult Dermal Wounds. Eur. J. Dermatol. 2001, 11, 424–431. [Google Scholar]
- Penn, J.W.; Grobbelaar, A.O.; Rolfe, K.J. The Role of the TGF-β Family in Wound Healing, Burns and Scarring: A Review. Int. J. Burns Trauma 2012, 2, 18–28. [Google Scholar]
- Wang, R.; Ghahary, A.; Shen, Q.; Scott, P.G.; Roy, K.; Tredget, E.E. Hypertrophic Scar Tissues and Fibroblasts Produce More Transforming Growth Factor-Beta1 MRNA and Protein than Normal Skin and Cells. Wound Repair Regen. 2000, 8, 128–137. [Google Scholar] [CrossRef]
- Schmid, P.; Itin, P.; Cherry, G.; Bi, C.; Cox, D.A. Enhanced Expression of Transforming Growth Factor-Beta Type I and Type II Receptors in Wound Granulation Tissue and Hypertrophic Scar. Am. J. Pathol. 1998, 152, 485–493. [Google Scholar] [PubMed]
- Smeland, E.B.; Blomhoff, H.K.; Holte, H.; Ruud, E.; Beiske, K.; Funderud, S.; Godal, T.; Ohlsson, R. Transforming Growth Factor Type Beta (TGF Beta) Inhibits G1 to S Transition, but Not Activation of Human B Lymphocytes. Exp. Cell Res. 1987, 171, 213–222. [Google Scholar] [CrossRef]
- Stavnezer, J. Regulation of Antibody Production and Class Switching by TGF-Beta. J. Immunol. 1995, 155, 1647–1651. [Google Scholar]
- Ishikawa, K.; Nishimura, T.; DeSerres, S.; Meyer, A.A. The Effects of Transforming Growth Factor-Beta Neutralization on Postburn Humoral Immunity. J. Trauma 2004, 57, 529–536. [Google Scholar] [CrossRef] [PubMed]
- van Zanten, A.R.H.; Dhaliwal, R.; Garrel, D.; Heyland, D.K. Enteral Glutamine Supplementation in Critically Ill Patients: A Systematic Review and Meta-Analysis. Crit. Care 2015, 19, 294. [Google Scholar] [CrossRef] [Green Version]
- Rowan, M.P.; Cancio, L.C.; Elster, E.A.; Burmeister, D.M.; Rose, L.F.; Natesan, S.; Chan, R.K.; Christy, R.J.; Chung, K.K. Burn Wound Healing and Treatment: Review and Advancements. Crit. Care 2015, 19, 243. [Google Scholar] [CrossRef] [Green Version]
- Hoşnuter, M.; Gürel, A.; Babucçu, O.; Armutcu, F.; Kargi, E.; Işikdemir, A. The Effect of CAPE on Lipid Peroxidation and Nitric Oxide Levels in the Plasma of Rats Following Thermal Injury. Burns 2004, 30, 121–125. [Google Scholar] [CrossRef]
- Mayo, J.C.; Tan, D.-X.; Sainz, R.M.; Lopez-Burillo, S.; Reiter, R.J. Oxidative Damage to Catalase Induced by Peroxyl Radicals: Functional Protection by Melatonin and Other Antioxidants. Free Radic. Res. 2003, 37, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Tunali, T.; Sener, G.; Yarat, A.; Emekli, N. Melatonin Reduces Oxidative Damage to Skin and Normalizes Blood Coagulation in a Rat Model of Thermal Injury. Life Sci. 2005, 76, 1259–1265. [Google Scholar] [CrossRef]
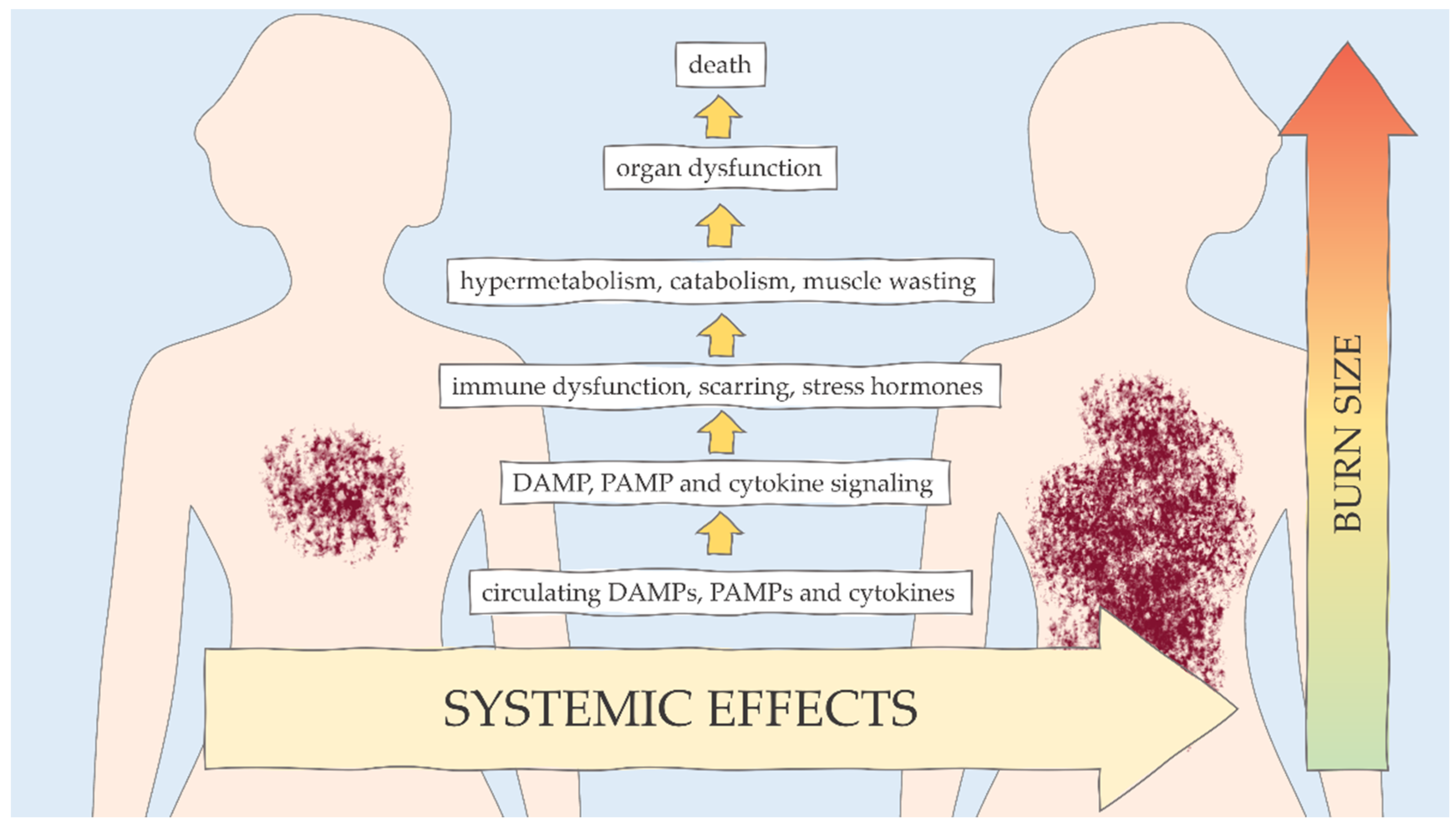
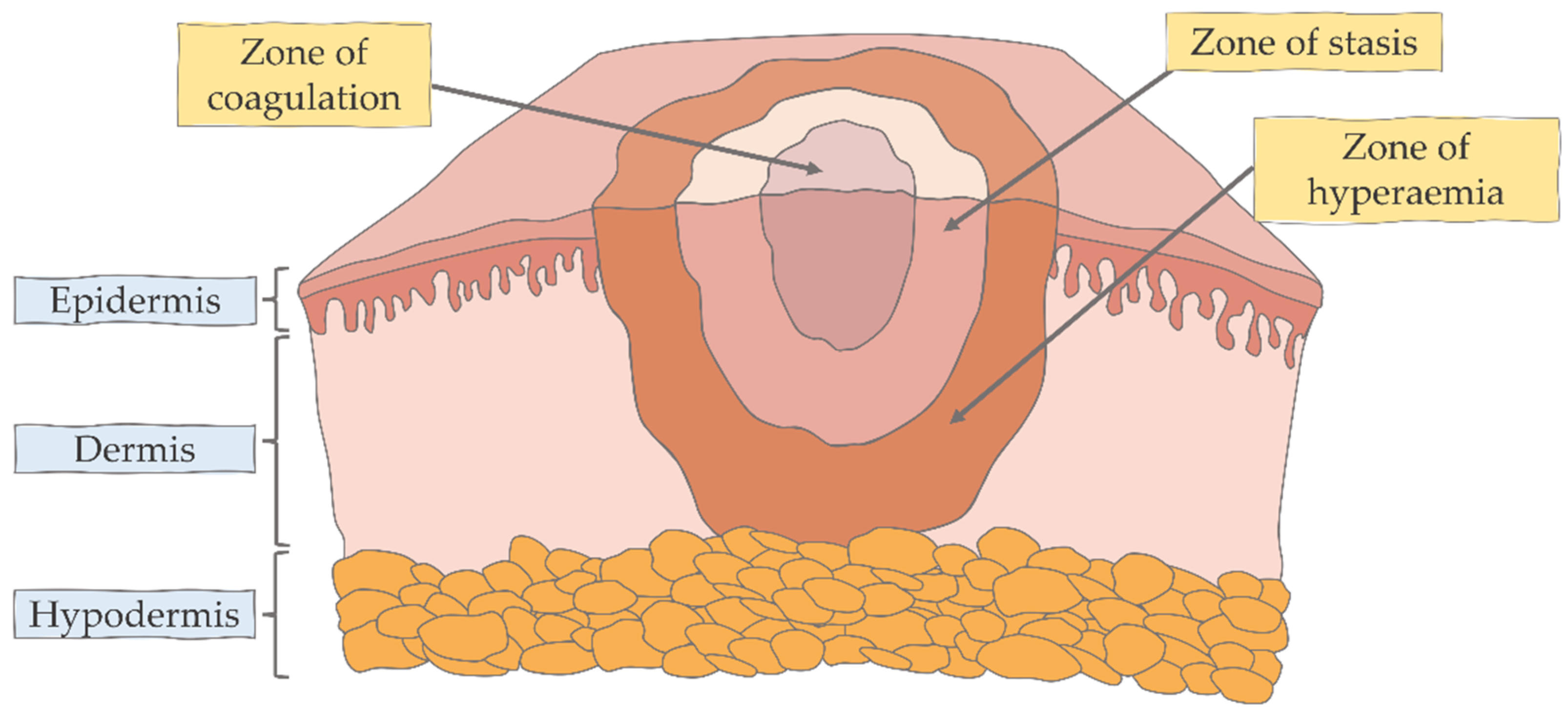

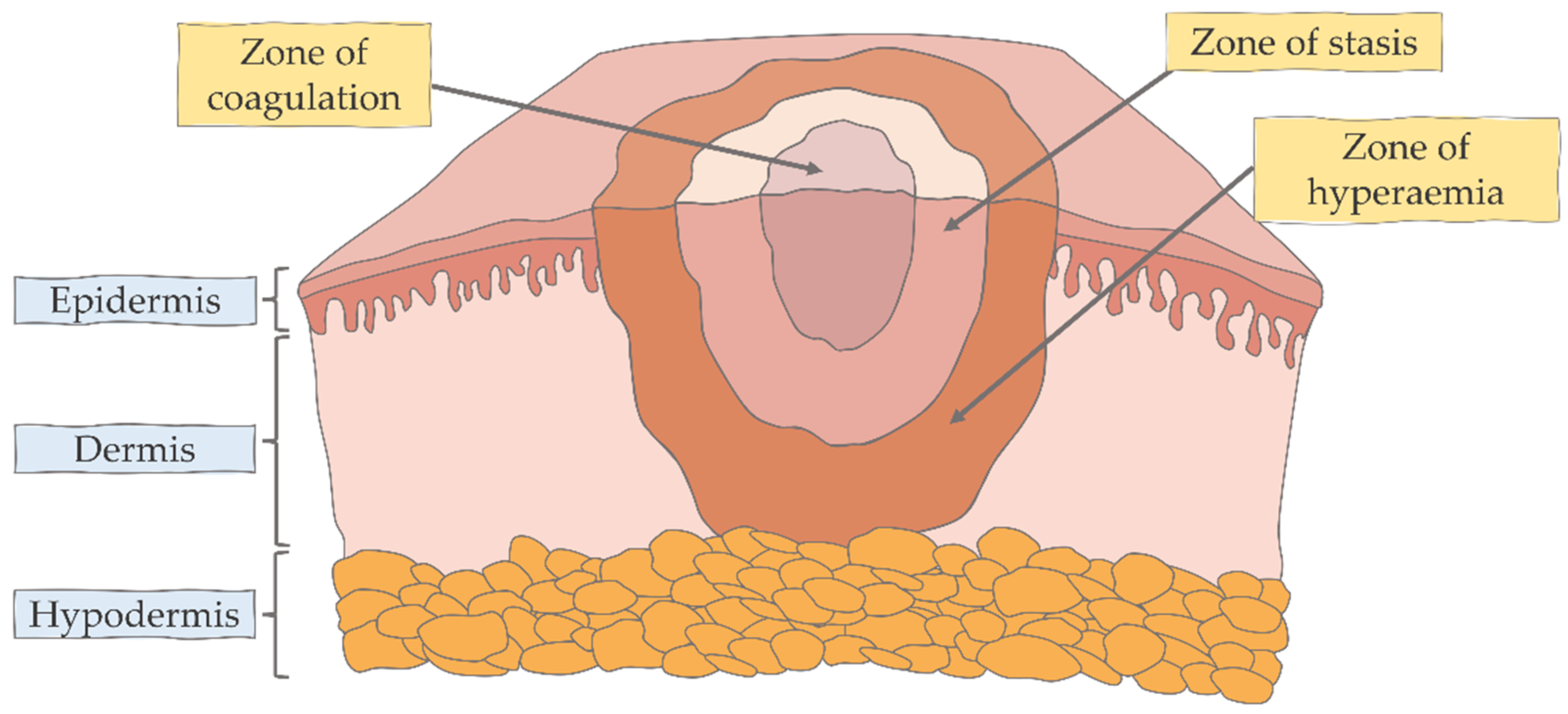
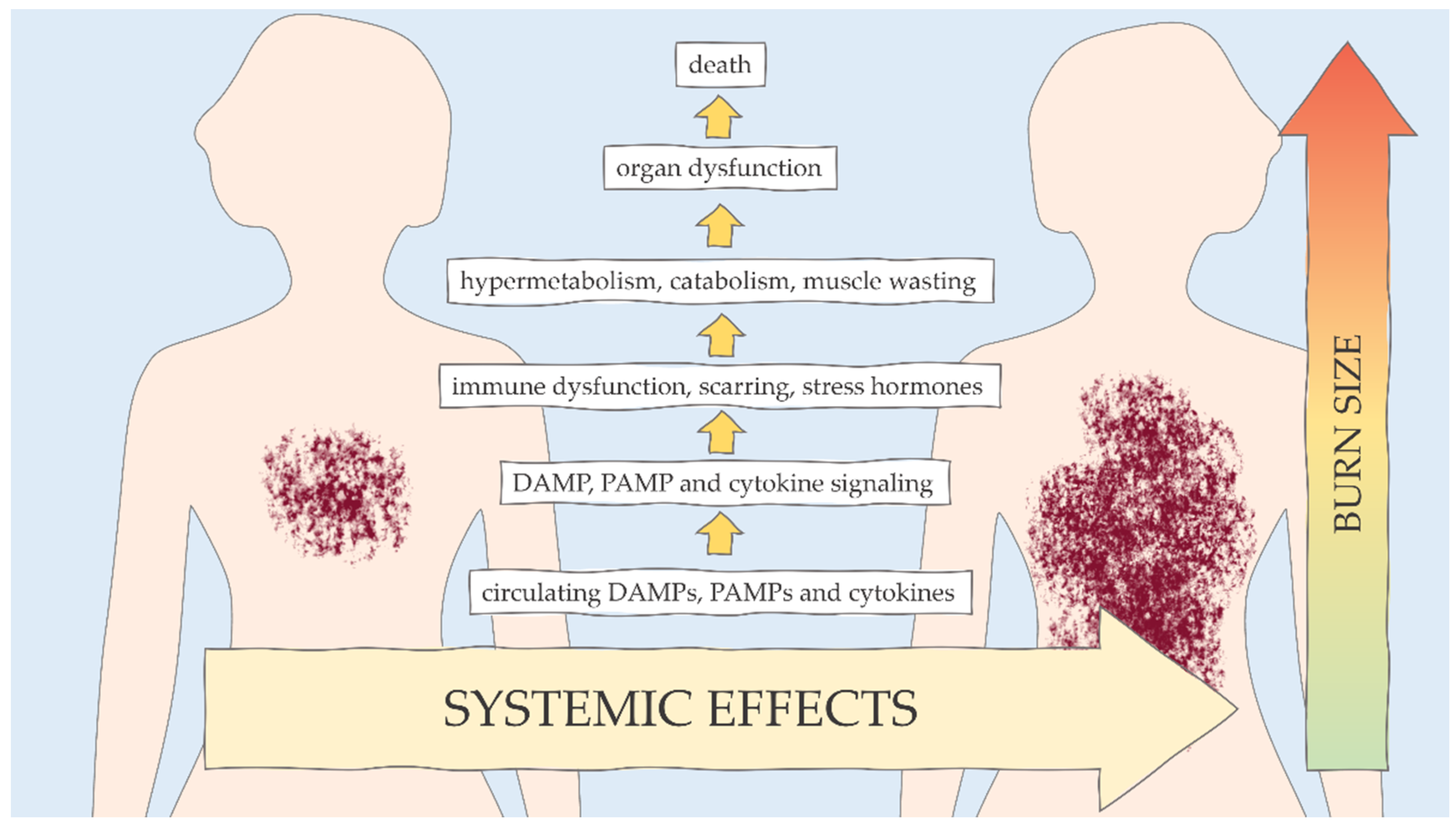{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sierawska, O.; Małkowska, P.; Taskin, C.; Hrynkiewicz, R.; Mertowska, P.; Grywalska, E.; Korzeniowski, T.; Torres, K.; Surowiecka, A.; Niedźwiedzka-Rystwej, P.; et al. Innate Immune System Response to Burn Damage—Focus on Cytokine Alteration. Int. J. Mol. Sci. 2022, 23, 716. https://doi.org/10.3390/ijms23020716
Sierawska O, Małkowska P, Taskin C, Hrynkiewicz R, Mertowska P, Grywalska E, Korzeniowski T, Torres K, Surowiecka A, Niedźwiedzka-Rystwej P, et al. Innate Immune System Response to Burn Damage—Focus on Cytokine Alteration. International Journal of Molecular Sciences. 2022; 23(2):716. https://doi.org/10.3390/ijms23020716
Chicago/Turabian StyleSierawska, Olga, Paulina Małkowska, Cansel Taskin, Rafał Hrynkiewicz, Paulina Mertowska, Ewelina Grywalska, Tomasz Korzeniowski, Kamil Torres, Agnieszka Surowiecka, Paulina Niedźwiedzka-Rystwej, and et al. 2022. "Innate Immune System Response to Burn Damage—Focus on Cytokine Alteration" International Journal of Molecular Sciences 23, no. 2: 716. https://doi.org/10.3390/ijms23020716
APA StyleSierawska, O., Małkowska, P., Taskin, C., Hrynkiewicz, R., Mertowska, P., Grywalska, E., Korzeniowski, T., Torres, K., Surowiecka, A., Niedźwiedzka-Rystwej, P., & Strużyna, J. (2022). Innate Immune System Response to Burn Damage—Focus on Cytokine Alteration. International Journal of Molecular Sciences, 23(2), 716. https://doi.org/10.3390/ijms23020716