
De Novo ACTG1 Variant Expands the Phenotype and Genotype of Partial Deafness and Baraitser–Winter Syndrome

 , , ,
, , ,  ,
,
Abstract
:1. Introduction
2. Results
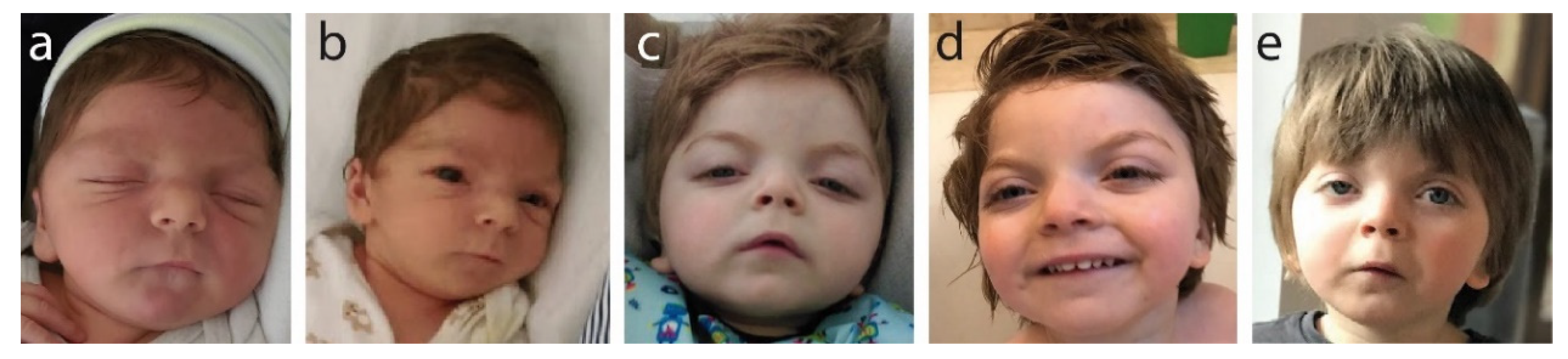
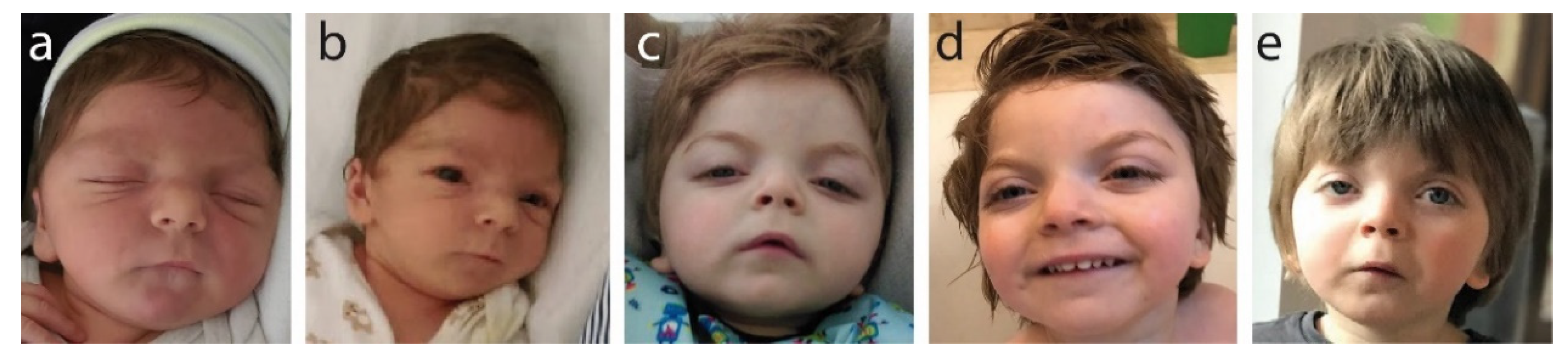
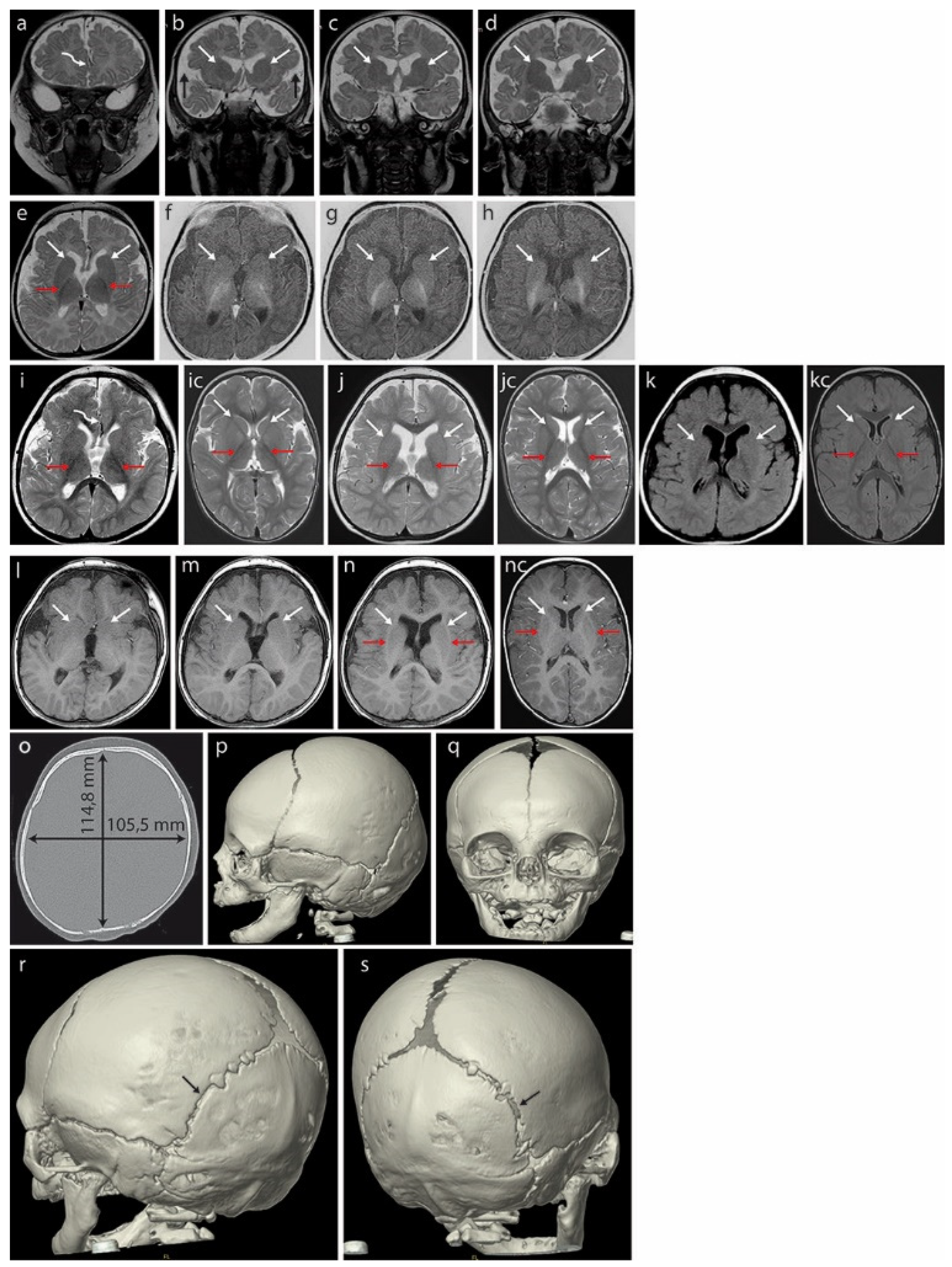
2.1. Patient Parameters at Birth
2.2. Sensorineural Deafness
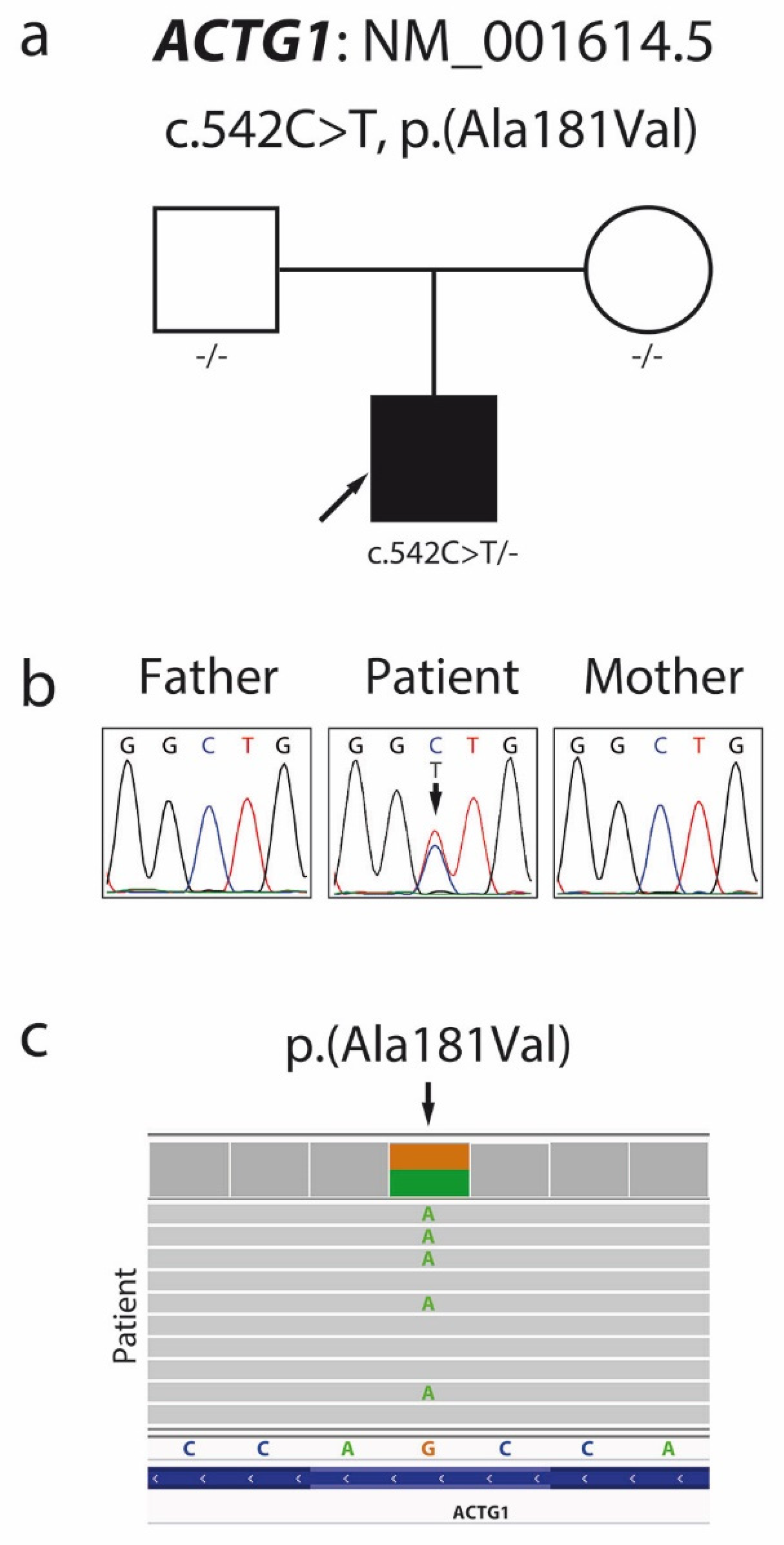
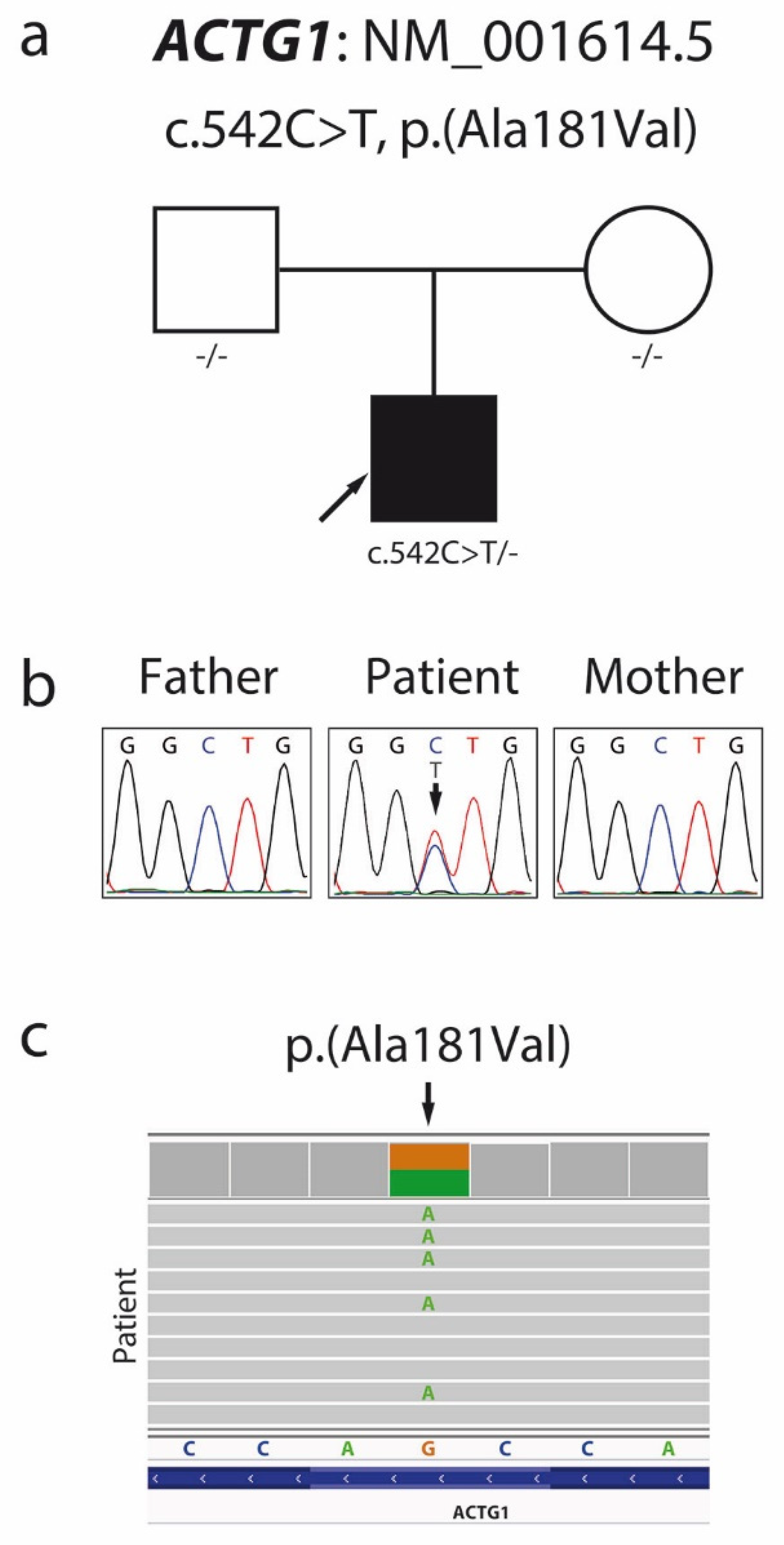
2.3. Molecular Analyses
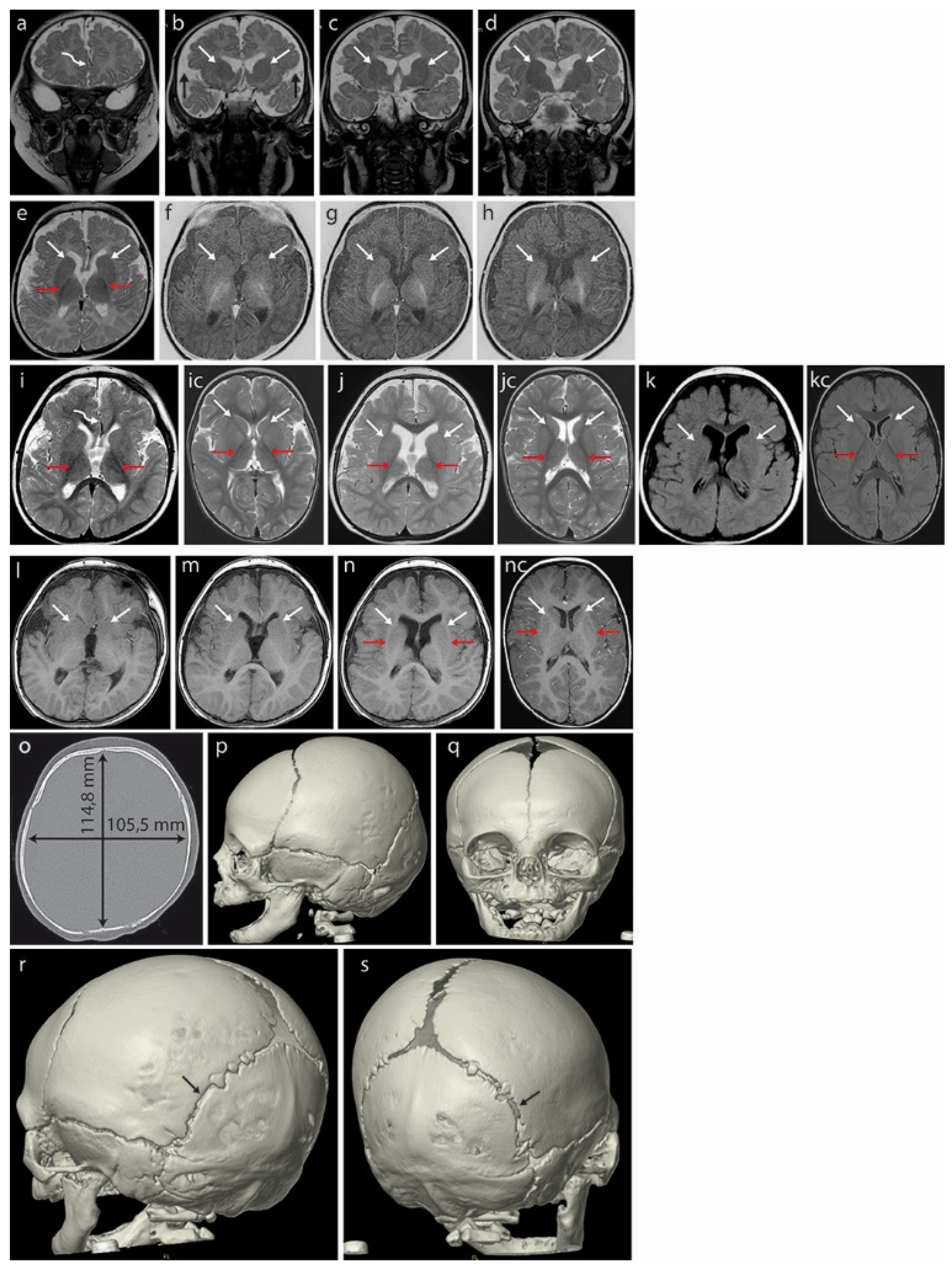
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pollard, T.D.; Cooper, J.A. Actin and actin-binding proteins. A critical evaluation of mechanisms and functions. Annu. Rev. Biochem. 1986, 55, 987–1035. [Google Scholar] [CrossRef]
- Erba, H.P.; Eddy, R.; Shows, T.; Kedes, L.; Gunning, P. Structure, chromosome location, and expression of the human gamma-actin gene: Differential evolution, location, and expression of the cytoskeletal beta- and gamma-actin genes. Mol. Cell Biol. 1988, 8, 1775–1789. [Google Scholar] [CrossRef]
- Höfer, D.; Ness, W.; Drenckhahn, D. Sorting of actin isoforms in chicken auditory hair cells. J. Cell Sci. 1997, 110, 765–770. [Google Scholar] [CrossRef]
- Hudspeth, A.J. How the ear’s works work. Nature 1989, 341, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Pollard, T.D.; Blanchoin, L.; Mullins, R.D. Molecular mechanisms controlling actin filament dynamics in nonmuscle cells. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 545–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilney, L.G.; Egelman, E.H.; DeRosier, D.J.; Saunder, J.C. Actin filaments, stereocilia, and hair cells of the bird cochlea. II. Packing of actin filaments in the stereocilia and in the cuticular plate and what happens to the organization when the stereocilia are bent. J. Cell Biol. 1983, 96, 822–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirokawa, N.; Tilney, L.G. Interactions between actin filaments and between actin filaments and membranes in quick-frozen and deeply etched hair cells of the chick ear. J. Cell Biol. 1982, 95, 249–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verloes, A.; Di Donato, N.; Masliah-Planchon, J.; Jongmans, M.; Abdul-Raman, O.A.; Albrecht, B.; Allanson, J.; Brunner, H.; Bertola, D.; Chassaing, N.; et al. Baraitser-Winter cerebrofrontofacial syndrome: Delineation of the spectrum in 42 cases. Eur. J. Hum. Genet. 2015, 23, 292–301. [Google Scholar] [CrossRef]
- Di Donato, N.; Kuechler, A.; Vergano, S.; Heinritz, W.; Bodurtha, J.; Merchant, S.R.; Breningstall, G.; Ladda, R.; Sell, S.; Altmüller, J.; et al. Update on the ACTG1-associated Baraitser-Winter cerebrofrontofacial syndrome. Am. J. Med. Genet. A 2016, 170, 2644–2651. [Google Scholar] [CrossRef]
- Kemerley, A.; Sloan, C.; Pfeifer, W.; Smith, R.; Drack, A. A novel mutation in ACTG1 causing Baraitser-Winter syndrome with extremely variable expressivity in three generations. Ophthalmic Genet. 2017, 38, 152–156. [Google Scholar] [CrossRef]
- Accogli, A.; Severino, M.; Riva, A.; Madia, F.; Balagura, G.; Iacomino, M.; Carlini, B.; Baldassari, S.; Giacomini, T.; Croci, C.; et al. Targeted re-sequencing in malformations of cortical development: Genotype-phenotype correlations. Seizure 2020, 80, 145–152. [Google Scholar] [CrossRef]
- Wang, H.; Guan, J.; Lan, L.; Yu, L.; Xie, L.; Liu, X.; Yang, J.; Zhao, C.; Wang, D.; Wang, Q. A novel de novo mutation of ACTG1 in two sporadic non-syndromic hearing loss cases. Sci. China Life Sci. 2018, 61, 729–732. [Google Scholar] [CrossRef] [PubMed]
- Miyajima, H.; Moteki, H.; Day, T.; Nishio, S.Y.; Murata, T.; Ikezono, T.; Takeda, H.; Abe, S.; Iwasaki, S.; Takahashi, M.; et al. Novel, ACTG1 mutations in patients identified by massively parallel DNA sequencing cause progressive hearing loss. Sci. Rep. 2020, 10, 7056. [Google Scholar] [CrossRef]
- Posey, J.E.; Harel, T.; Liu, P.; Rosenfeld, J.A.; James, R.A.; Coban Akdemir, Z.H.; Walkiewicz, M.; Bi, W.; Xiao, R.; Ding, Y.; et al. Resolution of disease phenotypes resulting from multilocus genomic variation. N. Engl. J. Med. 2017, 376, 21–31. [Google Scholar] [CrossRef]
- Miyagawa, M.; Nishio, S.Y.; Ichinose, A.; Iwasaki, S.; Murata, T.; Kitajiri, S.; Usami, S. Mutational spectrum and clinical features of patients with ACTG1 mutations identified by massively parallel DNA sequencing. Ann. Otol. Rhinol. Laryngol. 2015, 124, 84S–93S. [Google Scholar] [CrossRef] [PubMed]
- De Heer, A.M.; Huygen, P.L.; Collin, R.W.; Oostrik, J.; Kremer, H.; Cremers, C.W. Audiometric and vestibular features in a second Dutch, DFNA20/26 family with a novel mutation in ACTG1. Ann. Otol. Rhinol. Laryngol. 2009, 118, 382–390. [Google Scholar] [CrossRef]
- Chacon-Camacho, O.F.; Barragán-Arévalo, T.; Villarroel, C.E.; Almanza-Monterrubio, M.; Zenteno, J.C. Previously undescribed phenotypic findings and novel ACTG1 gene pathogenic variants in Baraitser-Winter cerebrofrontofacial syndrome. Eur. J. Med. Genet. 2020, 63, 103877. [Google Scholar] [CrossRef] [PubMed]
- Morgan, A.; Lenarduzzi, S.; Cappellani, S.; Pecile, V.; Morgutti, M.; Orzan, E.; Ghiselli, S.; Ambrosetti, U.; Brumat, M.; Gajendrarao, P.; et al. Genomic studies in a large cohort of hearing impaired haract patients revealed several new alleles, a rare case of uniparental disomy (UPD) and the importance to search for copy number variations. Front Genet. 2018, 9, 681. [Google Scholar] [CrossRef] [PubMed]
- Rainger, J.; Williamson, K.A.; Soares, D.C.; Truch, J.; Kurian, D.; Gillessen-Kaesbach, G.; Seawright, A.; Prendergast, J.; Halachev, M.; Wheeler, A.; et al. A recurrent de novo mutation in ACTG1 causes isolated ocular coloboma. Hum. Mutat. 2017, 38, 942–946. [Google Scholar] [CrossRef] [Green Version]
- Gieldon, L.; Mackenroth, L.; Kahlert, A.K.; Lemke, J.R.; Porrmann, J.; Schallner, J.; von der Hagen, M.; Markus, S.; Weidensee, S.; Novotna, B.; et al. Diagnostic value of partial exome sequencing in developmental disorders. PLoS ONE 2018, 13, e0201041. [Google Scholar] [CrossRef]
- Poirier, K.; Martinovic, J.; Laquerrière, A.; Cavallin, M.; Fallet-Bianco, C.; Desguerre, I.; Valence, S.; Grande-Goburghun, J.; Francannet, C.; Deleuze, J.F.; et al. Rare, ACTG1 variants in fetal microlissencephaly. Eur. J. Med. Genet. 2015, 58, 416–418. [Google Scholar] [CrossRef] [PubMed]
- Sloan-Heggen, C.M.; Bierer, A.O.; Shearer, A.E.; Kolbe, D.L.; Nishimura, C.J.; Frees, K.L.; Ephraim, S.S.; Shibata, S.B.; Booth, K.T.; Campbell, C.A.; et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum. Genet. 2016, 135, 441–450. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.; Yang, T.; Wei, S.; DeWan, A.T.; Morell, R.J.; Elfenbein, J.L.; Fisher, R.A.; Leal, S.M.; Smith, R.J.; Friderici, K.H. Mutations in the gamma-actin gene (ACTG1) are associated with dominant progressive deafness (DFNA20/26). Am. J. Hum. Genet. 2003, 73, 1082–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morín, M.; Bryan, K.E.; Mayo-Merino, F.; Goodyear, R.; Mencía, A.; Modamio-Høybjør, S.; del Castillo, I.; Cabalka, J.M.; Richardson, G.; Moreno, F.; et al. In vivo and in vitro effects of two novel gamma-actin (ACTG1) mutations that cause DFNA20/26 hearing impairment. Hum. Mol. Genet. 2009, 18, 3075–3089. [Google Scholar] [CrossRef] [Green Version]
- Rivière, J.B.; van Bon, B.W.; Hoischen, A.; Kholmanskikh, S.S.; O’Roak, B.J.; Gilissen, C.; Gijsen, S.; Sullivan, C.T.; Christian, S.L.; Abdul-Rahman, O.A.; et al. De novo mutations in the actin genes ACTB and ACTG1 cause Baraitser-Winter syndrome. Nat. Genet. 2012, 44, 440–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Li, H.; Ren, X.; Mao, H.; Zhu, Q.; Zhu, Z.; Yang, R.; Yuan, W.; Liu, J.; Wang, Q.; et al. Novel, ACTG1 mutation causing autosomal dominant non-syndromic hearing impairment in a Chinese family. J. Genet. Genom. 2008, 35, 553–558. [Google Scholar] [CrossRef]
- Turner, T.N.; Wilfert, A.B.; Bakken, T.E.; Bernier, R.A.; Pepper, M.R.; Zhang, Z.; Torene, R.I.; Retterer, K.; Eichler, E.E. Sex-based analysis of de novo variants in neurodevelopmental disorders. Am. J. Hum. Genet. 2019, 105, 1274–1285. [Google Scholar] [CrossRef] [PubMed]
- Cabanillas, R.; Diñeiro, M.; Cifuentes, G.A.; Castillo, D.; Pruneda, P.C.; Álvarez, R.; Sánchez-Durán, N.; Capín, R.; Plasencia, A.; Viejo-Díaz, M.; et al. Comprehensive genomic diagnosis of non-syndromic and syndromic hereditary hearing loss in Spanish patients. BMC Med. Genom. 2018, 11, 58. [Google Scholar] [CrossRef] [Green Version]
- Baux, D.; Vaché, C.; Blanchet, C.; Willems, M.; Baudoin, C.; Moclyn, M.; Faugère, V.; Touraine, R.; Isidor, B.; Dupin-Deguine, D.; et al. Combined genetic approaches yield a 48% diagnostic rate in a large cohort of French hearing-impaired patients. Sci. Rep. 2017, 7, 16783. [Google Scholar] [CrossRef] [Green Version]
- Stranneheim, H.; Lagerstedt-Robinson, K.; Magnusson, M.; Kvarnung, M.; Nilsson, D.; Lesko, N.; Engvall, M.; Anderlid, B.M.; Arnell, H.; Johansson, C.B.; et al. Integration of whole genome sequencing into a healthcare setting: High diagnostic rates across multiple clinical entities in 3219 rare disease patients. Genome Med. 2021, 13, 40. [Google Scholar] [CrossRef]
- Miyagawa, M.; Naito, T.; Nishio, S.Y.; Kamatani, N.; Usami, S. Targeted exon sequencing successfully discovers rare causative genes and clarifies the molecular epidemiology of Japanese deafness patients. PLoS ONE 2013, 8, e71381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longoni, M.; High, F.A.; Qi, H.; Joy, M.P.; Hila, R.; Coletti, C.M.; Wynn, J.; Loscertales, M.; Shan, L.; Bult, C.J.; et al. Genome-wide enrichment of damaging de novo variants in patients with isolated and complex congenital diaphragmatic hernia. Hum. Genet. 2017, 136, 679–691. [Google Scholar] [CrossRef] [PubMed]
- Baek, J.I.; Oh, S.K.; Kim, D.B.; Choi, S.Y.; Kim, U.K.; Lee, K.Y.; Lee, S.H. Targeted massive parallel sequencing: The effective detection of novel causative mutations associated with hearing loss in small families. Orphanet J. Rare Dis. 2012, 7, 60. [Google Scholar] [CrossRef] [Green Version]
- Retterer, K.; Juusola, J.; Cho, M.T.; Vitazka, P.; Millan, F.; Gibellini, F.; Vertino-Bell, A.; Smaoui, N.; Neidich, J.; Monaghan, K.G.; et al. Clinical application of whole-exome sequencing across clinical indications. Genet. Med. 2016, 18, 696–704. [Google Scholar] [CrossRef] [Green Version]
- Vontell, R.; Supramaniam, V.G.; Davidson, A.; Thornton, C.; Marnerides, A.; Holder-Espinasse, M.; Lillis, S.; Yau, S.; Jansson, M.; Hagberg, H.E.; et al. Post-mortem characterization of a case with an ACTG1 variant, agenesis of the corpus callosum and neuronal heterotopia. Front Physiol. 2019, 10, 623. [Google Scholar] [CrossRef] [PubMed]
- Stutterd, C.A.; Brock, S.; Stouffs, K.; Fanjul-Fernandez, M.; Lockhart, P.J.; McGillivray, G.; Mandelstam, S.; Pope, K.; Delatycki, M.B.; Jansen, A.; et al. Genetic heterogeneity of polymicrogyria: Study of 123 patients using deep sequencing. Brain Commun. 2020, 3, fcaa221. [Google Scholar] [CrossRef]
- Thiffault, I.; Farrow, E.; Zellmer, L.; Berrios, C.; Miller, N.; Gibson, M.; Caylor, R.; Jenkins, J.; Faller, D.; Soden, S.; et al. Clinical genome sequencing in an unbiased pediatric cohort. Genet. Med. 2019, 21, 303–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, T.; Imaizumi, T.; Yamamoto-Shimojima, K.; Lu, Y.; Yanagishita, T.; Shimada, S.; Chong, P.F.; Kira, R.; Ueda, R.; Ishiyama, A.; et al. Genomic backgrounds of Japanese patients with undiagnosed neurodevelopmental disorders. Brain Dev. 2019, 41, 776–782. [Google Scholar] [CrossRef]
- Yuan, Y.; Gao, X.; Huang, B.; Lu, J.; Wang, G.; Lin, X.; Qu, Y.; Dai, P. Phenotypic heterogeneity in a DFNA20/26 family segregating a novel ACTG1 mutation. BMC Genet. 2016, 17, 33. [Google Scholar] [CrossRef] [Green Version]
- Homma, T.K.; Freire, B.L.; Honjo Kawahira, R.S.; Dauber, A.; Funari, M.F.A.; Lerario, A.M.; Nishi, M.Y.; Albuquerque, E.V.; Vasques, G.A.; Collett-Solberg, P.F.; et al. Genetic disorders in prenatal onset syndromic short stature identified by exome sequencing. J. Pediatr. 2019, 215, 192–198. [Google Scholar] [CrossRef]
- Jin, S.C.; Homsy, J.; Zaidi, S.; Lu, Q.; Morton, S.; DePalma, S.R.; Zeng, X.; Qi, H.; Chang, W.; Sierant, M.C.; et al. Contribution of rare inherited and de novo variants in 2, 871 congenital heart disease probands. Nat. Genet. 2017, 49, 1593–1601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zazo Seco, C.; Wesdorp, M.; Feenstra, I.; Pfundt, R.; Hehir-Kwa, J.Y.; Lelieveld, S.H.; Castelein, S.; Gilissen, C.; de Wijs, I.J.; Admiraal, R.J.; et al. The diagnostic yield of whole-exome sequencing targeting a gene panel for hearing impairment in The Netherlands. Eur. J. Hum. Genet. 2017, 25, 308–314. [Google Scholar] [CrossRef] [Green Version]
- Mutai, H.; Suzuki, N.; Shimizu, A.; Torii, C.; Namba, K.; Morimoto, N.; Kudoh, J.; Kaga, K.; Kosaki, K.; Matsunaga, T. Diverse spectrum of rare deafness genes underlies early-childhood hearing loss in Japanese patients: A cross-sectional, multi-center next-generation sequencing study. Orphanet J. Rare Dis. 2013, 8, 172. [Google Scholar] [CrossRef] [Green Version]
- Van Wijk, E.; Krieger, E.; Kemperman, M.H.; de Leenheer, E.M.; Huygen, P.L.; Cremers, C.W.; Cremers, F.P.; Kremer, H. A mutation in the gamma actin 1 (ACTG1) gene causes autosomal dominant hearing loss (DFNA20/26). J. Med. Genet. 2003, 40, 879–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyagawa, M.; Nishio, S.Y.; Ikeda, T.; Fukushima, K.; Usami, S. Massively parallel DNA sequencing successfully identifies new causative mutations in deafness genes in patients with cochlear implantation and EAS. PLoS ONE 2013, 8, e75793. [Google Scholar] [CrossRef] [PubMed]
- Park, G.; Gim, J.; Kim, A.R.; Han, K.H.; Kim, H.S.; Oh, S.H.; Park, T.; Park, W.Y.; Choi, B.Y. Multiphasic analysis of whole exome sequencing data identifies a novel mutation of ACTG1 in a nonsyndromic hearing loss family. BMC Genom. 2013, 14, 191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Q.; Zhu, H.; Qian, X.; Chen, Z.; Yao, J.; Lu, Y.; Cao, X.; Xing, G. Targeted genomic capture and massively parallel sequencing to identify novel variants causing Chinese hereditary hearing loss. J. Transl. Med. 2014, 12, 311. [Google Scholar] [CrossRef] [Green Version]
- Vona, B.; Müller, T.; Nanda, I.; Neuner, C.; Hofrichter, M.A.; Schröder, J.; Bartsch, O.; Läßig, A.; Keilmann, A.; Schraven, S.; et al. Targeted next-generation sequencing of deafness genes in hearing-impaired individuals uncovers informative mutations. Genet. Med. 2014, 16, 945–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakuma, N.; Moteki, H.; Takahashi, M.; Nishio, S.Y.; Arai, Y.; Yamashita, Y.; Oridate, N.; Usami, S. An effective screening strategy for deafness in combination with a next-generation sequencing platform: A consecutive analysis. J. Hum. Genet. 2016, 61, 253–261. [Google Scholar] [CrossRef]
- Rendtorff, N.D.; Zhu, M.; Fagerheim, T.; Antal, T.L.; Jones, M.; Teslovich, T.M.; Gillanders, E.M.; Barmada, M.; Teig, E.; Trent, J.M.; et al. A novel missense mutation in ACTG1 causes dominant deafness in a Norwegian, DFNA20/26 family, but ACTG1 mutations are not frequent among families with hereditary hearing impairment. Eur. J. Hum. Genet. 2006, 14, 1097–1105. [Google Scholar] [CrossRef]
- Sommen, M.; Schrauwen, I.; Vandeweyer, G.; Boeckx, N.; Corneveaux, J.J.; van den Ende, J.; Boudewyns, A.; de Leenheer, E.; Janssens, S.; Claes, K.; et al. DNA diagnostics of hereditary hearing loss: A targeted resequencing approach combined with a mutation classification system. Hum. Mutat. 2016, 37, 812–819. [Google Scholar] [CrossRef] [PubMed]
- Zarrei, M.; Burton, C.L.; Engchuan, W.; Young, E.J.; Higginbotham, E.J.; MacDonald, J.R.; Trost, B.; Chan, A.J.S.; Walker, S.; Lamoureux, S.; et al. A large data resource of genomic copy number variation across neurodevelopmental disorders. NPJ Genom. Med. 2019, 4, 26. [Google Scholar] [CrossRef] [Green Version]
- Sorrentino, U.; Piccolo, C.; Rigon, C.; Brasson, V.; Trevisson, E.; Boaretto, F.; Martini, A.; Cassina, M. DFNA20/26 and other ACTG1-associated phenotypes: A case report and review of the literature. Audiol. Res. 2021, 11, 582–593. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Park, J.E.; Lee, C.; Kim, A.R.; Kim, B.J.; Park, W.Y.; Ki, C.S.; Lee, J. Genomic analysis of korean patient with microcephaly. Front. Genet. 2021, 11, 543528. [Google Scholar] [CrossRef] [PubMed]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, C.; Mou, C.; Dong, Y.; Tu, Y. dbNSFP v4, a comprehensive database of transcript-specific functional predictions and annotations for human nonsynonymous and splice-site SNVs. Genome Med. 2020, 12, 103. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| No. | HGMD Number | Nucleotide Change | Amino Acid Change | Variant Class | Reported Phenotype |
|---|---|---|---|---|---|
| 1 | CM1611295 | c.34A > G | p.Asn12Asp | DM | B-WS [9] |
| 2 | CM2015269 | c.88G > T | p.Val30Leu | DM | Pachygyria [11] |
| 3 | CM181678 | c.94C > T | p.Pro32Ser | DM | Hearing loss, non-syndromic [12] |
| 4 | CM208651 | c.102C > G | p.Ile34Met | DM? | Hearing loss [13] |
| 5 | CM208650 | c.110G > A | p.Arg37His | DM? | Hearing loss [13] |
| 6 | CM171610 | c.118C > T | p.His40Tyr | DM? | B-WS [14] |
| 7 | CM153972 | c.142G > C | p.Gly48Arg | DM | Deafness, dominant progressive [15] |
| 8 | CM094470 | c.151G > A | p.Asp51Asn | DM | Deafness, dominant progressive [16] |
| 9 | CM175231 | c.173C > T | p.Ala58Val | DM | B-WS [10] |
| 10 | CM208234 | c.176A > G | p.Gln59Arg | DM | B-WS [17] |
| 11 | CM1821877 | c.197C > T | p.Thr66Ile | DM | Hearing loss [18] |
| 12 | CM1710251 | c.209C > T | p.Pro70Leu | DM | Ocular coloboma [19] |
| 13 | CM1827026 | c.221G > T | p.Gly74Val | DM | Intellectual disability [20] |
| 14 | CM157593 | c.223A > C | p.Ile75Leu | DM? | Microlissencephaly [21] |
| 15 | CM208652 | c.246G > A | p.Met82Ile (↓) | DM? | Hearing loss [13] |
| 16 | CM164938 | c.244A > T | p.Met82Leu (↑) | DM | Hearing loss [22] |
| 17 | CM032825 | c.266C > T | p.Thr89Ile | DM | Deafness, dominant progressive [23] |
| 18 | CM094417 | c.354G > C | p.Lys118Asn (↓) | DM | Deafness, dominant progressive [24] |
| 19 | CM032826 | c.353A > T | p.Lys118Met (↑) | DM | Deafness, dominant progressive [23] |
| 20 | CM122513 | c.359C > T | p.Thr120Ile | DM | B-WS [25] |
| 21 | CM085221 | c.364A > G | p.Ile122Val | DM | Deafness, dominant progressive [26] |
| 22 | CM122514 | c.404C > T | p.Ala135Val | DM | B-WS [25] |
| 23 | CM1931694 | c.429C > T | p.Tyr143Tyr | DM? | Autism [27] |
| 24 | CM189438 | c.434C > G | p.Ser145Cys (↓) | DM? | Sensorineural deafness, non-syndromic [28] |
| 25 | CM1724902 | c.434C > T | p.Ser145Phe (↑) | DM | Hearing loss [29] |
| 26 | CM214253 | c.439C > T | p.Arg147Cys | DM | B-WS [30] |
| 27 | CM157618 | c.459G > C | p.Met153Ile | DM? | Microlissencephaly [21] |
| 28 | CM122515 | c.464C > T | p.Ser155Phe | DM | B-WS [25] |
| 29 | CM1310523 | c.485C > T | p.Thr162Met | DM | Deafness [31] |
| 30 | CM208653 | c.493A > G | p.Ile165Val | DM? | Hearing loss [13] |
| 31 | CM175615 | c.499G > A | p.Glu167Lys | DM? | Diaphragmatic hernia, congenital [32] |
| 32 | CM1611293 | c.535G > T | p.Asp179Tyr | DM | B-WS [9] |
| 33 | CM164939 | c.542C > G | p.Ala181Gly (↓) | DM | Hearing loss [22] |
| 34 | This report | c.542C > T | p.Ala181Val (↑) | DM | B-WS |
| 35 | CM189439 | c.548G > A | p.Arg183Gln | DM? | Sensorineural deafness, non-syndromic [28] |
| 36 | CM127916 | c.559G > C | p.Asp187His | DM | Hearing loss [33] |
| 37 | CM1618747 | c.574A > T | p.Ile192Phe | DM | Multiple congenital anomalies [34] |
| 38 | CM122516 | c.608C > A | p.Thr203Lys (↓) | DM | B-WS [25] |
| 39 | CM199153 | c.608C > T | p.Thr203Met (↑) | DM | B-WS [17,35] |
| 40 | CM215619 | c.616C > T | p.Arg206Trp | DM | Polymicrogyria [36] |
| 41 | CM1823137 | c.628C > T | p.Arg210Cys (↓) | DM | B-WS [37] |
| 42 | CM1915915 | c.628C > G | p.Arg210Gly (↑) | DM | B-WS [38] |
| 43 | CM161501 | c.638A > G | p.Lys213Arg | DM | Hearing impairment, non-syndromic [39] |
| 44 | CM1918057 | c.640G > A | p.Glu214Lys | DM | B-WS [40] |
| 45 | CM094418 | c.721G > A | p.Glu241Lys | DM | Deafness, dominant progressive [24] |
| 46 | CM157614 | c.728C > T | p.Pro243Leu | DM? | Microlissencephaly [21] |
| 47 | CM1722894 | c.757G > A | p.Glu253Lys | DM? | Congenital heart disease [41] |
| 48 | CM122517 | c.760C > T | p.Arg254Trp | DM | B-WS [25] |
| 49 | CM2015268 | c.767G > A | p.Arg256Gln (↓) | DM | Pachygyria [11] |
| 50 | CM122518 | c.766C > T | p.Arg256Trp (↑) | DM | B-WS [25] |
| 51 | CM171720 | c.773C > T | p.Pro258Leu | DM | Hearing impairment [42] |
| 52 | CM032827 | c.791C > T | p.Pro264Leu | DM | Deafness, dominant progressive [23] |
| 53 | CM1311076 | c.802G > A | p.Gly268Ser | DM? | Hearing loss, early childhood [43] |
| 54 | CM208654 | c.823C > T | p.His275Tyr | DM? | Hearing loss [13] |
| 55 | CM033588 | c.833C > T | p.Thr278Ile | DM | Deafness, dominant progressive [44] |
| 56 | CM189440 | c.848T > C | p.Met283Thr (↓) | DM? | Sensorineural deafness, non-syndromic [28] |
| 57 | CM1821805 | c.847A > G | p.Met283Val (↑) | DM | Hearing loss [18] |
| 58 | CM1310318 | c.895C > G | p.Leu299Val | DM | Deafness [45] |
| 59 | CM132288 | c.914T > C | p.Met305Thr | DM | Hearing loss, non-syndromic [46] |
| 60 | CM1412647 | c.946G > A | p.Glu316Lys | DM | Hearing lose [47] |
| 61 | CM1412838 | c.974T > A | p.Met325Lys | DM | Hearing loss [48] |
| 62 | CM032828 | c.994C > G | p.Pro332Ala (↓) | DM | Deafness, dominant progressive [23] |
| 63 | CM163638 | c.994C > T | p.Pro332Ser (↑) | DM | Hearing loss, sensorineural [49] |
| 64 | CM1611294 | c.1000G > C | p.Glu334Gln | DM | B-WS [9] |
| 65 | CM1611296 | c.1004G > A | p.Arg335His | DM | B-WS [9] |
| 66 | CM164940 | c.1045C > A | p.Leu349Met | DM | Hearing loss [22] |
| 67 | CM063834 | c.1109T > C | p.Val370Ala | DM | Deafness, dominant progressive [50] |
| 68 | CD168453 | c.626_632delTGCGCGA | p.Val209Alafs*73 | DM | Hearing loss, non-syndromic [51] |
| 69 | CN1927117 | Duplication of 859 kb including the entire gene + 37 others | DM? | Autism spectrum disorder [52] | |
| Algorithm | Raw Score | Prediction |
|---|---|---|
| SIFT4G | 0.001 | Damaging |
| Polyphen2 HDIV | 0.347 | Benign |
| Polyphen2 HVAR | 0.179 | Benign |
| LRT | 0.000 | Deleterious |
| MutationTaster | 1.000 | Disease causing |
| MutationAssessor | 4.780 | High |
| FATHMM | −4.870 | Damaging |
| PROVEAN | −3.140 | Damaging |
| MetaSVM | 1.132 | Damaging |
| MetaLR | 0.961 | Damaging |
| MetaRNN | 0.964 | Damaging |
| M-CAP | 0.965 | Damaging |
| REVEL | 0.954 | Pathogenic |
| MutPred | 0.822 | Pathogenic |
| MVP | 0.955 | Pathogenic |
| PrimateAI | 0.841 | Damaging |
| DEOGEN2 | 0.978 | Damaging |
| BayesDel addAF | 0.568 | Damaging |
| BayesDel noAF | 0.578 | Damaging |
| ClinPred | 0.998 | Damaging |
| LIST S2 | 0.968 | Damaging |
| FATHMM MKL | 0.969 | Damaging |
| FATHMM XF | 0.961 | Damaging |
| EIGEN | 0.610 | Pathogenic |
| EIGEN PC | 0.600 | Pathogenic |
| CADD | 26.7 | - |
| DANN | 0.981 | - |
| # | Nucleotide Change | Amino Acid Change | Inheritance | Sex | Population | Short Stature | ID | Hearing Loss | Absence of Speech | Seizures | Micro-Cephaly | Trigonocephaly | Brachycephaly | Hypertelorism | High-arched Eyebrows | Ptosis | Iris or Retina Coloboma | Central Nervous System |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | c.34A > G | p.Asn12Asp | de novo | F | nd | + | + | − | − | − | − | nd | nd | nd | nd | + | − | No MRI [9] |
| 2 | c.118C > T | p.His40Tyr | de novo | M | nd | nd | nd | nd | nd | nd | nd | nd | nd | nd | nd | nd | nd | nd [14] |
| 3 | c.173C > T | p.Ala58Val | parental | F* | nd | nd | nd | + | nd | nd | nd | nd | nd | nd | nd | − | − | nd [10] |
| 4 | c.173C > T | p.Ala58Val | nd | F∆ | nd | nd | nd | + | nd | nd | nd | nd | nd | nd | nd | − | − | nd [10] |
| 5 | c.173C > T | p.Ala58Val | nd | M□ | nd | nd | nd | + | nd | nd | nd | nd | nd | nd | nd | + | + (iris and retina) | nd [10] |
| 6 | c.176A > G | p.Gln59Arg | de novo | M1 | Mexican | + | + | nd | − | nd | − | nd | nd | + | + | + | + (iris, retina, optic nerve head) | Generalized decrease in the cerebral sulci and gyri compatible with pachygyria [17] |
| 7 | c.359C > T | p.Thr120Ile | de novo | F | nd | − | + | + | nd | + | − | + | nd | + | + | + | − | Pachygyria [25] |
| 8 | c.404C > T | p.Ala135Val | de novo | F | nd | + | + | + | nd | + | + | + | nd | − | + | + | + | Pachygyria [25] |
| 9 | c.439C > T | p.Arg147Cys | de novo | nd | nd | nd | nd | nd | nd | nd | nd | nd | nd | nd | nd | nd | nd | nd [30] |
| 10 | c.464C > T | p.Ser155Phe | de novo | M | nd | − | nd | nd | nd | + | − | + | nd | + | nd | + | nd | Pachygyria [25] |
| 11 | c.464C > T | p.Ser155Phe | de novo | F | nd | + | + | − | nd | + | + | + | nd | + | + | + | + | Pachygyria [25] |
| 12 | c.464C > T | p.Ser155Phe | nd | F | nd | nd | nd | nd | nd | + | nd | nd | nd | + | + | + | + | Pachygyria [25] |
| 13 | c.535G > T | Asp179Tyr | nd | F | nd | + | + | + | − | − | nd | nd | nd | nd | nd | − | − | Anterior-predominant pachygyria, posterior band heterotopias, enlarged ventricles, prominent perivascular spaces [9] |
| 14 | c.542C > T | p.Ala181Val | de novo | M | Polish | + | + | + R | + | − | + | − | + | + | + | + | − | Delayed myelination in frontal brain; gyri of the right cerebral hemisphere the cross brain midline |
| 15 | c.608C > A | p.Thr203Lys | de novo | M | nd | − | nd | + | nd | + | − | + | nd | + | + | + | − | Pachygyria [25] |
| 16 | c.608C > T | p.Thr203Met | de novo | F | nd | nd | na | na | na | na | + | nd | nd | nd | nd | na | nd | Post-mortem fetal investigation: agenesis of the CC, colpocephaly, bilateral posterior dilatation of the lateral ventricles, incomplete operculization of the sylvian fissures [35] |
| 17 | c.608C > T | p.Thr203Met | de novo | M2 | Mexican | + | + | nd | nd | nd | + | nd | nd | + | + | + | + (iris) | Cortical dysplasia with several areas of pachygyria, short and thick CC with rostral agenesis and hypoplastic cerebellar vermis [17] |
| 18 | c.628C > T | p.Arg210Cys | de novo | F | Korean | nd | − | nd | nd | nd | + | nd | nd | nd | nd | nd | nd | nd [54] |
| 19 | c.628C > T | p.Arg210Cys | de novo | nd | nd | nd | + | + | nd | nd | + | nd | nd | nd | nd | nd | nd | Brain anomalies (not specified) [37] |
| 20 | c.628C > G | p.Arg210Gly | de novo | F | Japanese | − | + | nd | nd | − | − | nd | nd | nd | nd | nd | nd | nd [38] |
| 21 | c.640G > A | p.Glu214Lys | de novo | M | nd | + | nd | nd | nd | nd | nd | nd | nd | nd | nd | nd | nd | nd [40] |
| 22 | c.760C > T | p.Arg254Trp | nd | M | nd | + | + | nd | − | − | nd | nd | nd | nd | nd | − | − | Anterior-predominant pachygyria, prominent perivascular spaces [9] |
| 23 | c.760C > T | p.Arg254Trp | de novo | M | nd | − | + | + | nd | − | + | + | nd | + | + | + | + | Pachygyria [25] |
| 24 | c.766C > T | p.Arg256Trp | nd | M | nd | + | + | − | − | − | + | nd | nd | nd | nd | − | − | Anterior-predominant pachygyria, posterior band heterotopias, agenesis of the CC, enlarged ventricles, prominent perivascular spaces [9] |
| 25 | c.766C > T | p.Arg256Trp | nd | M | nd | + | + | − | − | − | nd | nd | nd | nd | nd | + | − | Anterior-predominant pachygyria, posterior band heterotopias, mega-CC, enlarged ventricles, prominent perivascular spaces [9] |
| 26 | c.766C > T | p.Arg256Trp | de novo | M | nd | + | + | + | nd | + | + | + | nd | + | + | + | + | Pachygyria [25] |
| 27 | c.1000G > C | p.Glu334Gln | nd | M | nd | − | + | + (20–40 dB) | − | − | − | nd | nd | nd | nd | + | − | Frontal dysgyria, enlarged ventricles, prominent perivascular spaces (mild) [9] |
| 28 | c.1004G > A | p.Arg335His | de novo | F | nd | − | + | + | − | − | − | nd | nd | nd | nd | − | − | Frontal dysgyria, enlarged ventricles [9] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dawidziuk, M.; Kutkowska-Kazmierczak, A.; Bukowska-Olech, E.; Jurek, M.; Kalka, E.; Guilbride, D.L.; Furmanek, M.I.; Bekiesinska-Figatowska, M.; Bal, J.; Gawlinski, P. De Novo ACTG1 Variant Expands the Phenotype and Genotype of Partial Deafness and Baraitser–Winter Syndrome. Int. J. Mol. Sci. 2022, 23, 692. https://doi.org/10.3390/ijms23020692
Dawidziuk M, Kutkowska-Kazmierczak A, Bukowska-Olech E, Jurek M, Kalka E, Guilbride DL, Furmanek MI, Bekiesinska-Figatowska M, Bal J, Gawlinski P. De Novo ACTG1 Variant Expands the Phenotype and Genotype of Partial Deafness and Baraitser–Winter Syndrome. International Journal of Molecular Sciences. 2022; 23(2):692. https://doi.org/10.3390/ijms23020692
Chicago/Turabian StyleDawidziuk, Mateusz, Anna Kutkowska-Kazmierczak, Ewelina Bukowska-Olech, Marta Jurek, Ewa Kalka, Dorothy Lys Guilbride, Mariusz Ireneusz Furmanek, Monika Bekiesinska-Figatowska, Jerzy Bal, and Pawel Gawlinski. 2022. "De Novo ACTG1 Variant Expands the Phenotype and Genotype of Partial Deafness and Baraitser–Winter Syndrome" International Journal of Molecular Sciences 23, no. 2: 692. https://doi.org/10.3390/ijms23020692
APA StyleDawidziuk, M., Kutkowska-Kazmierczak, A., Bukowska-Olech, E., Jurek, M., Kalka, E., Guilbride, D. L., Furmanek, M. I., Bekiesinska-Figatowska, M., Bal, J., & Gawlinski, P. (2022). De Novo ACTG1 Variant Expands the Phenotype and Genotype of Partial Deafness and Baraitser–Winter Syndrome. International Journal of Molecular Sciences, 23(2), 692. https://doi.org/10.3390/ijms23020692