
Synthesis, Crystallographic, Quantum Chemical, Antitumor, and Molecular Docking/Dynamic Studies of 4-Hydroxycoumarin-Neurotransmitter Derivatives

 ,
,  ,
,  ,
,  ,
,  , ,
, ,  ,
, 

Abstract
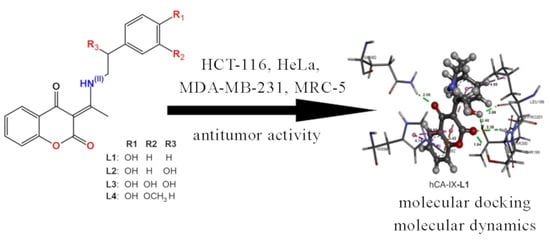
:
1. Introduction
2. Results
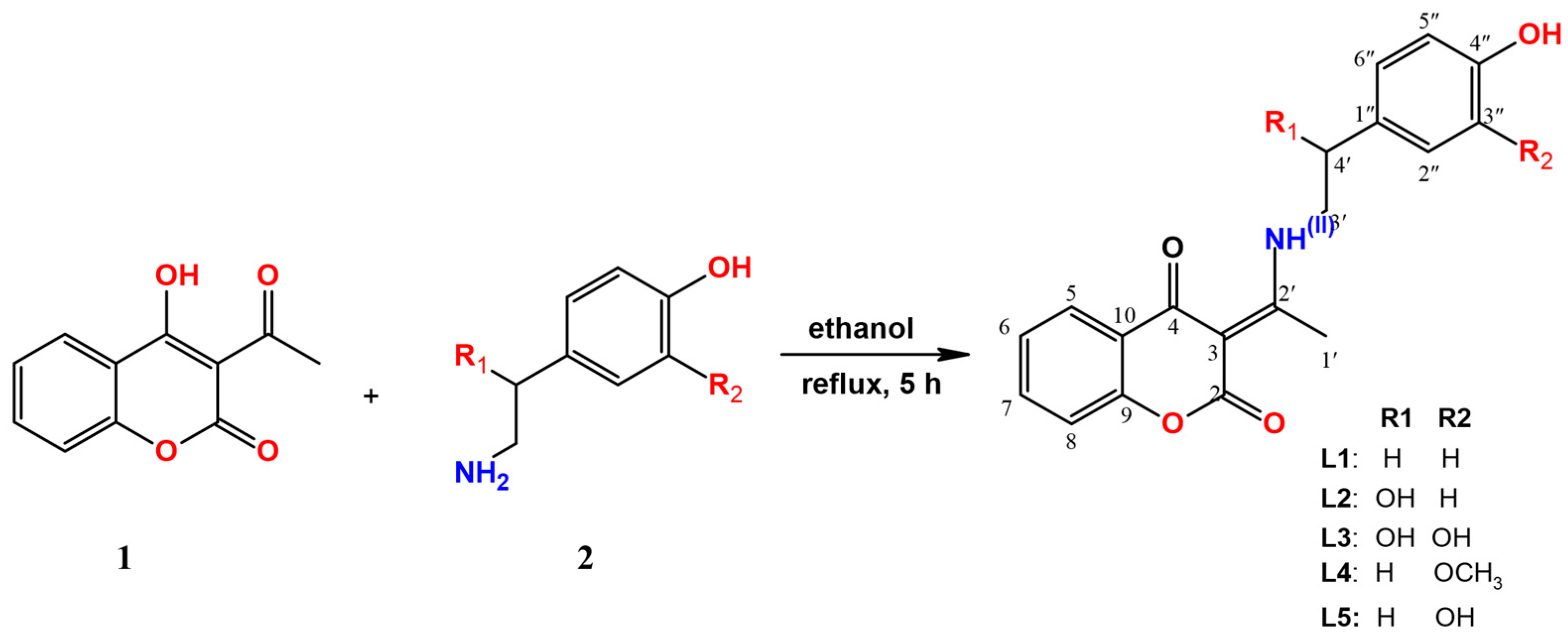
2.1. Chemistry
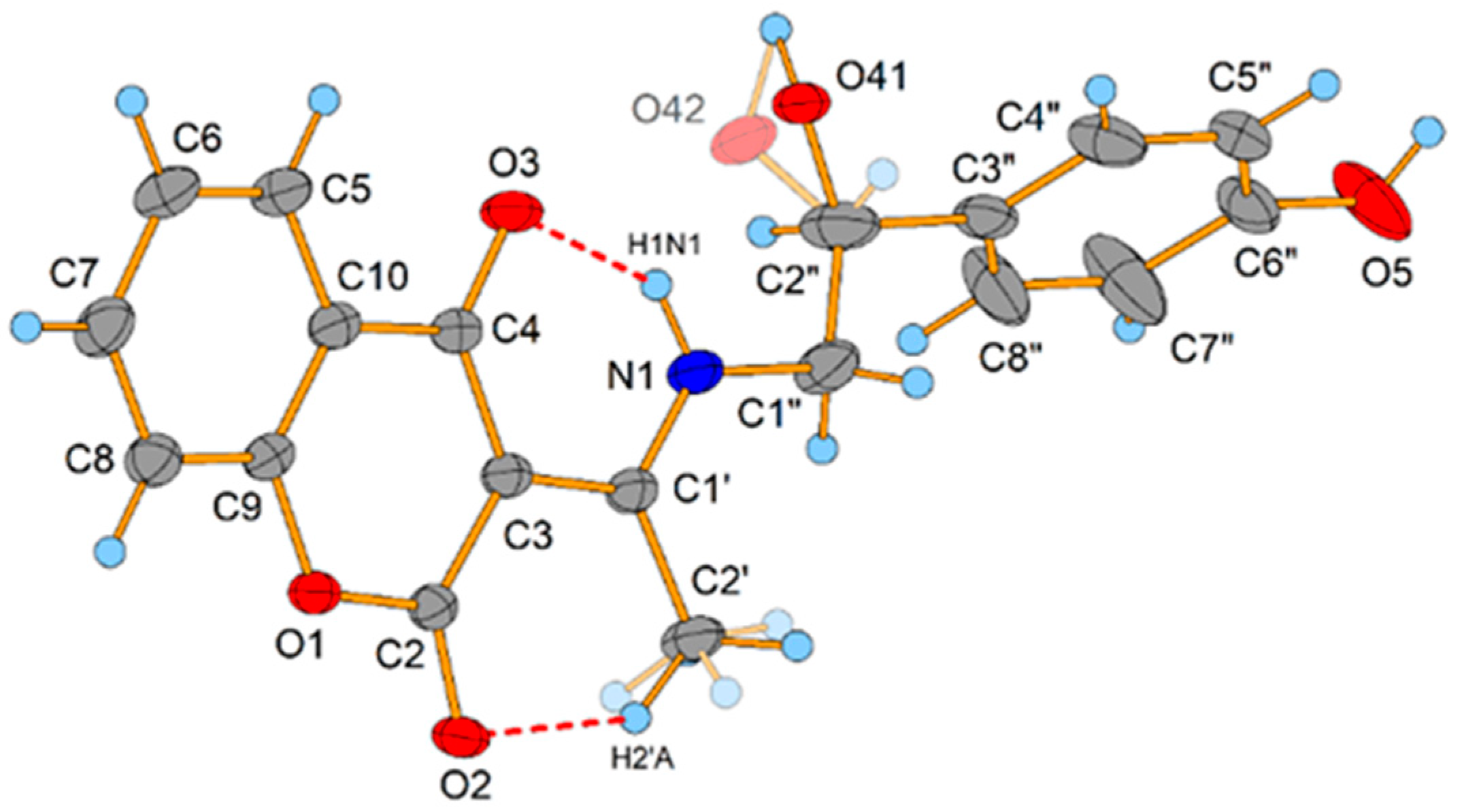
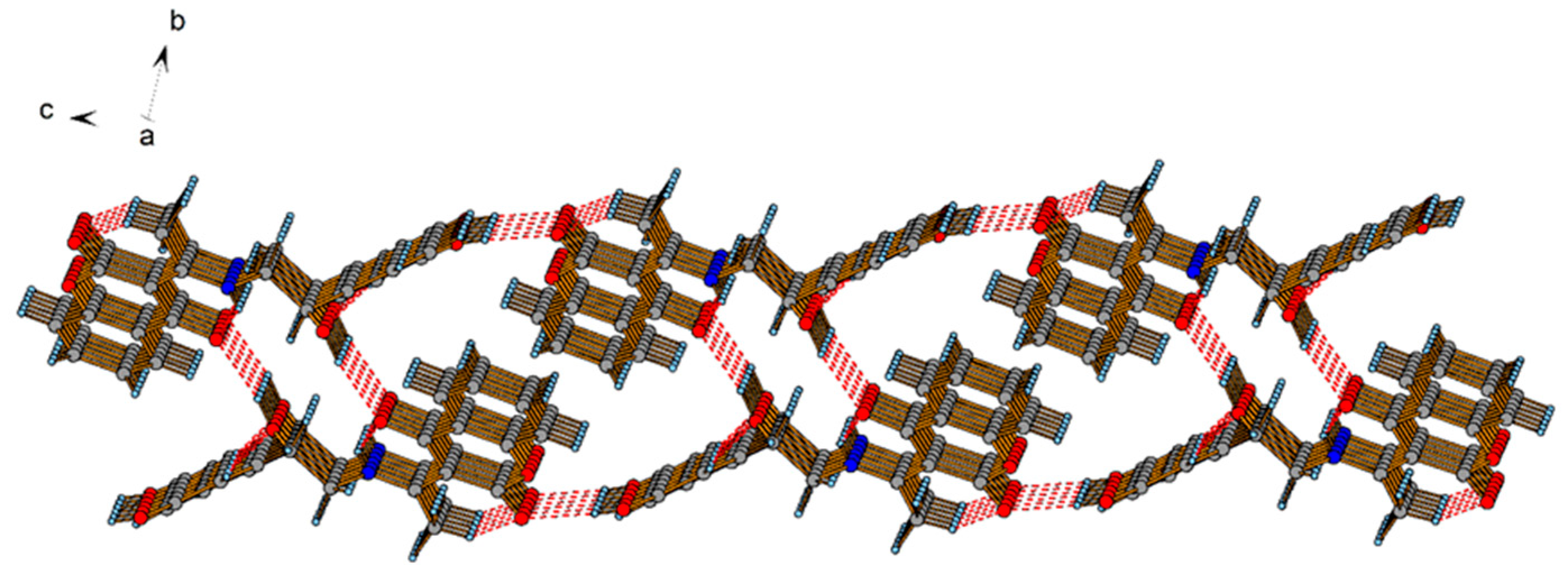
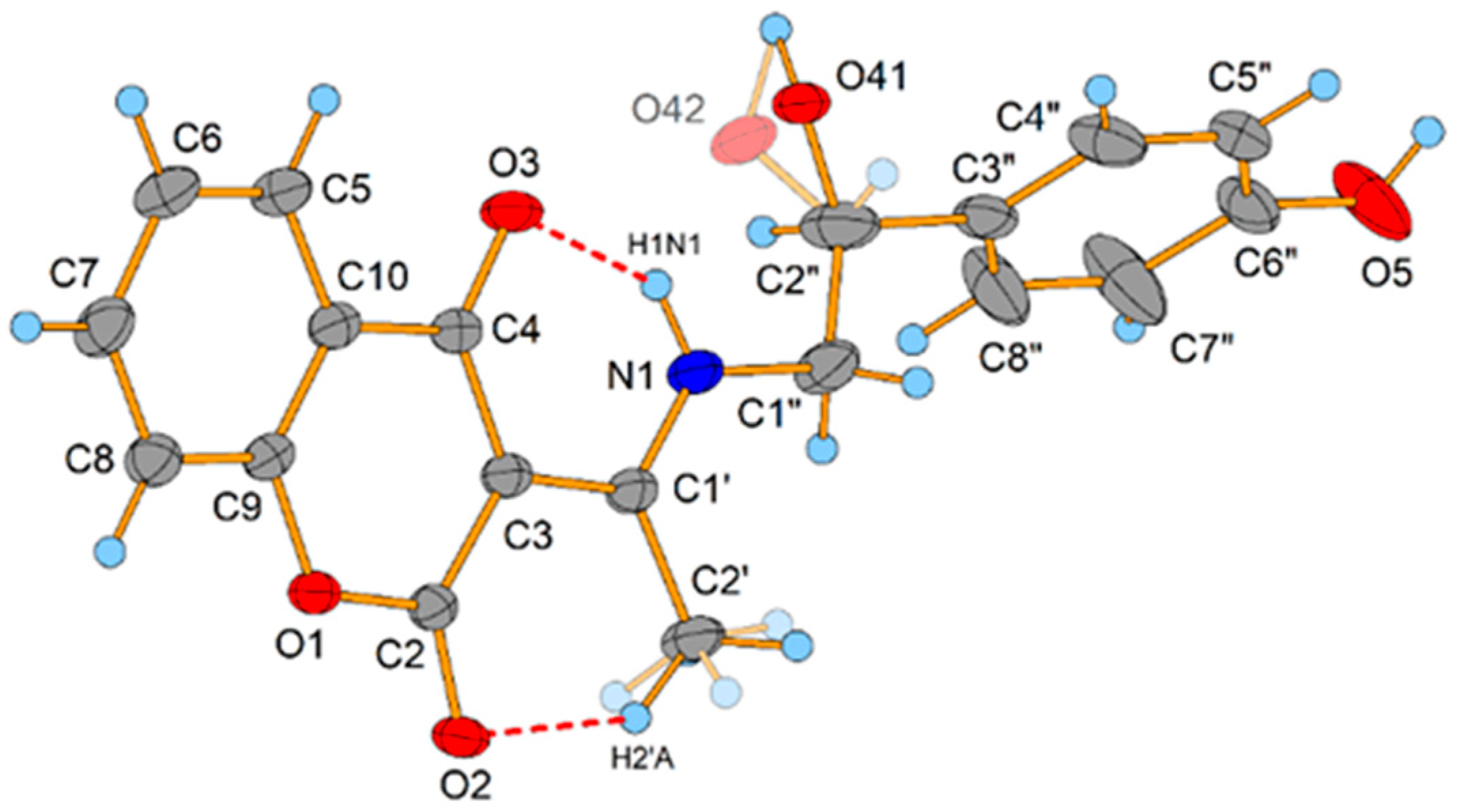
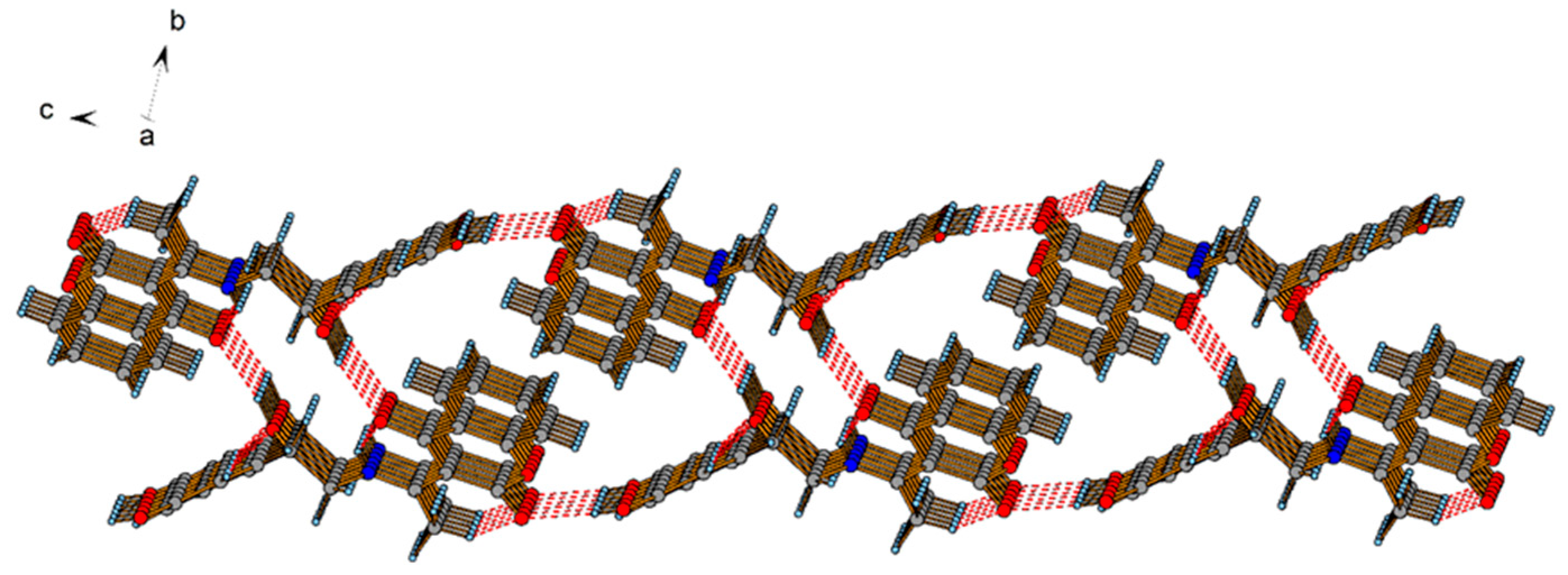
2.2. Crystallographic Structure of L2
2.3. Optimization of an L2 Structure
2.4. Comparison of Experimental and Theoretical NMR Spectra of L2
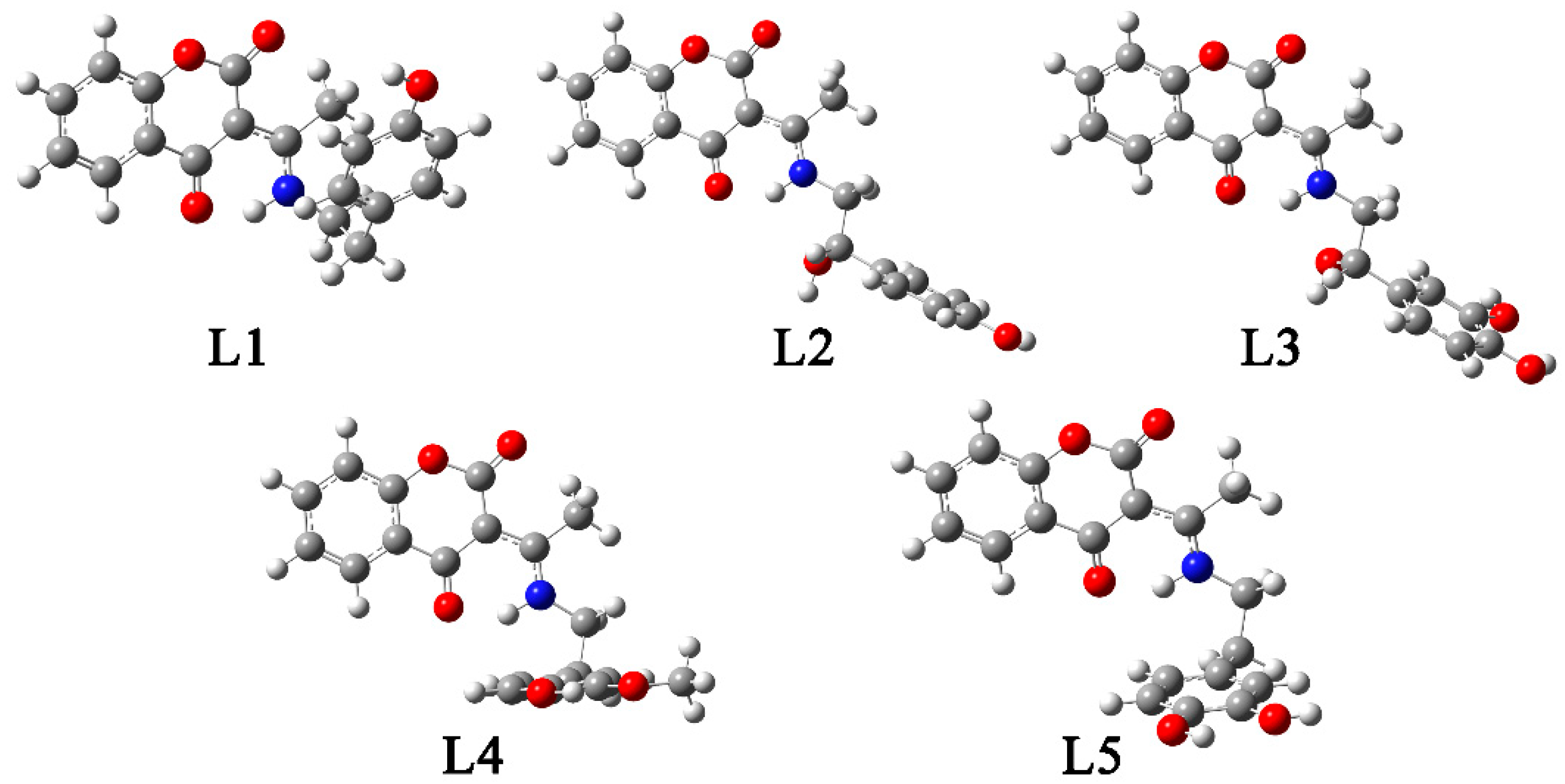
2.5. Optimization of Other Structures, NBO, and QTAIM Analyses
2.6. Reactivity Descriptors
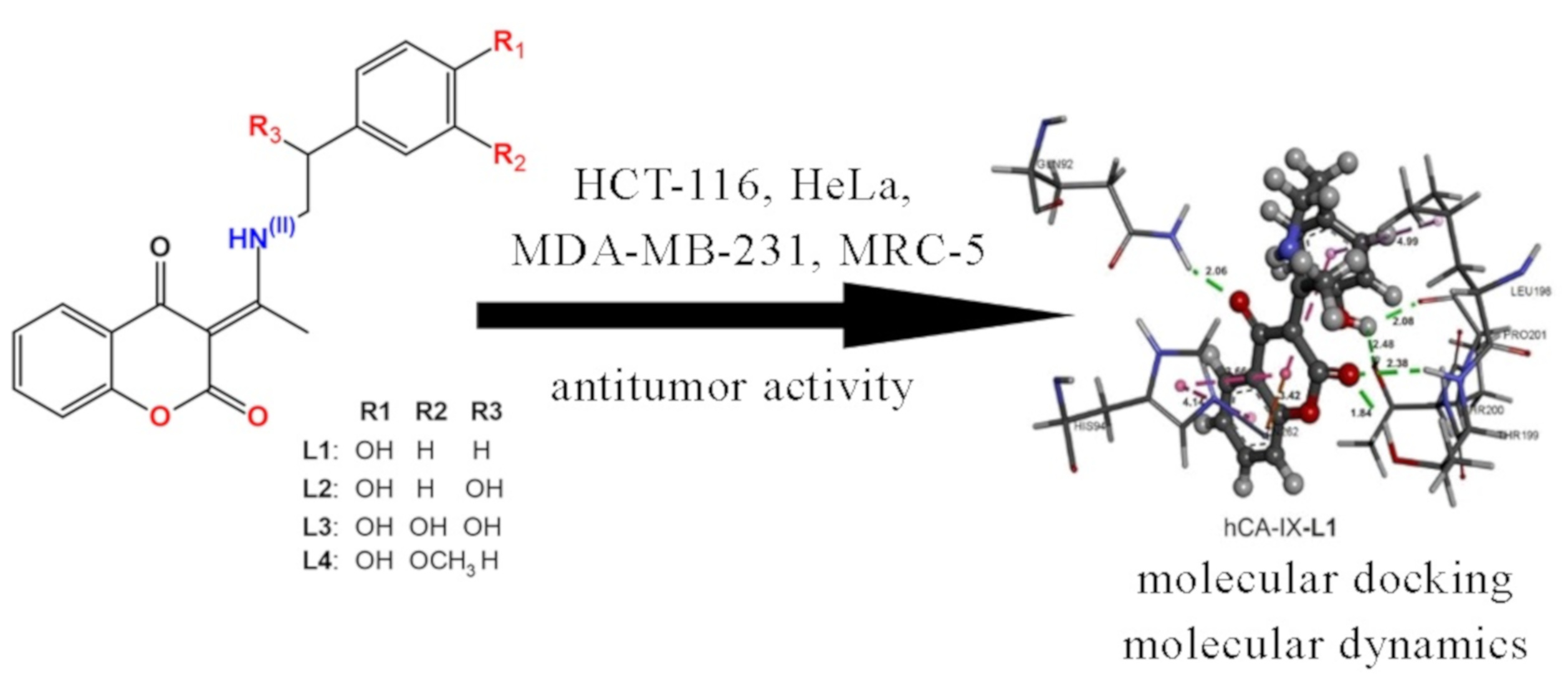
2.7. Cytotoxic Activity
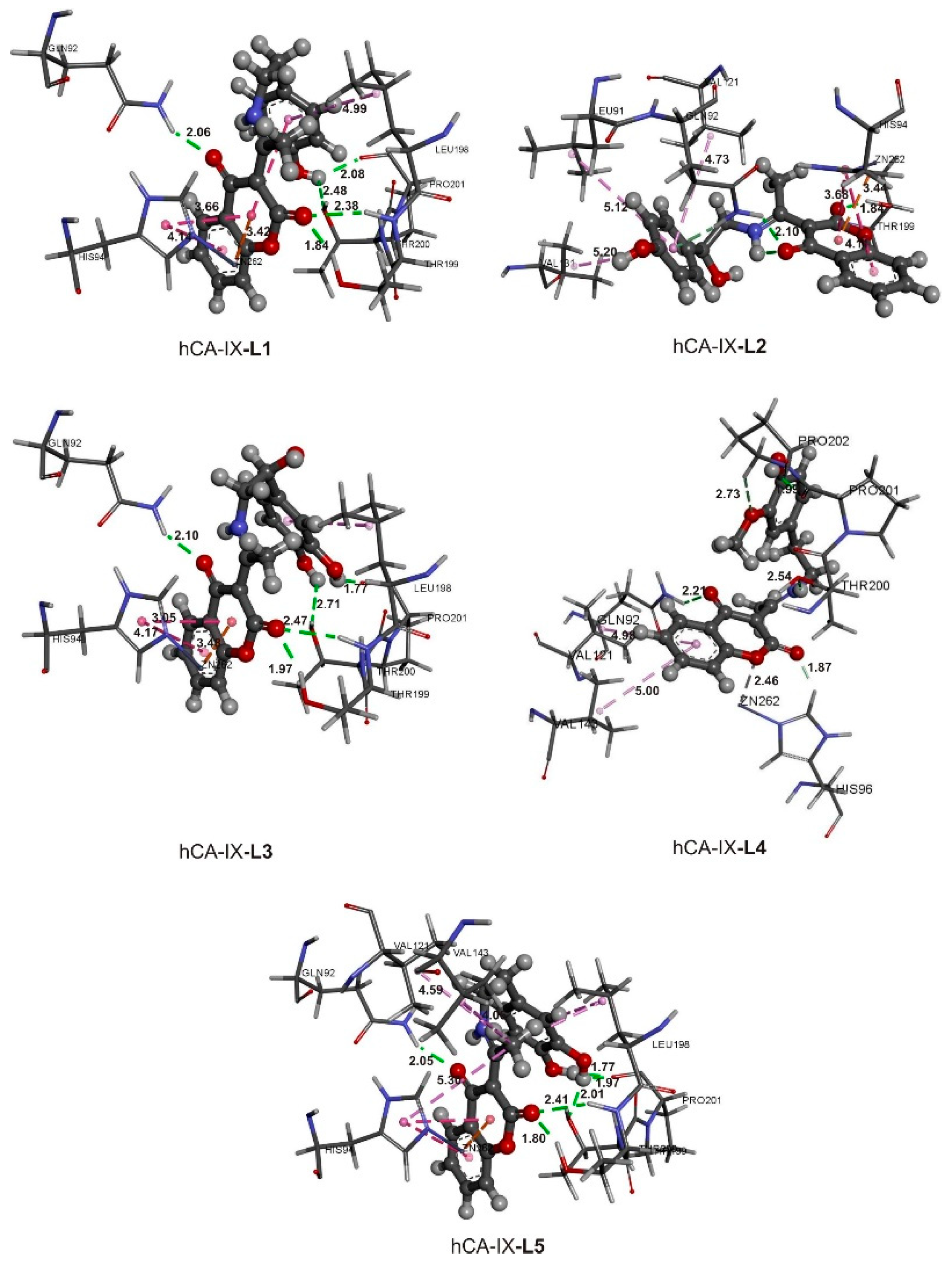
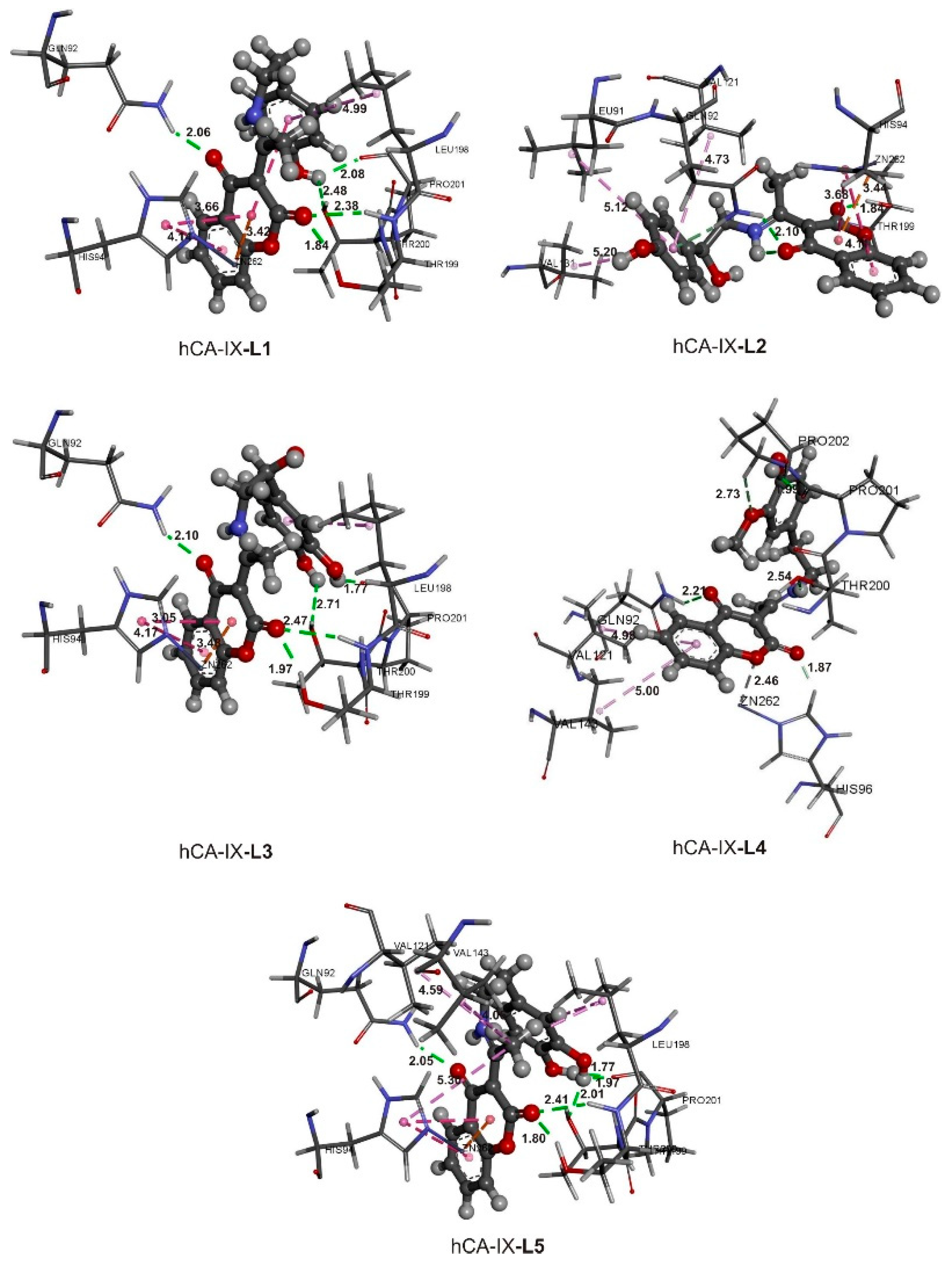
2.8. Active Site Confirmation and Molecular Docking Analysis
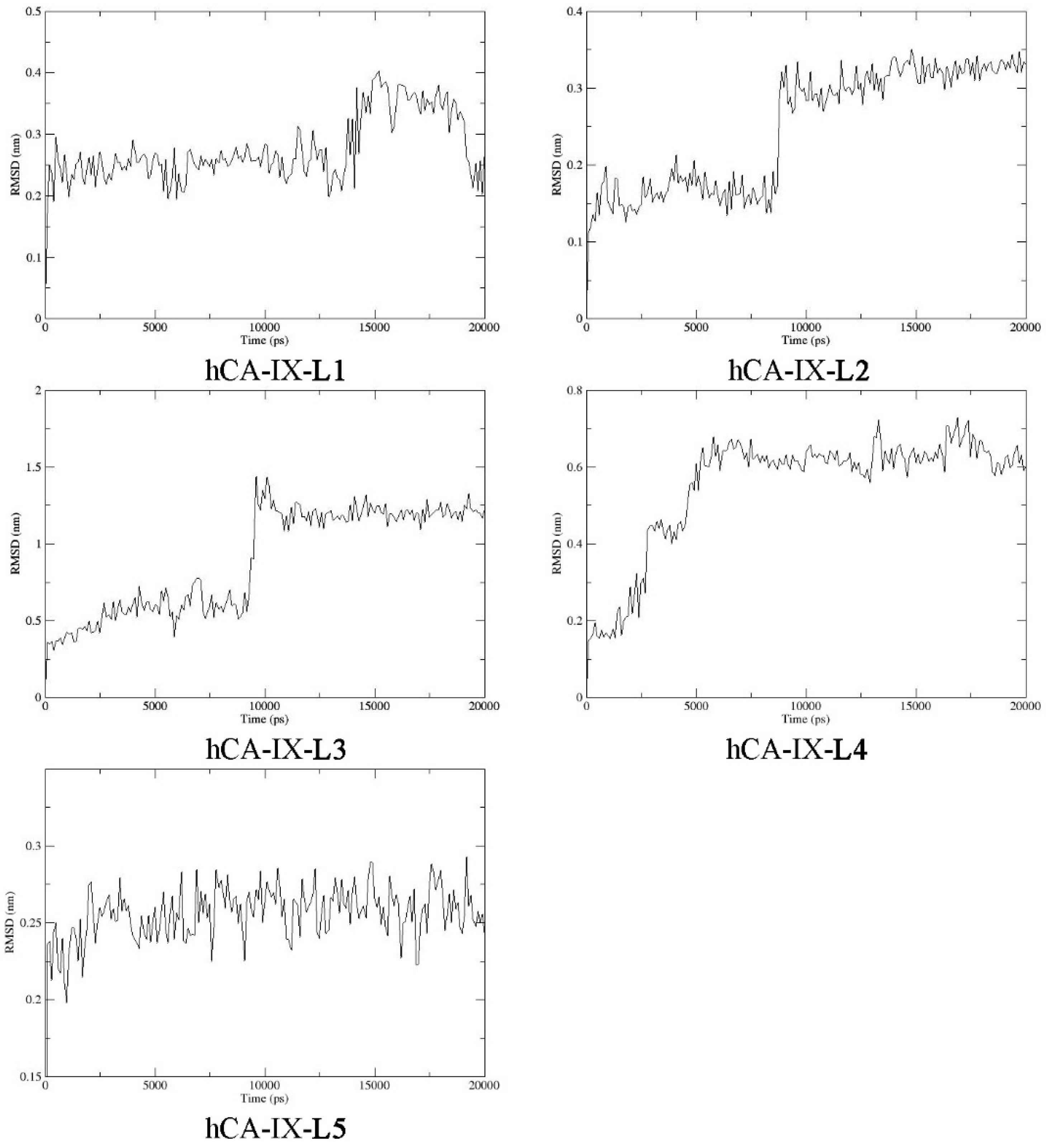
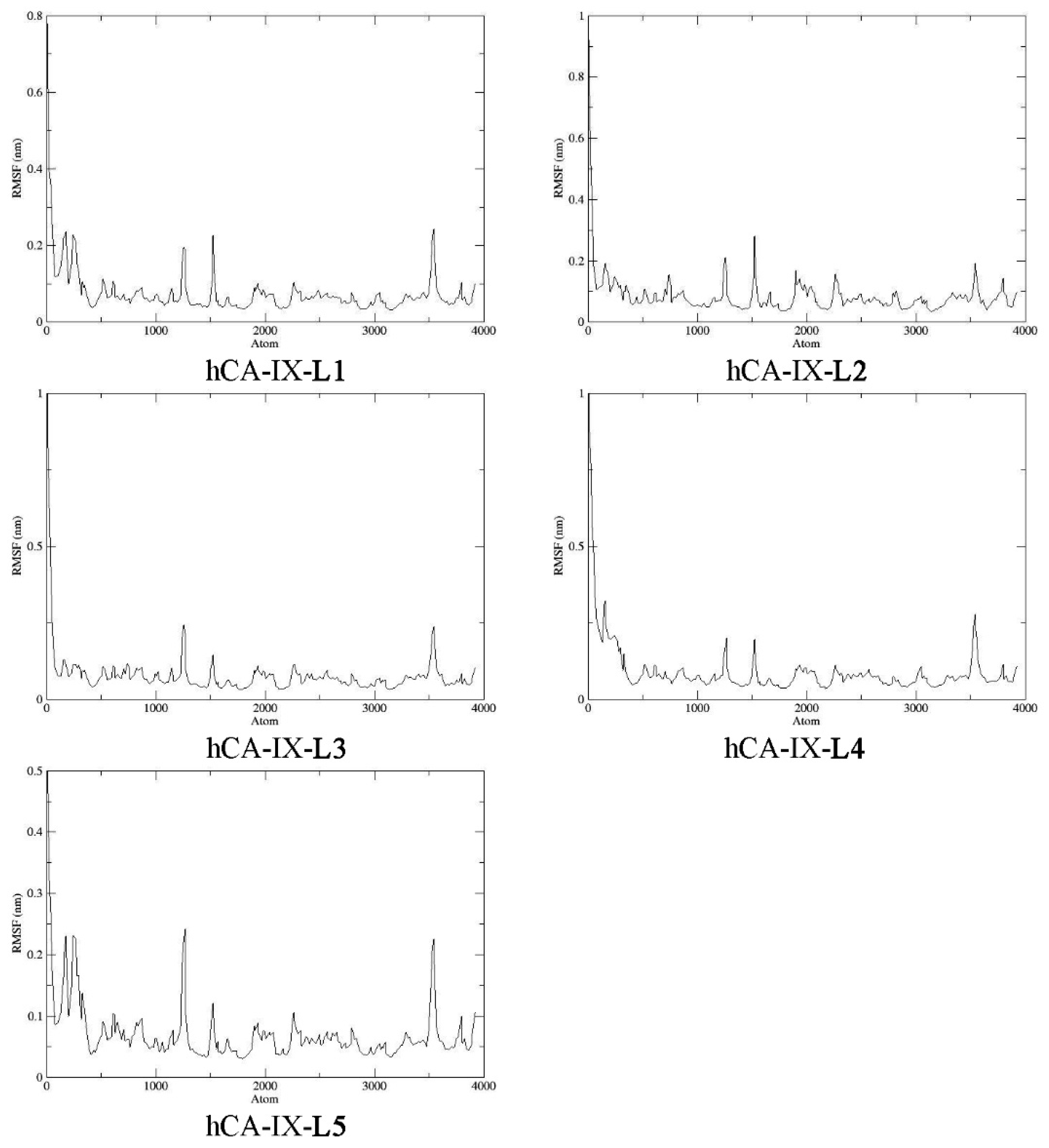
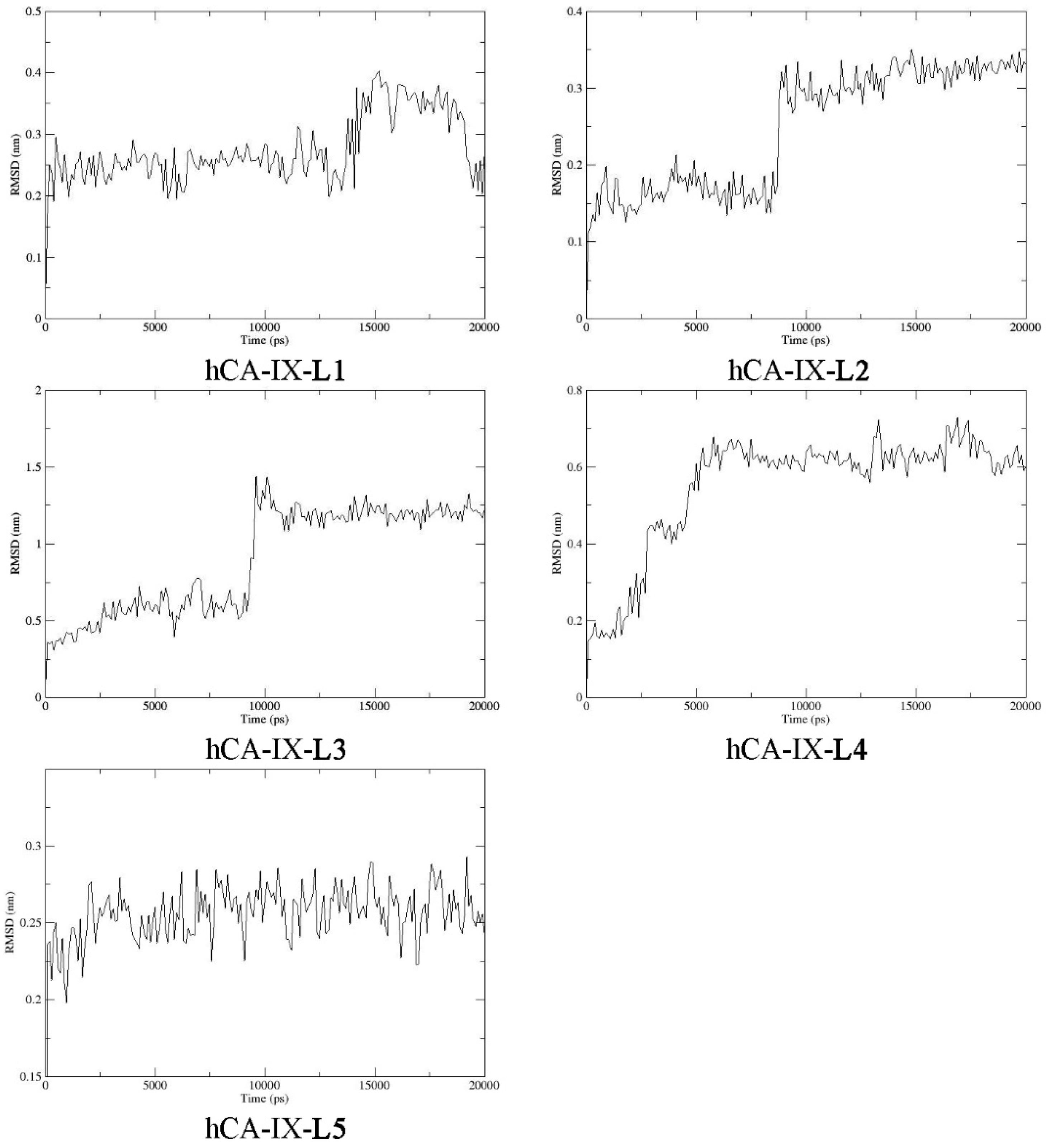
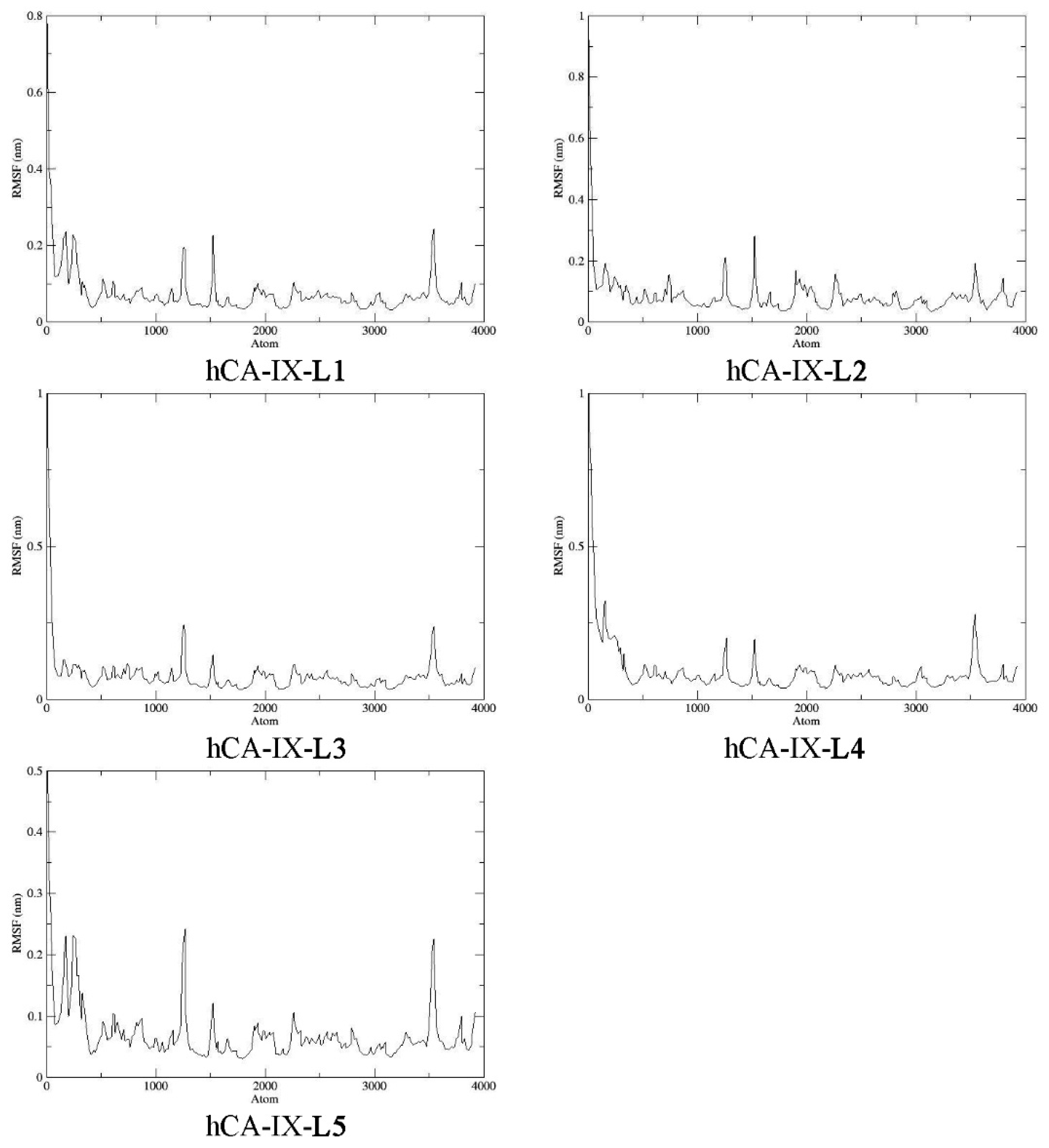
2.9. Molecular Dynamics
2.10. Ecotoxicology Assessment
3. Materials and Methods
3.1. Chemical Reagents and Instruments
3.2. Synthesis of Coumarin- Neurotransmitters Derivatives
3.2.1. (E)-3-(1-((4-hydroxyphenethyl)amino)ethylidene)chromane-2,4-dione (L1)
3.2.2. (E)-3-(1-((2-hydroxy-2-(4-hydroxyphenyl)ethyl)amino)ethylidene)chromane-2,4-dione (L2)
3.2.3. (E)-3-(1-((2-(3,4-dihydroxyphenyl)-2-hydroxyethyl)amino)ethylidene)chromane-2,4-dione (L3)
3.2.4. (E)-3-(1-((4-hydroxy-3-methoxyphenethyl)amino)ethylidene)chromane-2,4-dione (L4)
3.3. UHPLC-DAD Analysis
3.4. X-ray Data Collection and Structure Refinement
3.5. Cell Culturing
3.6. Cytotoxicity Assay
3.7. Computational Methods
3.8. Fukui Functions
3.9. Molecular Docking
3.10. Molecular Dynamics
3.11. Ecotoxicology Assessment
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Franz, K.J.; Metzler-Nolte, N. Introduction: Metals in Medicine. Chem. Rev. 2019, 119, 727–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnstone, T.C.; Suntharalingam, K.; Lippard, S.J. The Next Generation of Platinum Drugs: Targeted Pt(II) Agents, Nanoparticle Delivery, and Pt(IV) Prodrugs. Chem. Rev. 2016, 116, 3436–3486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonijević, M.R.; Simijonović, D.M.; Avdović, E.H.; Ćirić, A.; Petrović, Z.D.; Marković, J.D.; Stepanić, V.; Marković, Z.S. Green One-Pot Synthesis of Coumarin-Hydroxybenzohydrazide Hybrids and Their Antioxidant Potency. Antioxidants 2021, 10, 1106. [Google Scholar] [CrossRef]
- Touisni, N.; Maresca, A.; McDonald, P.C.; Lou, Y.; Scozzafava, A.; Dedhar, S.; Winum, J.Y.; Supuran, C.T. Glycosyl coumarin carbonic anhydrase IX and XII inhibitors strongly attenuate the growth of primary breast tumors. J. Med. Chem. 2011, 54, 8271–8277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, M.Z.; Osman, H.; Ali, M.A.; Ahsan, M.J. Therapeutic potential of coumarins as antiviral agents. Eur. J. Med. Chem. 2016, 123, 236–255. [Google Scholar] [CrossRef]
- de Souza, L.G.; Rennã, M.N.; Figueroa-Villar, J.D. Coumarins as cholinesterase inhibitors: A review. Chem. Biol. Interact. 2016, 254, 11–23. [Google Scholar] [CrossRef]
- Zhang, L.; Xu, Z. Coumarin-containing hybrids and their anticancer activities. Eur. J. Med. Chem. 2019, 181, 111587. [Google Scholar] [CrossRef]
- Montanari, S.; Allarà, M.; Scalvini, L.; Kostrzewa, M.; Belluti, F.; Gobbi, S.; Naldi, M.; Rivara, S.; Bartolini, M.; Ligresti, A.; et al. New Coumarin Derivatives as Cholinergic and Cannabinoid System Modulators. Molecules 2021, 26, 3254. [Google Scholar] [CrossRef]
- Menezes, J.C.J.M.D.S.; Diederich, M. Translational role of natural coumarins and their derivatives as anticancer agents. Future Med. Chem. 2019, 11, 1057–1082. [Google Scholar] [CrossRef] [PubMed]
- Berman, R.M.; Narasimhan, M.; Miller, H.L.; Anand, A.; Cappiello, A.; Oren, D.A.; Heninger, G.R.; Charney, D.S. Transient depressive relapse induced by catecholamine depletion: Potential phenotypic vulnerability marker? Arch. Gen. Psychiatry 1999, 56, 395–403. [Google Scholar] [CrossRef] [Green Version]
- Dimić, D.; Milenković, D.; Marković, J.D.; Marković, Z. Antiradical activity of catecholamines and metabolites of dopamine: Theoretical and experimental study. Phys. Chem. Chem. Phys. 2017, 128, 16655–16663. [Google Scholar] [CrossRef] [PubMed]
- Zeiadeh, I.; Najjar, A.; Karaman, R. Strategies for Enhancing the Permeation of CNS-Active Drugs through the Blood-Brain Barrier: A Review. Molecules 2018, 23, 1289. [Google Scholar] [CrossRef] [Green Version]
- Denora, N.; Laquintana, V.; Lopedota, A.; Serra, M.; Dazzi, L.; Biggio, G.; Pal, D.; Mitra, A.K.; Latrofa, A.; Trapani, G.; et al. Novel L-Dopa and dopamine prodrugs containing a 2-phenyl-imidazopyridine moiety. Pharm. Res. 2007, 24, 1309–1324. [Google Scholar] [CrossRef] [PubMed]
- Fernández, C.; Nieto, O.; Fontenla, J.A.; Rivas, E.; De Ceballos, M.L.; Fernandez-Mayoralas, A. Synthesis of glycosyl derivatives as dopamine prodrugs: Interaction with glucose carrier GLUT-1. Org. Biomol. Chem. 2003, 1, 767–771. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.S.; Ding, Y.; Liu, Y.; Kang, P.; Hou, J.W.; Zhang, X.Y.; Xie, S.S.; Zhang, T. Design, synthesis and biological evaluation of novel coumarin-N-benzyl pyridinium hybrids as multi-target agents for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2017, 139, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Dimić, D.S.; Marković, Z.S.; Saso, L.; Avdović, E.H.; Đorović, J.R.; Petrović, I.P.; Stanisavljević, D.D. Stevanović, M.J.; Potočňák, I.; Samoľová, E.; Trifunović, S.R.; et al. Synthesis and characterization of 3-(1-((3,4-dihydroxyphenethyl)amino)ethylidene)-chroman-2,4-dione as potentional anti-tumor agent. Oxid. Med. Cell. Longev. 2019, 2019, 2069250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemr, M.T.M.; AboulMagd, A.M.; Hassan, H.M.; Hamed, A.A.; Hamed, M.I.A.; Elsaadi, M.T. Design, synthesis and mechanistic study of new benzenesulfonamide derivatives as anticancer and antimicrobial agents via carbonic anhydrase IX inhibition. RSC Adv. 2021, 11, 26241–26257. [Google Scholar] [CrossRef]
- Alterio, V.; Hilvo, M.; Di Fiore, A.; Supuran, C.T.; Pan, P.; Parkkila, S.; Scaloni, A.; Pastorek, J.; Pastorekova, S.; Pedone, C.; et al. Crystal structure of the catalytic domain of the tumor-associated human carbonic anhydrase IX. Proc. Natl. Acad. Sci. USA 2009, 106, 16233–16238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.P.; Zhong, Y.; Han, Z.B.; Zhou, L.; Su, H.S.; Wang, J.; Liu, Y.; Cheng, M.S. Synthesis, molecular docking analysis and biological evaluations of saccharide-modified thiadiazole sulfonamide derivatives. Int. J. Mol. Sci. 2021, 22, 5482. [Google Scholar] [CrossRef]
- Salmas, R.E.; Senturk, M.; Yurtsever, M.; Durdagi, S. Discovering novel carbonic anhydrase type IX (CA IX) inhibitors from seven million compounds using virtual screening and in vitro analysis. J. Enzym. Inhib. Med. Chem. 2016, 31, 425–453. [Google Scholar] [CrossRef]
- Bozdag, M.; Ferraroni, M.; Carta, F.; Vullo, D.; Lucarini, L.; Orlandini, E.; Rossello, A.; Nuti, E.; Scozzafava, A.; Masini, E.; et al. Structural insights on carbonic anhydrase inhibitory action, isoform selectivity, and potency of sulfonamides and coumarins incorporating arylsulfonylureido groups. J. Med. Chem. 2014, 57, 9152–9167. [Google Scholar] [CrossRef] [PubMed]
- Chandak, N.; Ceruso, M.; Supuran, C.T.; Sharma, P.K. Novel sulfonamide bearing coumarin scaffolds as selective inhibitors of tumor associated carbonic anhydrase isoforms IX and XII. Bioorganic Med. Chem. 2016, 24, 2882–2886. [Google Scholar] [CrossRef] [PubMed]
- Thacker, P.S.; Alvala, M.; Arifuddin, M.; Angeli, A.; Supuran, C.T. Design, synthesis and biological evaluation of coumarin-3-carboxamides as selective carbonic anhydrase IX and XII inhibitors. Bioorg. Chem. 2019, 86, 386–392. [Google Scholar] [CrossRef]
- Avdović, E.H.; Stojković, D.L.J.; Jevtić, V.V.; Kosić, M.; Ristić, B.; Harhaji-Trajković, L.; Vukić, M.; Vuković, N.; Marković, Z.S.; Potočňák, I.; et al. Synthesis, characterization and cytotoxicity of a new palladium(II) complex with a coumarin-derived ligand 3-(1-(3-hydroxypropylamino)ethylidene)chroman-2,4-dione. Crystal structure of the 3-(1-(3-hydroxypropylamino)ethylidene)-chroman-2,4-dione. Inorg. Chim. Acta 2017, 466, 188–196. [Google Scholar] [CrossRef]
- Avdović, E.H.; Milenković, D.; Marković, J.M.D.; Đorović, J.; Vuković, N.; Vukić, M.D.; Jevtić, V.V.; Trifunović, S.R.; Potočňák, I.; Marković, Z. Synthesis, spectroscopic characterization (FT-IR, FT-Raman, and NMR), quantum chemical studies and molecular docking of 3-(1-(phenylamino)ethylidene)-chroman-2,4-dione. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2018, 195, 31–40. [Google Scholar] [CrossRef]
- Avdović, E.H.; Dimić, D.S.; Fronc, M.; Kožišek, J.; Klein, E.; Milanović, Ž.B.; Kesić, A.; Marković, Z.M. Structural and theoretical analysis, molecular docking/dynamics investigation of 3-(1-m-chloridoethylidene)-chromane-2,4-dione: The role of chlorine atom. J. Mol. Struct. 2021, 1231, 129962. [Google Scholar] [CrossRef]
- Milanovic, Ž.B.; Markovic, Z.S.; Dimic, D.S.; Klisuric, O.R.; Radojevic, I.D.; Šeklic, D.S.; Živanovic, M.N.; Markovic, J.D.; Radulovic, M.; Avdovic, E.H. Synthesis, structural characterization, biological activity and molecular docking study of 4,7-dihydroxycoumarin modified by aminophenol derivatives. Comptes Rendus Chim. 2021, 24, 215–232. [Google Scholar] [CrossRef]
- Avdović, E.H.; Milanović, Ž.B.; Živanović, M.N.; Šeklić, D.S.; Radojević, I.D.; Čomić, L.R.; Trifunović, S.R.; Amić, A.; Marković, Z.S. Synthesis, spectroscopic characterization, biological activity, DFT and molecular docking study of novel 4-hydroxycoumarine derivatives and corresponding palladium(II) complexes. Inorg. Chim. Acta 2020, 504, 119465. [Google Scholar] [CrossRef]
- Bernstein, J.; Davis, R.E.; Shimoni, L.; Chang, N.-L. Patterns in Hydrogen Bonding: Functionality and Graph Set Analysis in Crystals. Angew. Chem. Int. Ed. Engl. 1995, 34, 1555–1573. [Google Scholar] [CrossRef]
- Gilli, G.; Bellucci, F.; Ferretti, V.; Bertolasi, V. Evidence for resonance-assisted hydrogen bonding from crystal-structure correlations on the enol form of the .beta.-diketone fragment. J. Am. Chem. Soc. 1989, 111, 1023–1028. [Google Scholar] [CrossRef]
- Avdović, E.H.; Stojković, D.L.; Jevtić, V.V.; Milenković, D.; Marković, Z.S.; Vuković, N.; Potočňák, I.; Radojević, I.D.; Čomić, L.R.; Trifunović, S.R. Preparation and antimicrobial activity of a new palladium(II) complexes with a coumarin-derived ligands. Crystal structures of the 3-(1-(o-toluidino)ethylidene)-chroman-2,4-dione and 3-(1-(m-toluidino) ethylidene)-chroman-2,4-dione. Inorg. Chim. Acta 2019, 484, 52–59. [Google Scholar] [CrossRef]
- Małecka, M.; Grabowski, S.J.; Budzisz, E. Crystal and molecular structures of 3-[1-(2-hydroxyethylamino)-ethylidene]-chroman-2,4-dione and 2-methoxy-3-[1-(benzylamino)-ethylidene]-2,3-dihydro-2,4-dioxo-2λ5-benzo[e][1,2]oxaphosphinane and DFT study of intramolecular H-bonds of related compounds. Chem. Phys. 2004, 297, 235–244. [Google Scholar] [CrossRef]
- Stojković, D.L.; Jevtić, V.V.; Vuković, N.; Vukić, M.; Potočňák, I.; Zelen, I.R.; Zarić, M.M.; Mišić, M.M.; Baskić, D.; Kaluđerović, G.N.; et al. Crystal and molecular structure of a new palladium(II) complex with a coumarin-valine derivate. J. Struct. Chem. 2017, 58, 550–557. [Google Scholar] [CrossRef]
- Małecka, M.; Budzisz, E. 3-[(1-Benzylamino)ethylidene]-2H-chromene-2,4(3H)-dione. Acta Crystallogr. Sect. E Struct. Rep. Online 2006, 62, o5058–o5060. [Google Scholar] [CrossRef] [Green Version]
- Brahmia, A.; Ben Ayed, T.; Ben Hassen, R. 3-[1-(2-Hydroxyanilino)ethylidene]-3H-chromen-2,4-dione. Acta Crystallogr. Sect. E Struct. Rep. Online 2013, 69, o1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Avdović, E.H.; Dimić, D.S.; Marković, J.M.D.; Vuković, N.; Radulović, M.Đ.; Živanović, M.N.; Filipović, N.D.; Đorović, J.R.; Trifunović, S.R.; Marković, Z.S. Spectroscopic and theoretical investigation of the potential anti-tumor and anti-microbial agent, 3-(1-((2-hydroxyphenyl)amino)ethylidene)chroman-2,4-dione. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2019, 206, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Petrović, A.Z.; Ćoćić, D.C.; Bockfeld, D.; Živanović, M.; Milivojević, N.; Virijević, K.; Janković, N.; Scheurer, A.; Vraneš, M.; Bogojeski, J.V. Biological activity of bis(pyrazolylpyridine) and terpiridine Os(ii) complexes in the presence of biocompatible ionic liquids. Inorg. Chem. Front. 2021, 8, 2749–2770. [Google Scholar] [CrossRef]
- Sieveking, I.; Thomas, P.; Estévez, J.C.; Quiñones, N.; Cuéllar, M.A.; Villena, J.; Espinosa-Bustos, C.; Fierro, A.; Tapia, R.A.; Maya, J.D.; et al. 2-Phenylaminonaphthoquinones and related compounds: Synthesis, trypanocidal and cytotoxic activities. Bioorg. Med. Chem. 2014, 22, 4609–4620. [Google Scholar] [CrossRef]
- Durdagi, S.; Korkmaz, N.; Işık, S.; Vullo, D.; Astley, D.; Ekinci, D.; Salmas, R.E.; Senturk, M.; Supuran, C.T. Kinetic and docking studies of cytosolic/tumor-associated carbonic anhydrase isozymes I, II and IX with some hydroxylic compounds. J. Enzym. Inhib. Med. Chem. 2016, 31, 1214–1220. [Google Scholar] [CrossRef] [Green Version]
- Supuran, C.T. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin. Drug Discov. 2017, 12, 61–88. [Google Scholar] [CrossRef]
- Abo-Ashour, M.F.; Eldehna, W.M.; Nocentini, A.; Ibrahim, H.S.; Bua, S.; Abdel-Aziz, H.A.; Abou-Seri, S.M.; Supuran, C.T. Novel synthesized SLC-0111 thiazole and thiadiazole analogues: Determination of their carbonic anhydrase inhibitory activity and molecular modeling studies. Bioorg. Chem. 2019, 87, 794–802. [Google Scholar] [CrossRef] [PubMed]
- Kokkinidis, M.; Glykos, N.M.; Fadouloglou, V.E. Protein flexibility and enzymatic catalysis. In Advances in Protein Chemistry and Structural Biology; Academic Press Inc.: Cambridge, MA, USA, 2012; Volume 87, pp. 181–218. [Google Scholar]
- Kalathiya, U.; Padariya, M.; Baginski, M. Structural, functional, and stability change predictions in human telomerase upon specific point mutations. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef]
- Avdović, E.H.; Petrović, I.P.; Stevanović, M.J.; Saso, L.; Marković, J.M.D.; Filipović, N.D.; Živić, M.; Antić, T.N.C.; Žižić, M.V.; Todorović, N.V.; et al. Synthesis and Biological Screening of New 4-Hydroxycoumarin Derivatives and Their Palladium(II) Complexes. Oxid. Med. Cell. Longev. 2021, 2021, 8849568. [Google Scholar] [CrossRef] [PubMed]
- CrysAlisPRO 2017, Oxford Diffraction/Agilent Technologies UK Ltd.: Yarnton, UK, 2017.
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Diamond—Crystal and Molecular Structure Visualization, Crystal Impact—Dr. H. Putz & Dr. K. Brandenburg GbR, Kreuzherrenstr. 102, 53227 Bonn, Germany. Available online: https://www.crystalimpact.de/diamond (accessed on 9 January 2022).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01, Gaussian, Inc.: Wallingford, CT, USA, 2009.
- Becke, A.D.; Johnson, E.R. A density-functional model of the dispersion interaction. J. Chem. Phys. 2005, 123, 154101. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Schultz, N.E.; Truhlar, D.G. Design of Density Functionals by Combining the Method of Constraint Satisfaction with Parametrization for Thermochemistry, Thermochemical Kinetics, and Noncovalent Interactions. J. Chem. Theory Comput. 2006, 2, 364–382. [Google Scholar] [CrossRef] [PubMed]
- Austin, A.; Petersson, G.A.; Frisch, M.J.; Dobek, F.J.; Scalmani, G.; Throssell, K. A Density Functional with Spherical Atom Dispersion Terms. J. Chem. Theory Comput. 2012, 8, 4989–5007. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Zieliński, R.; Szymusiak, H. Application of Dft B3Lyp/Giao and B3Lyp/Csgt Methods for Interpretation of Nmr Spectra of Flavonoids. Pol. J. Food Nutr. Sci. 2003, 12, 157–162. [Google Scholar]
- Bohmann, J.A.; Weinhold, F.; Farrar, T.C. Natural chemical shielding analysis of nuclear magnetic resonance shielding tensors from gauge-including atomic orbital calculations. J. Chem. Phys. 1997, 107, 1173. [Google Scholar] [CrossRef]
- Foster, J.P.; Weinhold, F. Natural hybrid orbitals. J. Am. Chem. Soc. 1980, 102, 7211–7218. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Keith, T.A. AIMAll 2016. Available online: http://aim.tkgristmill.com/ (accessed on 9 January 2022).
- Yang, W.; Mortier, W.J. The use of global and local molecular parameters for the analysis of the gas-phase basicity of amines. J. Am. Chem. Soc. 1986, 108, 5708–5711. [Google Scholar] [CrossRef] [PubMed]
- De Luca, G.; Sicilia, E.; Russo, N.; Mineva, T. On the Hardness Evaluation in Solvent for Neutral and Charged Systems. J. Am. Chem. Soc. 2002, 124, 1494–1499. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Ravindranath, P.A.; Forli, S.; Goodsell, D.S.; Olson, A.J.; Sanner, M.F. AutoDockFR: Advances in Protein-Ligand Docking with Explicitly Specified Binding Site Flexibility. PLOS Comput. Biol. 2015, 11, e1004586. [Google Scholar] [CrossRef] [Green Version]
- BIOVIA. Dassault Systèmes, Discovery Studio Modeling Environment, 2017, San Diego: Dassault Systèmes. 2017. Available online: https://www.3ds.com/products-services/biovia/products/molecular-modeling-simulation/biovia-discovery-studio/ (accessed on 9 January 2022).
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; MacKerell, A.D. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ1 and χ2 Dihedral Angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Bo, X.; Sun, J.; Mei, Q.; Wei, B.; An, Z.; Han, D.; Li, Z.; Xie, J.; Zhan, J.; He, M. Degradation of prosulfocarb by hydroxyl radicals in gas and aqueous phase: Mechanisms, kinetics and toxicity. Ecotoxicol. Environ. Saf. 2020, 191, 110175. [Google Scholar] [CrossRef] [PubMed]
- Mei, Q.; Sun, J.; Han, D.; Wei, B.; An, Z.; Wang, X.; Xie, J.; Zhan, J.; He, M. Sulfate and hydroxyl radicals-initiated degradation reaction on phenolic contaminants in the aqueous phase: Mechanisms, kinetics and toxicity assessment. Chem. Eng. J. 2019, 373, 668–676. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density functional approach to the frontier-electron theory of chemical reactivity. J. Am. Chem. Soc. 1984, 106, 4049–4050. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | L2 |
|---|---|
| Empirical formula | C19H17NO5 |
| Formula weight | 339.33 |
| Temperature | 95(2) K |
| Wavelength | 1.54184 Å |
| Crystal system Space group | Triclinic P-1 |
| Unit cell dimensions | a = 7.0726(2) Å → α = 99.978(2)° b = 9.8752(2) Å → β = 99.380(2)° c = 12.7283(3) Å → γ = 110.120(2)° |
| Volume | 797.94(4) Å3 |
| Z; density (calculated) | 2; 1.412 g cm−3 |
| Absorption coefficient | 0.855 mm−1 |
| F(000) | 356 |
| Crystal shape, color | prism, colorless |
| Crystal size | 0.234 × 0.066 × 0.044 mm3 |
| θ range for data collection | 3.633–74.514° |
| Index ranges | −7 ≤ h ≤ 8, −12 ≤ k ≤ 12, −15 ≤ l ≤ 15 |
| Reflections collected/independent | 12478/3213 [R(int) = 0.0218] |
| Data/restraints/parameters | 3213/0/248 |
| Goodness-of-fit on F2 | 1.027 |
| Final R indices [I >2σ(I)] | R1 = 0.0511, wR2 = 0.1265 |
| R indices (all data) | R1 = 0.0563, wR2 = 0.1309 |
| Largest diff. peak and hole | 0.394; −0.486 e Å−3 |
| D−H···A | d(D−H) | d(H···A) | d(D···A) | <(DHA) |
|---|---|---|---|---|
| C2′−H2’A···O2 | 0.98 | 1.90 | 2.709(2) | 138.5 |
| C7″−H7”···O41 i | 0.95 | 2.38 | 3.235(4) | 149.3 |
| N1−H1N1···O3 | 0.92(3) | 1.80(3) | 2.5909(19) | 142(2) |
| O5−H1O5···O2 ii | 0.83(3) | 1.92(4) | 2.7236(19) | 161(3) |
| O41−H1O4···O3 iii | 0.92(4) | 1.93(4) | 2.797(2) | 158(3) |
| δ(1H) (ppm) | δ(13C) (ppm) | ||||
|---|---|---|---|---|---|
| Experimental | Theoretical | Experimental | Theoretical | ||
| C2′–H(3) | 2.5 | 2.6 | C2′ | 18.6 | 22.2 |
| C1″–H(2) | 3.4 | 3.8 | C1″ | 51.6 | 53.8 |
| O5–H | 4.8 | 4.8 | C2″ | 70.2 | 75.2 |
| C2″–H | 5.8 | 5.1 | C3 | 96.1 | 98.1 |
| C7″–H | 6.7 | 7.1 | C7″ | 115.0 | 113.2 |
| C5″–H | 6.7 | 7.1 | C5″ | 115.0 | 114.4 |
| C6–H | 7.3 | 7.6 | C8 | 116.2 | 116.1 |
| C8–H | 7.3 | 7.6 | C10 | 120.4 | 119.9 |
| C8″–H | 7.3 | 7.6 | C6 | 123.6 | 122.7 |
| C7–H | 7.6 | 7.8 | C5 | 125.7 | 125.9 |
| C4″–H | 7.9 | 7.9 | C4″ | 127.3 | 127.4 |
| C5–H | 9.4 | 8.5 | C8″ | 127.3 | 129.3 |
| N1–H | 13.8 | 13.6 | C7 | 132.7 | 133.9 |
| MAE (ppm) | 0.3 | C3″ | 133.9 | 134.8 | |
| C9 | 153.1 | 153.6 | |||
| C6″ | 156.8 | 156.5 | |||
| C2 | 162.0 | 159.9 | |||
| C1′ | 176.0 | 174.7 | |||
| C4 | 179.4 | 177.4 | |||
| MAE (ppm) | 1.4 | ||||
| O–C–C–C–N–H | Aromatic Ring Substituents | |||
|---|---|---|---|---|
| Compound | Interaction Energy [kJ mol−1] | Electron Density [a.u.] | Laplacian [a.u.] | Interaction Energy [kJ mol−1] |
| L1 | LP(O)→σ(N–H), 102 | 0.057 | 0.166 | / |
| L2 | LP(O)→σ(N–H), 86 | 0.052 | 0.158 | / |
| L3 | LP(O)→σ(N–H), 87 | 0.052 | 0.158 | LP(O)→σ(O–H), 4 |
| L4 | LP(O)→σ(N–H), 93 | 0.055 | 0.162 | LP(O)→σ(O–H), 17 |
| L5 | LP(O)→σ(N–H), 102 | 0.056 | 0.163 | LP(O)→σ(O–H), 2 |
| IC₅₀, µM | HCT-116 | HeLa | MDA-MB-231 | MRC-5 | ||||
|---|---|---|---|---|---|---|---|---|
| 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | |
| L1 | >500 | 170 | >500 | 452 | >500 | >500 | ||
| L2 | 248 | 206 | ||||||
| L3 | 107 | 495 | ||||||
| L4 | 74 | 150 | ||||||
| L5 | 73 | 92 | ||||||
| Conformations | ΔGbind (kJ mol−1) | Ki (nM) | ΔGvdw+hbond+desolv (kJ mol−1) | ΔGelec (kJ mol−1) | ΔGtotal (kJ mol−1) | ΔGtor (kJ mol−1) | ΔGunb (kJ mol−1) |
|---|---|---|---|---|---|---|---|
| hCA-IX-AZA | −37.8 | 237.83 | −39.9 | 0.3 | −2.3 | 2.8 | 0.0 |
| hCA-IX-L1 | −41.2 | 60.4 | −38.8 | −0.5 | −6.7 | 4.8 | 0.0 |
| hCA-IX-L2 | −39.8 | 107.8 | −39.1 | −0.3 | −6.1 | 5.7 | 0.0 |
| hCA-IX-L3 | −41.9 | 45.2 | −39.5 | −0.3 | −8.7 | 6.7 | 0.0 |
| hCA-IX-L4 | −42.1 | 41.8 | −41.0 | 0.2 | −7.0 | 5.7 | 0.0 |
| hCA-IX-L5 | −43.7 | 21.9 | −39.5 | −1.0 | −9.0 | 5.7 | 0.0 |
| Complex | ΔEelec (kJ mol−1) | ΔEVDW (kJ mol−1) | ΔGpolar (kJ mol−1) | ΔGnonpolar (kJ mol−1) | ΔGbinding (kJ mol−1) |
|---|---|---|---|---|---|
| hCA-IX-L1 | −338.9 ± 27.7 | −105.9 ± 14.4 | 393.6 ± 27.0 | −17.2 ± 1.1 | −68.4 ± 52.2 |
| hCA-IX-L2 | −333.4 ± 21.2 | −116.0 ± 18.1 | 419.8 ± 29.3 | −18.3 ± 1.1 | −47.8 ± 39.8 |
| hCA-IX-L3 | −27.1 ± 27.0 | −94.9 ±16.4 | 59.0 ± 17.9 | −13.0 ± 1.2 | −75.9 ± 24.3 |
| hCA-IX-L4 | −167.9 ± 1.4 | −138.7 ± 1.2 | 141.1 ± 1.3 | −19.3 ± 0.1 | −184.9 ± 1.3 |
| hCA-IX-L5 | −351.0 ± 26.6 | −135.4 ± 16.5 | 316.1 ± 26.6 | −19.5 ± 1.2 | −189.7 ± 27.1 |
| Compound | LC50 (Fish, 96 h) | LC50 (Daphnia, 48 h) | EC50 (Green Algae, 96 h) | ChV (Fish, Chronic) | ChV (Daphnia, Chronic) | ChV (Green Algae, Chronic) |
|---|---|---|---|---|---|---|
| L1 | 424 | 96.3 | 35.3 | 36.3 | 8.2 | 53.4 |
| L2 | 5080 | 576 | 399 | 372 | 37.6 | 419 |
| L3 | 2150 | 66,400 | 109 | 2000 | 30,600 | 8.78 |
| L4 | 611 | 128 | 50.6 | 51.4 | 10.6 | 73.3 |
| L5 | 249 | 4170 | 31.5 | 184 | 1750 | 3.18 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dimić, D.S.; Kaluđerović, G.N.; Avdović, E.H.; Milenković, D.A.; Živanović, M.N.; Potočňák, I.; Samoľová, E.; Dimitrijević, M.S.; Saso, L.; Marković, Z.S.; et al. Synthesis, Crystallographic, Quantum Chemical, Antitumor, and Molecular Docking/Dynamic Studies of 4-Hydroxycoumarin-Neurotransmitter Derivatives. Int. J. Mol. Sci. 2022, 23, 1001. https://doi.org/10.3390/ijms23021001
Dimić DS, Kaluđerović GN, Avdović EH, Milenković DA, Živanović MN, Potočňák I, Samoľová E, Dimitrijević MS, Saso L, Marković ZS, et al. Synthesis, Crystallographic, Quantum Chemical, Antitumor, and Molecular Docking/Dynamic Studies of 4-Hydroxycoumarin-Neurotransmitter Derivatives. International Journal of Molecular Sciences. 2022; 23(2):1001. https://doi.org/10.3390/ijms23021001
Chicago/Turabian StyleDimić, Dušan S., Goran N. Kaluđerović, Edina H. Avdović, Dejan A. Milenković, Marko N. Živanović, Ivan Potočňák, Erika Samoľová, Milena S. Dimitrijević, Luciano Saso, Zoran S. Marković, and et al. 2022. "Synthesis, Crystallographic, Quantum Chemical, Antitumor, and Molecular Docking/Dynamic Studies of 4-Hydroxycoumarin-Neurotransmitter Derivatives" International Journal of Molecular Sciences 23, no. 2: 1001. https://doi.org/10.3390/ijms23021001
APA StyleDimić, D. S., Kaluđerović, G. N., Avdović, E. H., Milenković, D. A., Živanović, M. N., Potočňák, I., Samoľová, E., Dimitrijević, M. S., Saso, L., Marković, Z. S., & Dimitrić Marković, J. M. (2022). Synthesis, Crystallographic, Quantum Chemical, Antitumor, and Molecular Docking/Dynamic Studies of 4-Hydroxycoumarin-Neurotransmitter Derivatives. International Journal of Molecular Sciences, 23(2), 1001. https://doi.org/10.3390/ijms23021001