
Identifying Candidate Protein Markers of Acute Kidney Injury in Acute Decompensated Heart Failure

 , , , , ,
, , , , ,
Abstract
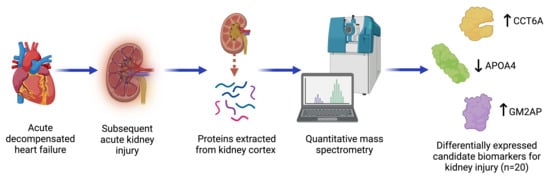
:
1. Introduction
2. Results
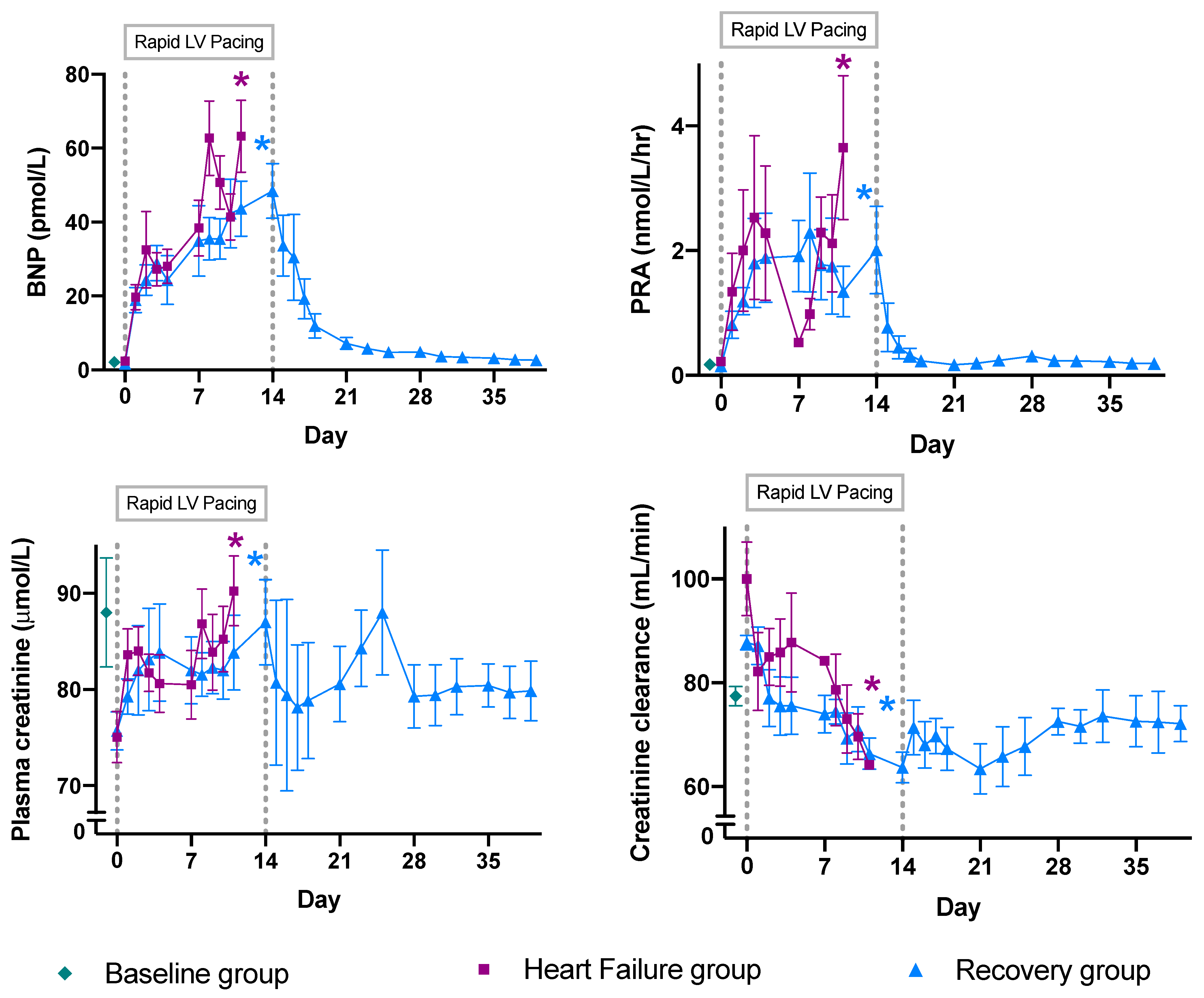
2.1. Physiological Results
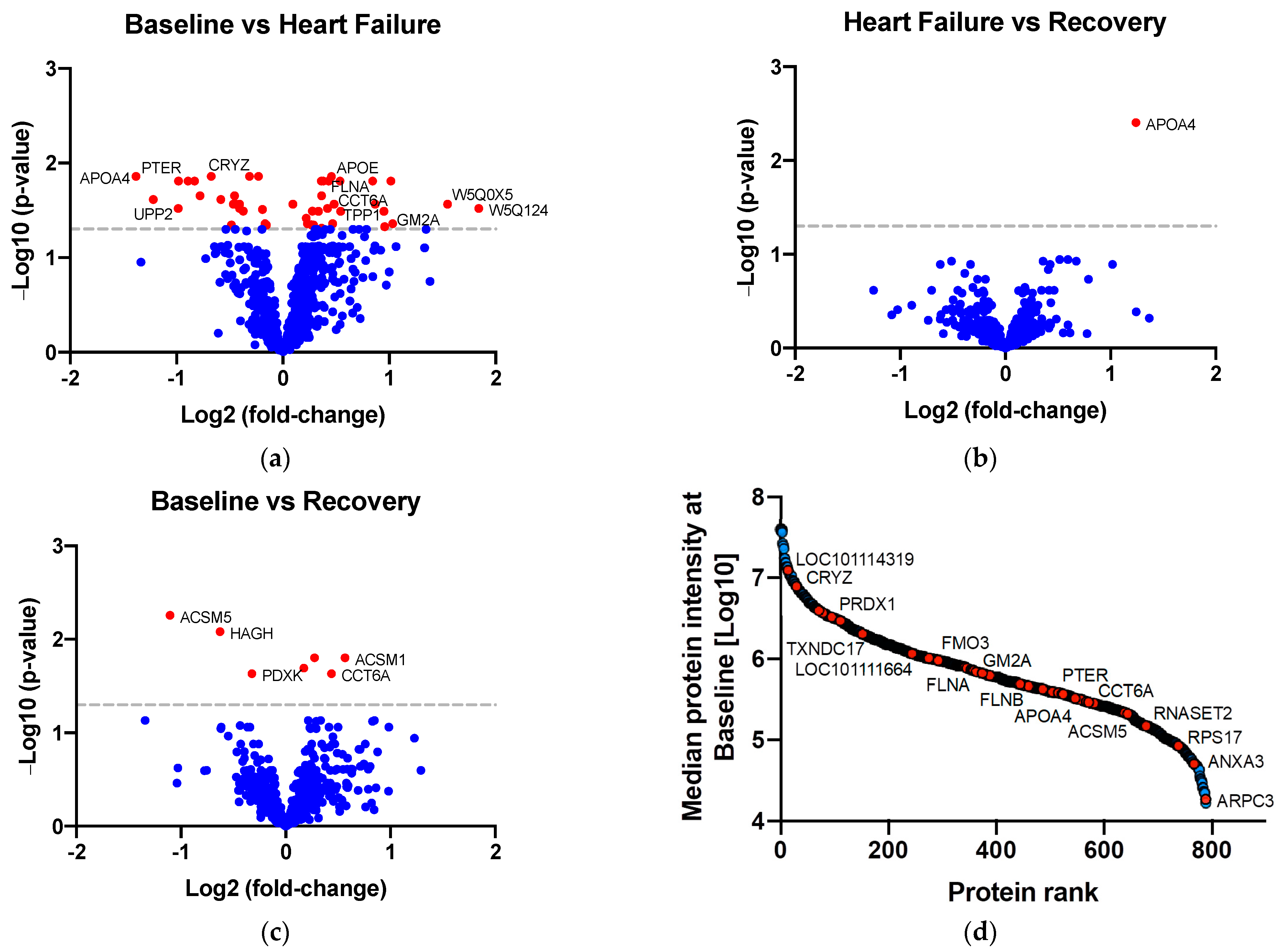
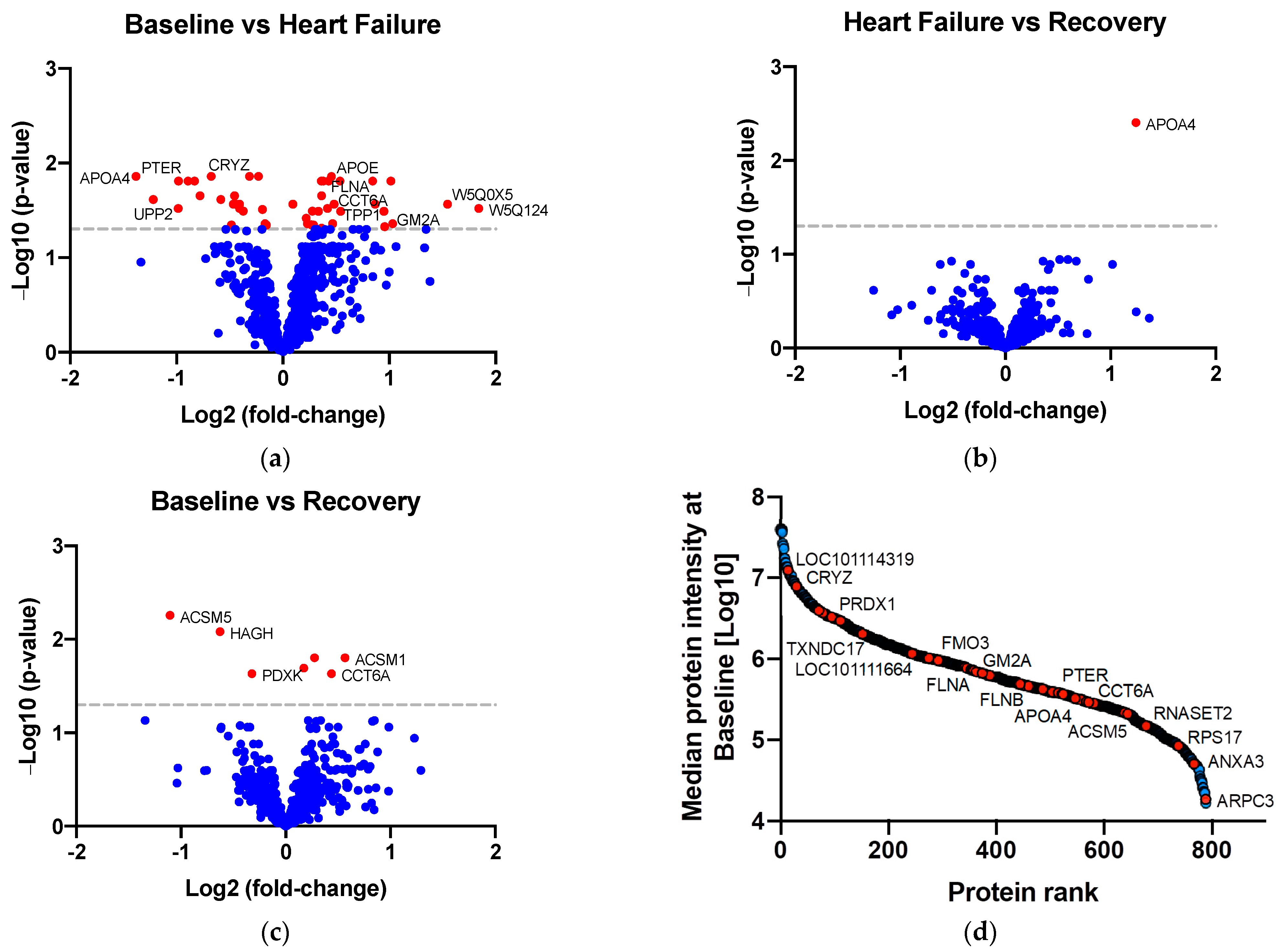
2.2. Identifying Proteins of Differential Abundance
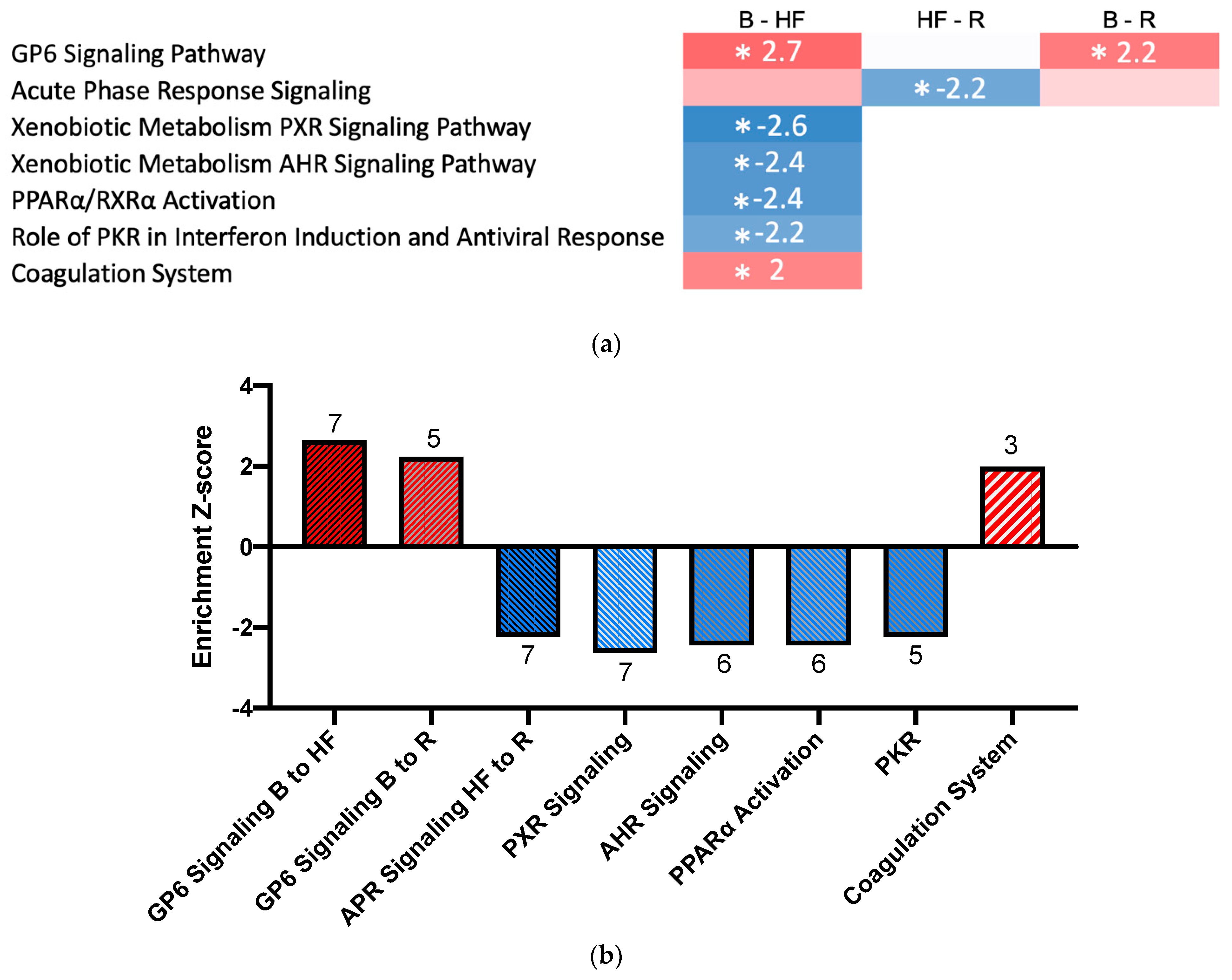
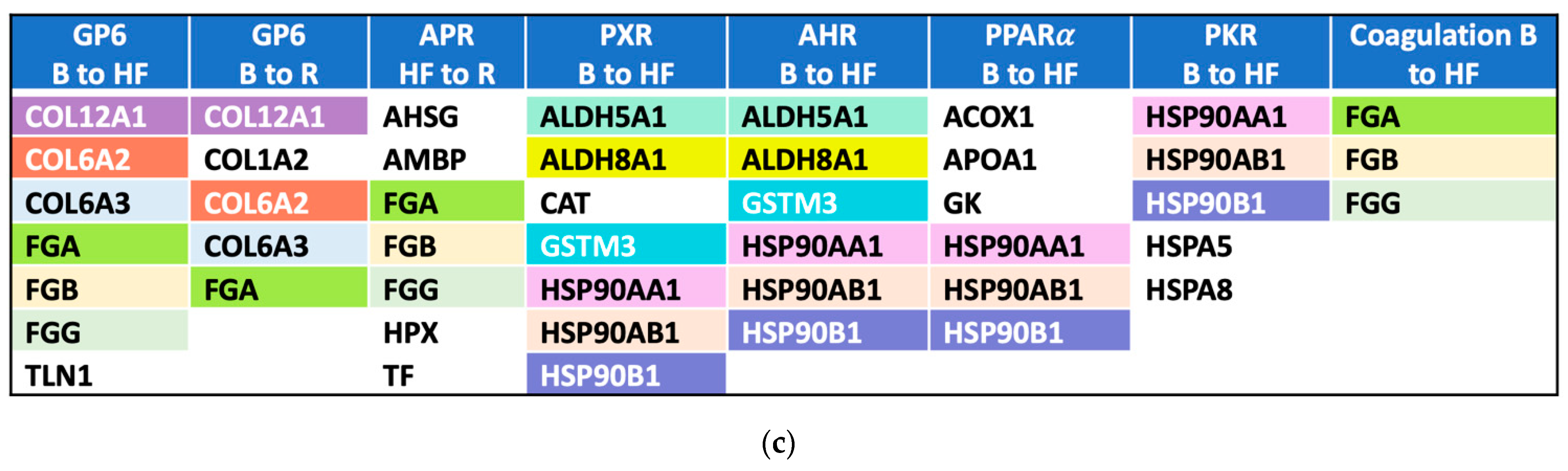
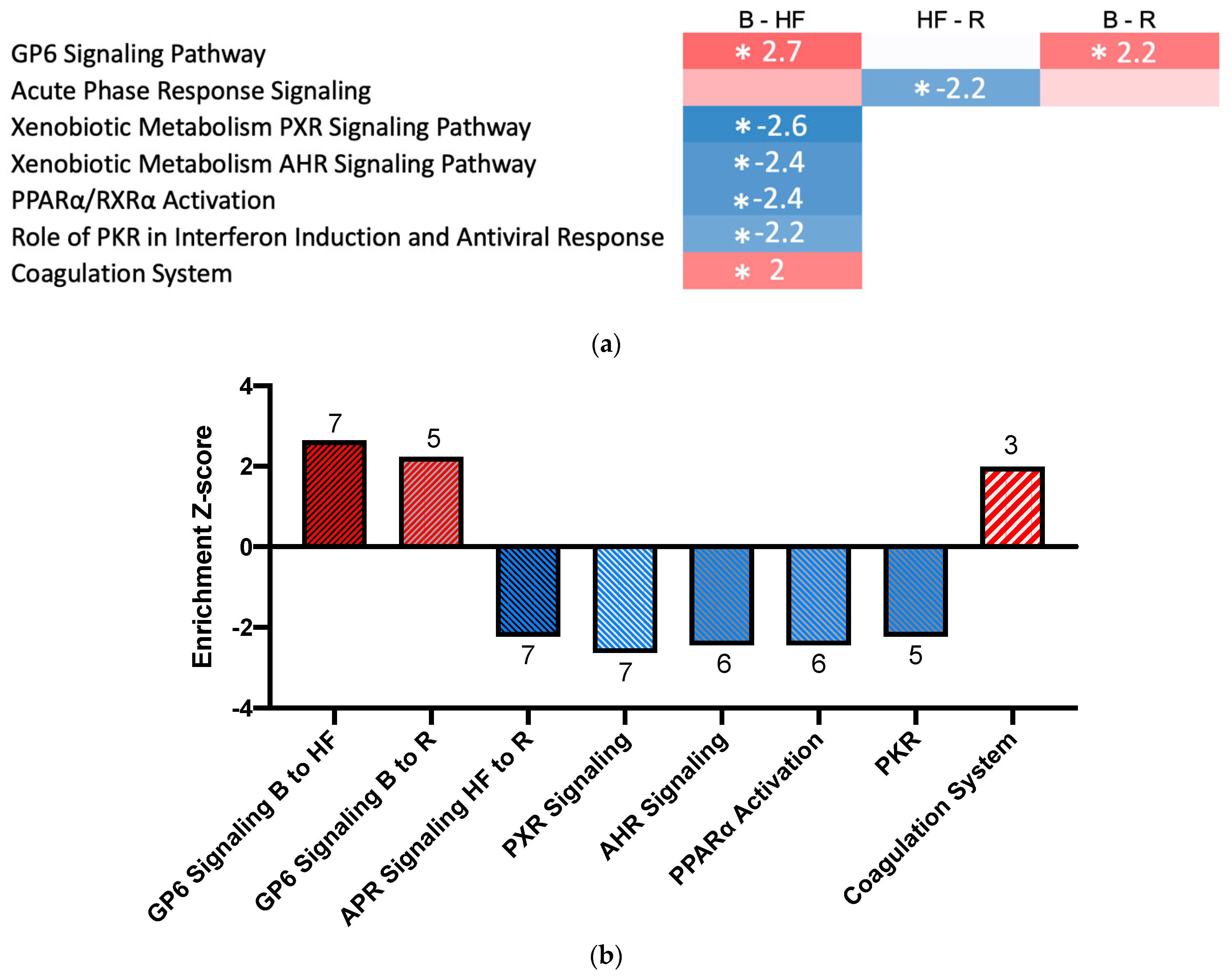
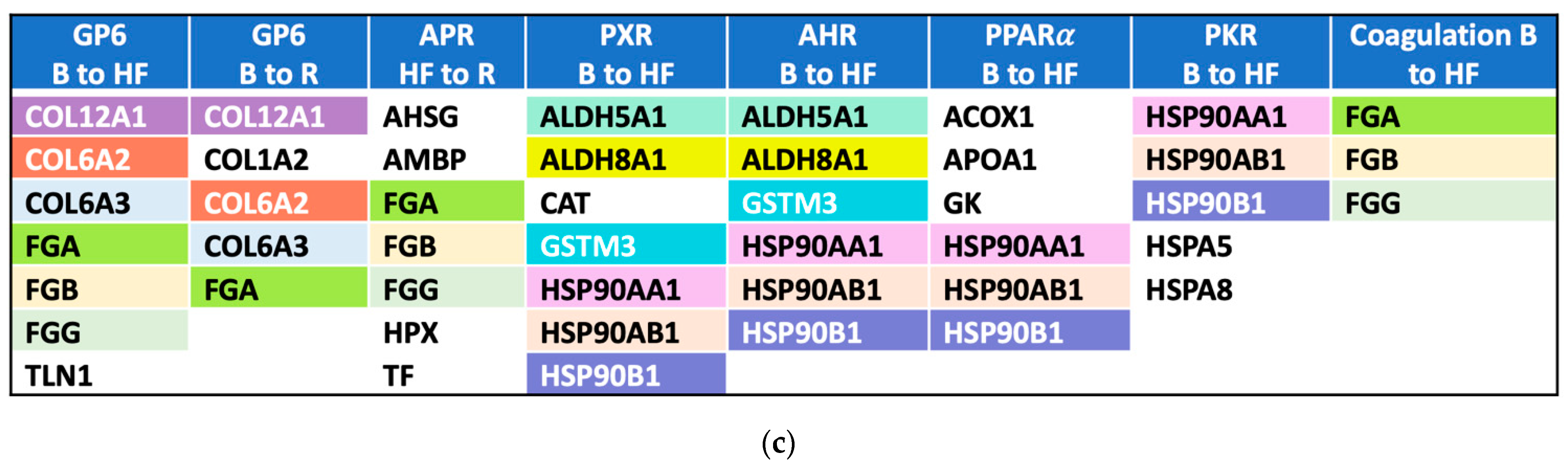
2.3. Pathway Activation and Inhibition
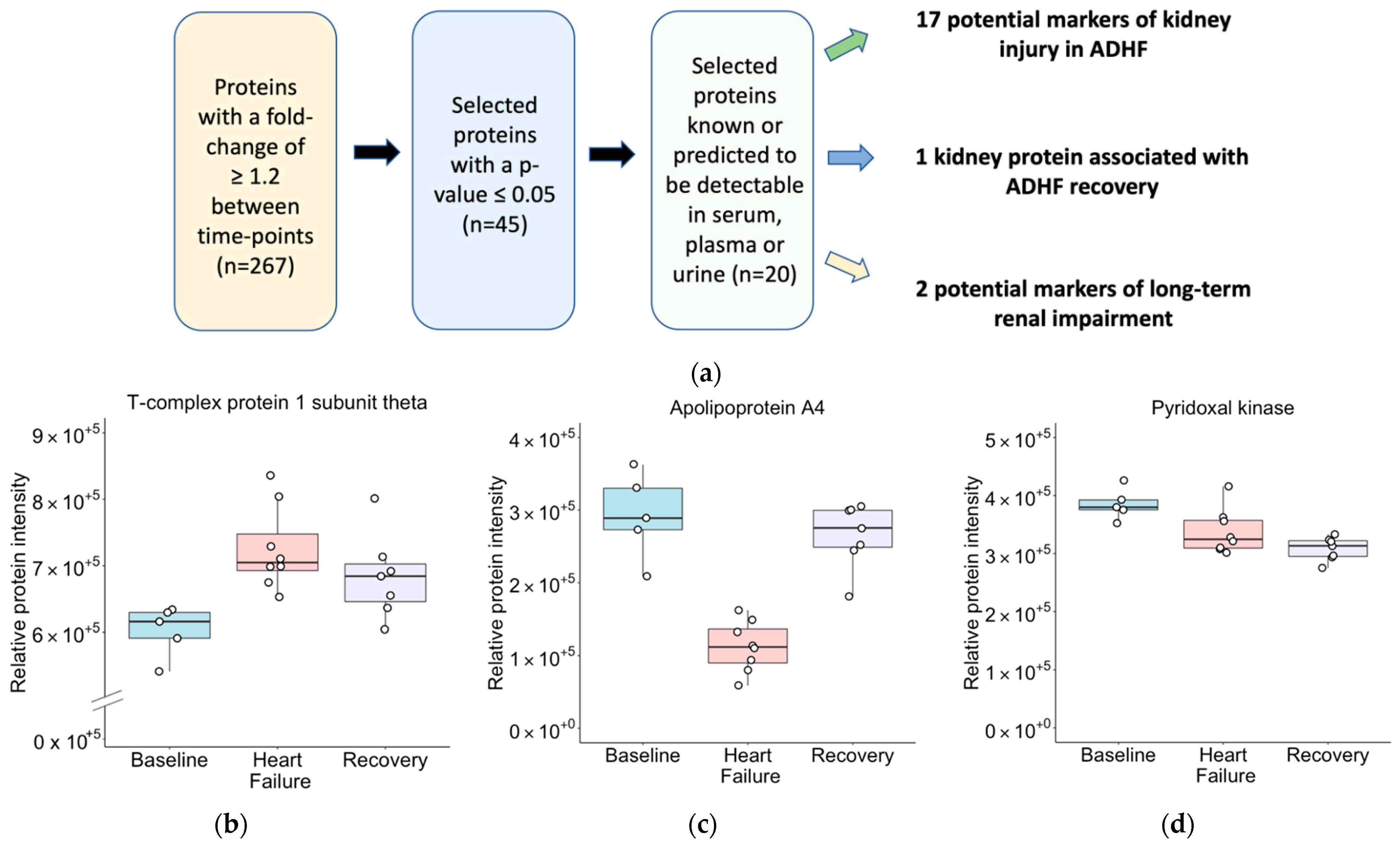
2.4. Biomarker Discovery
3. Discussion
4. Materials and Methods
4.1. Animals
4.1.1. Study Protocol for Control Sheep
4.1.2. Instrumentation of Paced Sheep
4.1.3. Study Protocol for Paced Sheep
4.1.4. Specimen Preparation, Handling, and Storage
4.2. Sample Preparation for Proteomics
4.3. Mass Spectrometry
4.4. SWATH Data Processing
4.5. Quality Control
4.6. Statistical Analysis
4.7. Comparison with AKI-Related Genes
4.8. Identifying Enriched and Activated/Repressed Pathways
4.9. Identifying Circulating or Urinary Biomarkers
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, L.; Eisen, H.J. Epidemiology of Heart Failure and Scope of the Problem. Cardiol. Clin. 2014, 32, 1–8. [Google Scholar] [CrossRef]
- Lippi, G.; Sanchis-Gomar, F. Global epidemiology and future trends of heart failure. AME Med. J. 2020, 5, 15. [Google Scholar] [CrossRef]
- McAlister, F.A.; Ezekowitz, J.; Tonelli, M.; Armstrong, P.W. Renal insufficiency and heart failure: Prognostic and therapeutic implications from a prospective cohort study. Circulation 2004, 109, 1004–1009. [Google Scholar] [CrossRef] [Green Version]
- De Silva, R.; Nikitin, N.P.; Witte, K.K.; Rigby, A.S.; Goode, K.; Bhandari, S.; Clark, A.L.; Cleland, J.G. Incidence of renal dysfunction over 6 months in patients with chronic heart failure due to left ventricular systolic dysfunction: Contributing factors and relationship to prognosis. Eur. Heart J. 2005, 27, 569–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottlieb, S.S.; Abraham, W.; Butler, J.; Forman, D.E.; Loh, E.; Massie, B.M.; O’Connor, C.M.; Rich, M.W.; Stevenson, L.W.; Young, J.; et al. The prognostic importance of different definitions of worsening renal function in congestive heart failure. J. Card. Fail. 2002, 8, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Richards, A.M. Biomarkers in Acute Heart Failure—Cardiac and Kidney. Card. Fail. Rev. 2015, 1, 107–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashani, K.; Cheungpasitporn, W.; Ronco, C. Biomarkers of acute kidney injury: The pathway from discovery to clinical adoption. Clin. Chem. Lab. Med. 2017, 55, 1074–1089. [Google Scholar] [CrossRef] [PubMed]
- Endre, Z.H.; Pickering, J.W.; Walker, R.J. Clearance and beyond: The complementary roles of GFR measurement and injury biomarkers in acute kidney injury (AKI). Am. J. Physiol. Physiol. 2011, 301, F697–F707. [Google Scholar] [CrossRef] [PubMed]
- Vanmassenhove, J.; Vanholder, R.; Nagler, E.; Van Biesen, W. Urinary and serum biomarkers for the diagnosis of acute kidney injury: An in-depth review of the literature. Nephrol. Dial. Transplant. 2012, 28, 254–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashani, K.; Al-Khafaji, A.; Ardiles, T.; Artigas, A.; Bagshaw, S.M.; Bell, M.; Bihorac, A.; Birkhahn, R.; Cely, C.M.; Chawla, L.S.; et al. Discovery and validation of cell cycle arrest biomarkers in human acute kidney injury. Crit. Care 2013, 17, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endre, Z.H.; Pickering, J.W. Acute kidney injury: Cell cycle arrest biomarkers win race for AKI diagnosis. Nat. Rev. Nephrol. 2014, 10, 683–685. [Google Scholar] [CrossRef] [PubMed]
- Pickering, J.W.; Endre, Z.H. Bench to bedside: The next steps for biomarkers in acute kidney injury. Am. J. Physiol. Physiol. 2016, 311, F717–F721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schanz, M.; Shi, J.; Wasser, C.; Alscher, M.D.; Kimmel, M. Urinary [TIMP-2] × [IGFBP7] for risk prediction of acute kidney injury in decompensated heart failure. Clin. Cardiol. 2017, 40, 485–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hishikari, K.; Hikita, H.; Nakamura, S.; Nakagama, S.; Mizusawa, M.; Yamamoto, T.; Doi, J.; Hayashi, Y.; Utsugi, Y.; Araki, M.; et al. Urinary Liver-Type Fatty Acid-Binding Protein Level as a Predictive Biomarker of Acute Kidney Injury in Patients with Acute Decompensated Heart Failure. Cardiorenal Med. 2017, 7, 267–275. [Google Scholar] [CrossRef]
- Shirakabe, A.; Hata, N.; Kobayashi, N.; Okazaki, H.; Matsushita, M.; Shibata, Y.; Nishigoori, S.; Uchiyama, S.; Asai, K.; Shimizu, W. Clinical Usefulness of Urinary Liver Fatty Acid-Binding Protein Excretion for Predicting Acute Kidney Injury during the First 7 Days and the Short-Term Prognosis in Acute Heart Failure Patients with Non-Chronic Kidney Disease. Cardiorenal Med. 2017, 7, 301–315. [Google Scholar] [CrossRef]
- Shirakabe, A.; Hata, N.; Kobayashi, N.; Okazaki, H.; Matsushita, M.; Shibata, Y.; Uchiyama, S.; Sawatani, T.; Asai, K.; Shimizu, W. Worsening renal failure in patients with acute heart failure: The importance of cardiac biomarkers. ESC Heart Fail. 2019, 6, 416–427. [Google Scholar] [CrossRef] [Green Version]
- Shirakabe, A.; Hata, N.; Kobayashi, N.; Okazaki, H.; Shinada, T.; Tomita, K.; Yamamoto, M.; Tsurumi, M.; Matsushita, M.; Yamamoto, Y.; et al. Serum Heart-Type Fatty Acid-Binding Protein Level Can Be Used to Detect Acute Kidney Injury on Admission and Predict an Adverse Outcome in Patients with Acute Heart Failure. Circ. J. 2014, 79, 119–128. [Google Scholar] [CrossRef] [Green Version]
- Maisel, A.S.; Wettersten, N.; van Veldhuisen, D.J.; Mueller, C.; Filippatos, G.; Nowak, R.; Hogan, C.; Kontos, M.C.; Cannon, C.M.; Müller, G.A.; et al. Neutrophil Gelatinase-Associated Lipocalin for Acute Kidney Injury During Acute Heart Failure Hospitalizations: The AKINESIS Study. J. Am. Coll. Cardiol. 2016, 68, 1420–1431. [Google Scholar] [CrossRef]
- Manguba, A.S., Jr.; Vela Parada, X.; Coca, S.G.; Lala, A. Synthesizing Markers of Kidney Injury in Acute Decompensated Heart Failure: Should We Even Keep Looking? Curr. Heart Fail. Rep. 2019, 16, 257–273. [Google Scholar] [CrossRef]
- Rademaker, M.T.; Pilbrow, A.P.; Ellmers, L.J.; Palmer, S.C.; Davidson, T.; Mbikou, P.; Scott, N.J.A.; Permina, E.; Charles, C.J.; Endre, Z.H.; et al. Acute Decompensated Heart Failure and the Kidney: Physiological, Histological and Transcriptomic Responses to Development and Recovery. J. Am. Heart Assoc. 2021, 10, e021312. [Google Scholar] [CrossRef] [PubMed]
- Berger, K.; Bangen, J.-M.; Hammerich, L.; Liedtke, C.; Floege, J.; Smeets, B.; Moeller, M.J. Origin of regenerating tubular cells after acute kidney injury. Proc. Natl. Acad. Sci. USA 2014, 111, 1533–1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isbir, S.C.; Tekeli, A.; Ergen, A.; Yilmaz, H.; Ak, K.; Civelek, A.; Zeybek, N.; Arsan, S. Genetic Polymorphisms Contribute to Acute Kidney Injury after Coronary Artery Bypass Grafting. Heart Surg. Forum 2007, 10, E439–E444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco-Gozalo, V.; Casanova, A.G.; Sancho-Martínez, S.M.; Prieto, M.; Quiros, Y.; Morales, A.I.; Martínez-Salgado, C.; Agüeros-Blanco, C.; Benito-Hernández, A.; Ramos-Barron, M.A.; et al. Combined use of GM2AP and TCP1-eta urinary levels predicts recovery from intrinsic acute kidney injury. Sci. Rep. 2020, 10, 11599. [Google Scholar] [CrossRef] [PubMed]
- Boes, E.; Fliser, D.; Ritz, E.; König, P.; Lhotta, K.; Mann, J.F.; Müller, G.A.; Neyer, U.; Riegel, W.; Riegler, P.; et al. Apolipoprotein A-IV Predicts Progression of Chronic Kidney Disease: The Mild to Moderate Kidney Disease Study. J. Am. Soc. Nephrol. 2005, 17, 528–536. [Google Scholar] [CrossRef]
- Kronenberg, F.; Kuen, E.; Ritz, E.; König, P.; Kraatz, G.; Lhotta, K.; Mann, J.F.E.; Müller, G.A.; Neyer, U.; Riegel, W.; et al. Apolipoprotein A-IV serum concentrations are elevated in patients with mild and moderate renal failure. J. Am. Soc. Nephrol. 2002, 13, 461–469. [Google Scholar] [CrossRef]
- Rouillard, A.D.; Gundersen, G.W.; Fernandez, N.F.; Wang, Z.; Monteiro, C.D.; McDermott, M.G.; Ma’Ayan, A. The harmonizome: A collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Jansen, M.P.B.; Florquin, S.; Roelofs, J.J.T.H. The role of platelets in acute kidney injury. Nat. Rev. Nephrol. 2018, 14, 457–471. [Google Scholar] [CrossRef] [PubMed]
- Rabb, H.; Griffin, M.D.; McKay, D.B.; Swaminathan, S.; Pickkers, P.; Rosner, M.H.; Kellum, J.A.; Ronco, C. Inflammation in AKI: Current Understanding, Key Questions, and Knowledge Gaps. J. Am. Soc. Nephrol. 2015, 27, 371–379. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Xu, M.; Meng, X.; Li, S.; Liu, Q.; Bai, M.; You, R.; Huang, S.; Yang, L.; Zhang, Y.; et al. Nuclear receptor PXR targets AKR1B7 to protect mitochondrial metabolism and renal function in AKI. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Okamura, M.; Shizu, R.; Abe, T.; Kodama, S.; Hosaka, T.; Sasaki, T.; Yoshinari, K. PXR Functionally Interacts with NF-kappaB and AP-1 to Downregulate the Inflammation-Induced Expression of Chemokine CXCL2 in Mice. Cells 2020, 9, 2296. [Google Scholar] [CrossRef] [PubMed]
- Tao, S.; Guo, F.; Ren, Q.; Liu, J.; Wei, T.; Li, L.; Ma, L.; Fu, P. Activation of aryl hydrocarbon receptor by 6-formylindolo[3,2-b]carbazole alleviated acute kidney injury by repressing inflammation and apoptosis. J. Cell. Mol. Med. 2021, 25, 1035–1047. [Google Scholar] [CrossRef]
- Larigot, L.; Juricek, L.; Dairou, J.; Coumoul, X. AhR signaling pathways and regulatory functions. Biochim. Open 2018, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Miao, J.; Liu, W.; Peng, L.; Chen, Y.; Zhong, Q. Formononetin protects against cisplatin induced acute kidney injury through activation of the PPARalpha/Nrf2/HO1/NQO1 pathway. Int. J. Mol. Med. 2021, 47, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Rakhshandehroo, M.; Knoch, B.; Müller, M.; Kersten, S. Peroxisome Proliferator-Activated Receptor Alpha Target Genes. PPAR Res. 2010, 2010, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Zhang, F.; Lin, H.; Quan, M.; Yang, Y.; Lv, Y.; He, Z.; Qian, Y. The Protein Kinase R Inhibitor C16 Alleviates Sepsis-Induced Acute Kidney Injury Through Modulation of the NF-kappaB and NLR Family Pyrin Domain-Containing 3 (NLPR3) Pyroptosis Signal Pathways. Med. Sci. Monit. 2020, 26, e926254. [Google Scholar] [CrossRef] [PubMed]
- García, M.A.; Meurs, E.F.; Esteban, M. The dsRNA protein kinase PKR: Virus and cell control. Biochimie 2007, 89, 799–811. [Google Scholar] [CrossRef]
- Scrascia, G.; Rotunno, C.; Simone, S.; Montemurno, E.; Amorese, L.; De Palo, M.; Castellano, G.; Pertosa, G.B.; Gesualdo, L.; Paparella, D. Acute kidney injury in high-risk cardiac surgery patients: Roles of inflammation and coagulation. J. Cardiovasc. Med. 2017, 18, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Chu, L.P.; Yu, Y.F.; Guo, L.C.; Peng, J.Q.; Zhou, L.F.; Wei, H.Y.; Du, P.F.; Wang, Y.; Jiang, D.H. Predictive value of complement and coagulation indicators in sepsis related acute kidney injury. Zhonghua Nei Ke Za Zhi 2020, 59, 854–859. [Google Scholar] [PubMed]
- Uhlen, M.; Karlsson, M.J.; Hober, A.; Svensson, A.S.; Scheffel, J.; Kotol, D.; Zhong, W.; Tebani, A.; Strandberg, L.; Edfors, F.; et al. The human secretome. Sci. Signal. 2019, 12, 609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, J.; Ko, C.-W.; Tso, P.; Bhargava, A. Apolipoprotein A-IV: A Multifunctional Protein Involved in Protection against Atherosclerosis and Diabetes. Cells 2019, 8, 319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potprommanee, L.; Ma, H.-T.; Shank, L.; Juan, Y.-H.; Liao, W.-Y.; Chen, S.-T.; Yu, C.-J. GM2-Activator Protein: A New Biomarker for Lung Cancer. J. Thorac. Oncol. 2015, 10, 102–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quiros, Y.; Ferreira, L.; Sancho-Martínez, S.M.; González-Buitrago, J.M.; López-Novoa, J.M.; López-Hernández, F.J. Sub-nephrotoxic doses of gentamicin predispose animals to developing acute kidney injury and to excrete ganglioside M2 activator protein. Kidney Int. 2010, 78, 1006–1015. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Shi, W.; Tian, K.; Kong, Y. Chaperonin containing t-complex polypeptide 1 subunit 6A correlates with lymph node metastasis, abnormal carcinoembryonic antigen and poor survival profiles in non-small cell lung carcinoma. World J. Surg. Oncol. 2020, 18, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Vallin, J.; Grantham, J. The role of the molecular chaperone CCT in protein folding and mediation of cytoskeleton-associated processes: Implications for cancer cell biology. Cell Stress Chaperones 2018, 24, 17–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, X.; Wang, Z.; Ren, J.; Zhuang, Z.; Liu, K.; Wang, K.; He, R. Overexpression of chaperonin containing T-complex polypeptide subunit zeta 2 (CCT6b) suppresses the functions of active fibroblasts in a rat model of joint contracture. J. Orthop. Surg. Res. 2019, 14, 125. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Hu, Z.-P.; Li, Z.; Gao, P.-J.; Zhu, J.-Y. Overexpression of chaperonin containing TCP1, subunit 3 predicts poor prognosis in hepatocellular carcinoma. World J. Gastroenterol. 2015, 21, 8588–8604. [Google Scholar] [CrossRef]
- Sancho-Martínez, S.M.; Sánchez-Juanes, F.; Blanco-Gozalo, V.; Fontecha-Barriuso, M.; Prieto-García, L.; Fuentes-Calvo, I.; González-Buitrago, J.M.; Morales, A.I.; Martínez-Salgado, C.; Ramos-Barron, M.A.; et al. Urinary TCP1-eta: A Cortical Damage Marker for the Pathophysiological Diagnosis and Prognosis of Acute Kidney Injury. Toxicol. Sci. 2019, 174, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Kinsey, G.R.; Li, L.; Okusa, M.D. Inflammation in Acute Kidney Injury. Nephron Exp. Nephrol. 2008, 109, e102–e107. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, D.; Bijol, V.; Krishnamoorthy, A.; Gonzalez, V.R.; Frendl, G.; Zhang, Q.; Goering, P.L.; Brown, R.P.; Waikar, S.S.; Vaidya, V.S. Fibrinogen Excretion in the Urine and Immunoreactivity in the Kidney Serves as a Translational Biomarker for Acute Kidney Injury. Am. J. Pathol. 2012, 181, 818–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zager, R.A.; Johnson, A.C.M.; Becker, K. Renal cortical hemopexin accumulation in response to acute kidney injury. Am. J. Physiol. Physiol. 2012, 303, F1460–F1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzpatrick, M.A.; Nicholls, M.G.; Espiner, E.A.; Ikram, H.; Bagshaw, P.; Yandle, T.G. Neurohormonal changes during onset and offset of ovine heart failure: Role of ANP. Am. J. Physiol. 1989, 256, H1052–H1059. [Google Scholar] [PubMed]
- Rademaker, M.T.; Charles, C.J.; Espiner, E.A.; Frampton, C.M.; Nicholls, M.G.; Richards, A.M. Natriuretic peptide responses to acute and chronic ventricular pacing in sheep. Am. J. Physiol. Circ. Physiol. 1996, 270, H594–H602. [Google Scholar] [CrossRef] [PubMed]
- Spannbauer, A.; Traxler-Weidenauer, D.; Zlabinger, K.; Gugerell, A.; Winkler, J.; Mester-Tonczar, J.; Lukovic, D.; Müller, C.; Riesenhuber, M.; Pavo, N.; et al. Large Animal Models of Heart Failure with Reduced Ejection Fraction (HFrEF). Front. Cardiovasc. Med. 2019, 6, 117. [Google Scholar] [CrossRef] [Green Version]
- Damman, K.; Testani, J.M. The kidney in heart failure: An update. Eur. Heart J. 2015, 36, 1437–1444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rademaker, M.T.; Ellmers, L.J.; Charles, C.J.; Richards, A.M. Urocortin 2 protects heart and kidney structure and function in an ovine model of acute decompensated heart failure: Comparison with dobutamine. Int. J. Cardiol. 2015, 197, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Doellinger, J.; Schneider, A.; Hoeller, M.; Lasch, P. Sample Preparation by Easy Extraction and Digestion (SPEED)—A Universal, Rapid, and Detergent-free Protocol for Proteomics Based on Acid Extraction. Mol. Cell. Proteom. 2020, 19, 209–222. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020; Available online: https://www.R-project.org/ (accessed on 11 December 2019).
- R Studio Team. RStudio: Integrated Development for R; RStudio, Inc.: Boston, MA, USA, 2018; Available online: http://www.rstudio.com/ (accessed on 2 November 2021).
- Ludwig, C.; Claassen, M.; Schmidt, A.; Aebersold, R. Estimation of Absolute Protein Quantities of Unlabeled Samples by Selected Reaction Monitoring Mass Spectrometry. Mol. Cell. Proteom. 2012, 11, 13987. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Buil, A.; Collins, B.; Gillet, L.; Blum, L.C.; Cheng, L.; Vitek, O.; Mouritsen, J.; Lachance, G.; Spector, T.D.; et al. Quantitative variability of 342 plasma proteins in a human twin population. Mol. Syst. Biol. 2015, 11, 786. [Google Scholar] [CrossRef] [PubMed]
- Niu, L.; Geyer, P.E.; Albrechtsen, N.J.W.; Gluud, L.L.; Santos, A.; Doll, S.; Treit, P.V.; Holst, J.J.; Knop, F.K.; Vilsbøll, T.; et al. Plasma proteome profiling discovers novel proteins associated with non-alcoholic fatty liver disease. Mol. Syst. Biol. 2019, 15, e8793. [Google Scholar] [CrossRef]
- Yoshida, K.; Bartel, A. tableone: Create ‘Table 1’ to Describe Baseline Characteristics with or without Propensity Score Weights 2021 R Package Version 0.13.0. Available online: https://CRAN.R-project.org/package=tableone (accessed on 2 November 2021).
- Anderson, N.L.; Polanski, M.; Pieper, R.; Gatlin, T.; Tirumalai, R.S.; Conrads, T.P.; Veenstra, T.D.; Adkins, J.N.; Pounds, J.G.; Fagan, R.; et al. The human plasma proteome: A nonredundant list developed by combination of four separate sources. Mol. Cell. Proteom. 2004, 3, 311–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omenn, G.S.; States, D.J.; Adamski, M.; Blackwell, T.W.; Menon, R.; Hermjakob, H.; Apweiler, R.; Haab, B.B.; Simpson, R.J.; Eddes, J.S.; et al. Overview of the HUPO Plasma Proteome Project: Results from the pilot phase with 35 collaborating laboratories and multiple analytical groups, generating a core dataset of 3020 proteins and a publicly-available database. Proteomics 2005, 5, 3226–3245. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
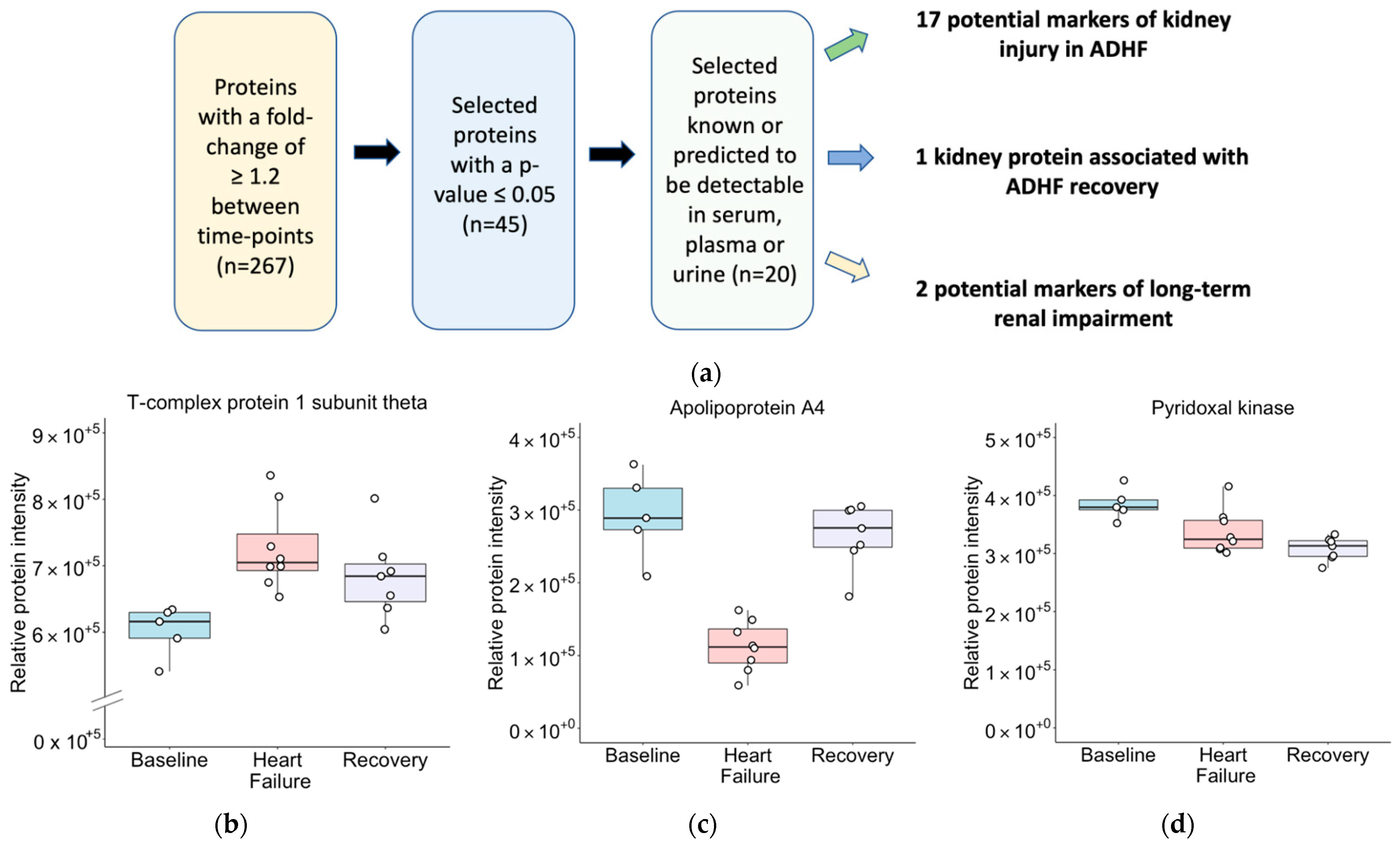
| Protein Name | Gene | Timepoint | Fold-Change | Adjusted p-Value | Previously Associated with AKI | Enriched in Kidney Cells 2 |
|---|---|---|---|---|---|---|
| Upregulated proteins | ||||||
| Annexin | ANXA3 | B-HF | ↑ 1.37 | 0.01 | Yes 1 [21] | |
| NTR domain-containing protein W5NUX8 | LOC101123672 | B-HF | ↑ 1.79 | 0.02 | No | |
| Uncharacterized protein W5P4A8 | RNASET2 | B-HF | ↑ 2.02 | 0.02 | No | |
| Filamin A | FLNA | B-HF | ↑ 1.35 | 0.02 | No | |
| Apolipoprotein E | APOE | B-HF | ↑ 1.45 | 0.02 | Yes 1 [22] | Enriched in proximal tubular cells |
| Talin 1 | TLN1 | B-HF | ↑ 1.30 | 0.02 | No | |
| T-complex protein 1 subunit delta | CCT4 | B-HF | ↑ 1.28 | 0.02 | No | |
| Ribosomal protein S17 | RPS17 | B-HF | ↑ 1.29 | 0.02 | No | |
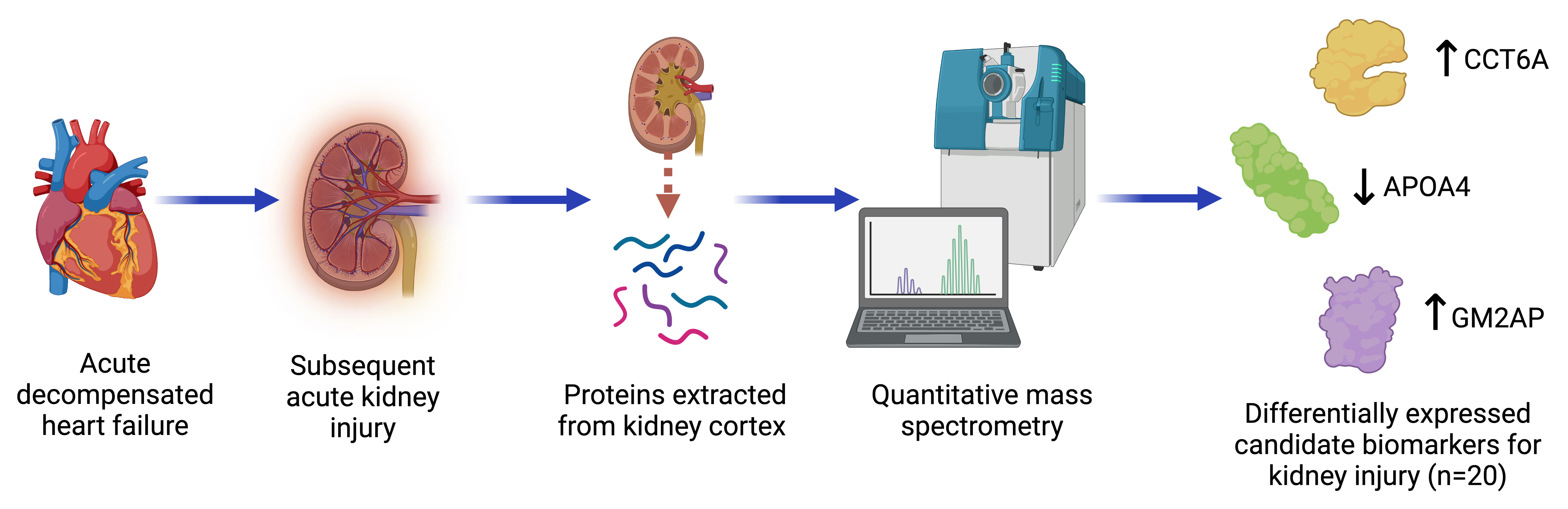
| Chaperonin containing TCP1 subunit 6A | CCT6A | B-HF | ↑ 1.40 | 0.03 | No | |
| B-R | ↑ 1.35 | 0.02 | ||||
| SERPIN domain-containing protein W5Q0X5 | LOC101115576 | B-HF | ↑ 2.92 | 0.03 | No | |
| Actin-related protein 2/3 complex subunit 3 | ARPC3 | B-HF | ↑ 1.83 | 0.03 | No | |
| Filamin B | FLNB | B-HF | ↑ 1.34 | 0.03 | No | |
| SERPIN domain-containing protein W5Q124 | LOC101119509 | B-HF | ↑ 3.58 | 0.03 | No | |
| Integrin beta | ITGB1 | B-HF | ↑ 1.21 | 0.03 | Yes 1 | |
| Clusterin | LOC101113728 | B-HF | ↑ 1.93 | 0.03 | No | |
| Tripeptidyl peptidase 1 | TPP1 | B-HF | ↑ 1.46 | 0.03 | No | |
| T-complex protein 1 subunit alpha | TCP1 | B-HF | ↑ 1.26 | 0.03 | No | |
| Heat shock protein 90 beta family member 1 | HSP90B1 | B-HF | ↑ 1.38 | 0.04 | Yes1 | |
| GM2 ganglioside activator | GM2A | B-HF | ↑ 2.05 | 0.04 | Yes 1 [23] | |
| T-complex protein 1 subunit theta | CCT8 | B-HF | ↑ 1.20 | 0.05 | No | |
| Thioredoxin domain containing 17 | TXNDC17 | B-HF | ↑ 1.22 | 0.05 | No | |
| GC vitamin D binding protein | GC | B-HF | ↑ 1.94 | 0.05 | No | |
| T-complex protein 1 subunit gamma | CCT3 | B-HF | ↑ 1.29 | 0.05 | No | |
| Profilin | PFN1 | B-R | ↑ 1.21 | 0.02 | No | |
| Acyl-CoA synthetase medium chain family member 1 | ACSM1 | B-R | ↑ 1.48 | 0.02 | No | |
| Downregulated proteins | ||||||
| Hydroxyacylglutathione hydrolase | HAGH | B-HF | ↓ 1.60 | 0.01 | No | Enriched in proximal tubular cells |
| B-R | ↓ 1.54 | 0.01 | ||||
| Apolipoprotein A4 | APOA4 | B-HF | ↓ 2.61 | 0.01 | Yes 1 [24,25] | |
| HF-R | ↑ 2.36 | 0.004 | ||||
| Crystallin zeta | CRYZ | B-HF | ↓ 1.24 | 0.01 | No | Enriched in distal tubular cells, proximal tubular cells, and collecting duct cells |
| Phosphotriesterase related | PTER | B-HF | ↓ 1.97 | 0.02 | No | Enriched in proximal tubular cells |
| Apolipoprotein A2 | APOA2 | B-HF | ↓ 1.86 | 0.02 | No | |
| Dimethylaniline monooxygenase [N-oxide-forming] | FMO3 | B-HF | ↓ 1.78 | 0.02 | No | |
| Glutathione transferase | LOC101111664 | B-HF | ↓ 1.72 | 0.02 | No | |
| Solute carrier family 27 member 2 | SLC27A2 | B-HF | ↓ 1.37 | 0.02 | Yes 1 | Enriched in proximal tubular cells |
| Aldo_ket_red domain-containing protein | LOC101109111 | B-HF | ↓ 2.33 | 0.02 | No | |
| Sulfurtransferase | TST | B-HF | ↓ 1.50 | 0.02 | No | Enriched in proximal tubular cells |
| Uncharacterized protein W5NYA7 | LOC101114319 | B-HF | ↓ 1.38 | 0.03 | No | |
| Solute carrier family 5 member 12 | SLC5A12 | B-HF | ↓ 1.36 | 0.03 | No | Enriched in proximal tubular cells |
| Acyl-CoA_dh_1 domain-containing protein W5Q8A9 | B-HF | ↓ 1.37 | 0.03 | No | ||
| Dehydrogenase/reductase 1 | DHRS1 | B-HF | ↓ 1.33 | 0.03 | No | |
| Uridine phosphorylase | UPP2 | B-HF | ↓ 1.98 | 0.03 | No | Enriched in distal and proximal tubular cells |
| Uncharacterized protein W5PN31 | B-HF | ↓ 1.33 | 0.03 | No | ||
| Peroxiredoxin | PRDX1 | B-HF | ↓ 1.29 | 0.03 | Yes 1 | |
| 3-hydroxybutyrate dehydrogenase 1 | BDH1 | B-HF | ↓ 1.40 | 0.05 | No | |
| Acyl-CoA synthetase medium chain family member 5 | ACSM5 | B-R | ↓ 2.15 | 0.01 | No | |
| Pyridoxal kinase | PDXK | B-R | ↓ 1.25 | 0.02 | No | |
| Protein Name | Gene | Timepoint | Fold-Change | Adjusted p-Value | Previously Associated with AKI |
|---|---|---|---|---|---|
| Potential markers of kidney injury in ADHF | |||||
| Upregulated | |||||
| Filamin A | FLNA | B-HF | ↑ 1.35 | 0.02 | No |
| Apolipoprotein E | APOE | B-HF | ↑ 1.45 | 0.02 | Yes 1 [22] |
| Talin 1 | TLN1 | B-HF | ↑ 1.30 | 0.02 | No |
| Filamin B | FLNB | B-HF | ↑ 1.34 | 0.03 | No |
| Integrin beta | ITGB1 | B-HF | ↑ 1.21 | 0.03 | Yes1 |
| Tripeptidyl peptidase 1 | TPP1 | B-HF | ↑ 1.46 | 0.03 | No |
| T-complex protein 1 subunit alpha | TCP1 | B-HF | ↑ 1.26 | 0.03 | No |
| Heat shock protein 90 beta family member 1 | HSP90B1 | B-HF | ↑ 1.38 | 0.04 | Yes 1 |
| GM2 ganglioside activator | GM2A | B-HF | ↑ 2.05 | 0.04 | Yes |
| T-complex protein 1 subunit theta | CCT8 | B-HF | ↑ 1.20 | 0.05 | No |
| Thioredoxin domain containing 17 | TXNDC17 | B-HF | ↑ 1.22 | 0.05 | No |
| GC vitamin D binding protein | GC | B-HF | ↑ 1.94 | 0.05 | No |
| T-complex protein 1 subunit gamma | CCT3 | B-HF | ↑ 1.29 | 0.05 | No |
| Downregulated | |||||
| Crystallin zeta | CRYZ | B-HF | ↓ 1.24 | 0.01 | No |
| Apolipoprotein A2 | APOA2 | B-HF | ↓ 1.86 | 0.02 | No |
| Uridine phosphorylase | UPP2 | B-HF | ↓ 1.98 | 0.03 | No |
| Peroxiredoxin | PRDX1 | B-HF | ↓ 1.29 | 0.03 | Yes1 |
| Kidney proteins associated with ADHF recovery | |||||
| Apolipoprotein A4 | APOA4 | B-HF | ↓ 2.61 | 0.01 | Yes 1 [24,25] |
| HF-R | ↑ 2.36 | 0.004 | |||
| Potential markers of long-term renal impairment | |||||
| Chaperonin containing TCP1 subunit 6A | CCT6A | B-HF | ↑ 1.40 | 0.03 | No |
| B-R | ↑ 1.35 | 0.02 | |||
| Pyridoxal kinase | PDXK | B-R | ↓ 1.25 | 0.02 | No |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Templeton, E.M.; Lassé, M.; Kleffmann, T.; Ellmers, L.J.; Palmer, S.C.; Davidson, T.; Scott, N.J.A.; Pickering, J.W.; Charles, C.J.; Endre, Z.H.; et al. Identifying Candidate Protein Markers of Acute Kidney Injury in Acute Decompensated Heart Failure. Int. J. Mol. Sci. 2022, 23, 1009. https://doi.org/10.3390/ijms23021009
Templeton EM, Lassé M, Kleffmann T, Ellmers LJ, Palmer SC, Davidson T, Scott NJA, Pickering JW, Charles CJ, Endre ZH, et al. Identifying Candidate Protein Markers of Acute Kidney Injury in Acute Decompensated Heart Failure. International Journal of Molecular Sciences. 2022; 23(2):1009. https://doi.org/10.3390/ijms23021009
Chicago/Turabian StyleTempleton, Evelyn M., Moritz Lassé, Torsten Kleffmann, Leigh J. Ellmers, Suetonia C. Palmer, Trent Davidson, Nicola J. A. Scott, John W. Pickering, Christopher J. Charles, Zoltan H. Endre, and et al. 2022. "Identifying Candidate Protein Markers of Acute Kidney Injury in Acute Decompensated Heart Failure" International Journal of Molecular Sciences 23, no. 2: 1009. https://doi.org/10.3390/ijms23021009
APA StyleTempleton, E. M., Lassé, M., Kleffmann, T., Ellmers, L. J., Palmer, S. C., Davidson, T., Scott, N. J. A., Pickering, J. W., Charles, C. J., Endre, Z. H., Cameron, V. A., Richards, A. M., Rademaker, M. T., & Pilbrow, A. P. (2022). Identifying Candidate Protein Markers of Acute Kidney Injury in Acute Decompensated Heart Failure. International Journal of Molecular Sciences, 23(2), 1009. https://doi.org/10.3390/ijms23021009