
Transcriptome-Guided Drug Repurposing for Aggressive SCCs

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
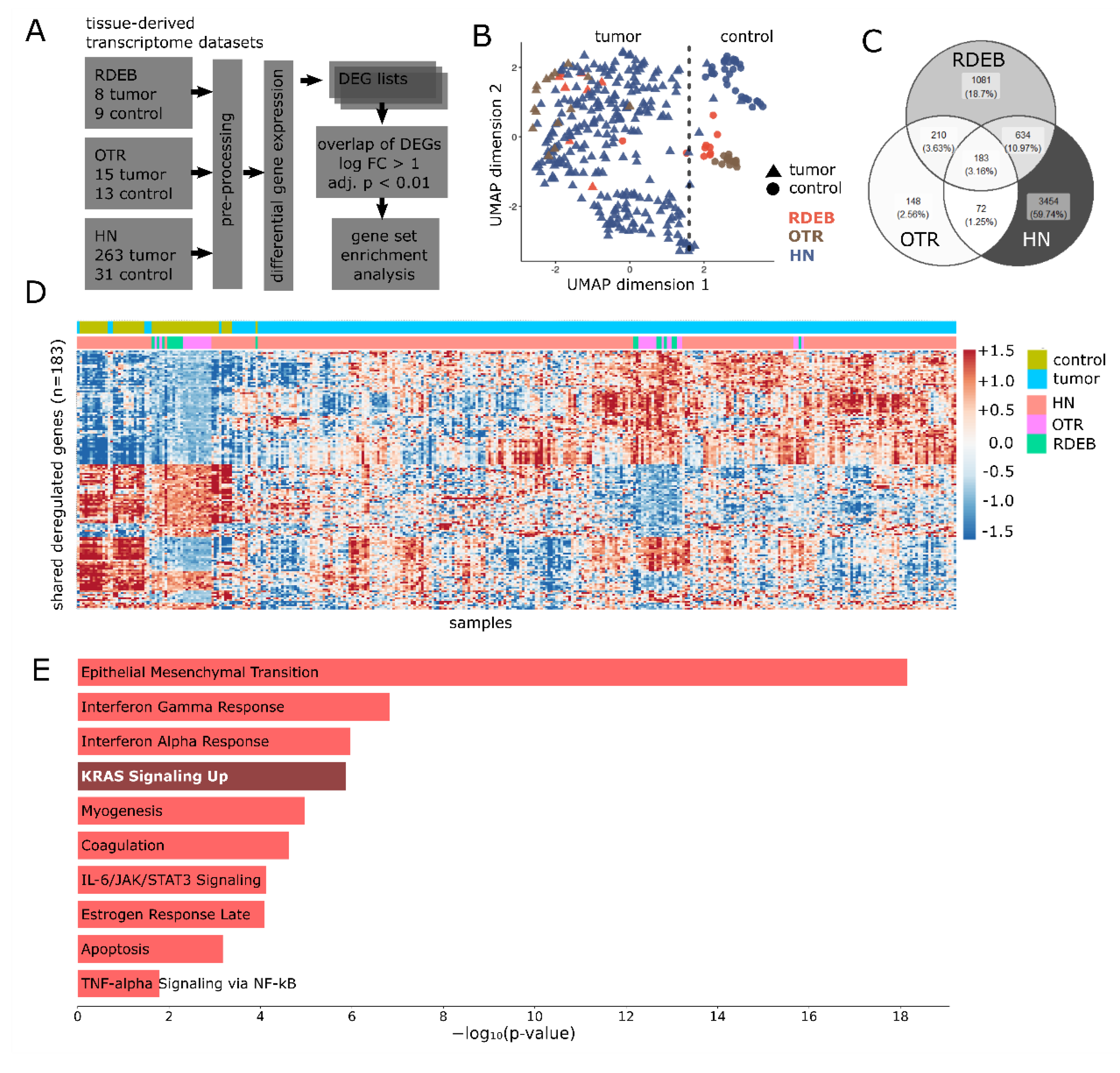
2.1. Gene Expression Overlaps between HN-, OTR-, and RDEB-SCC
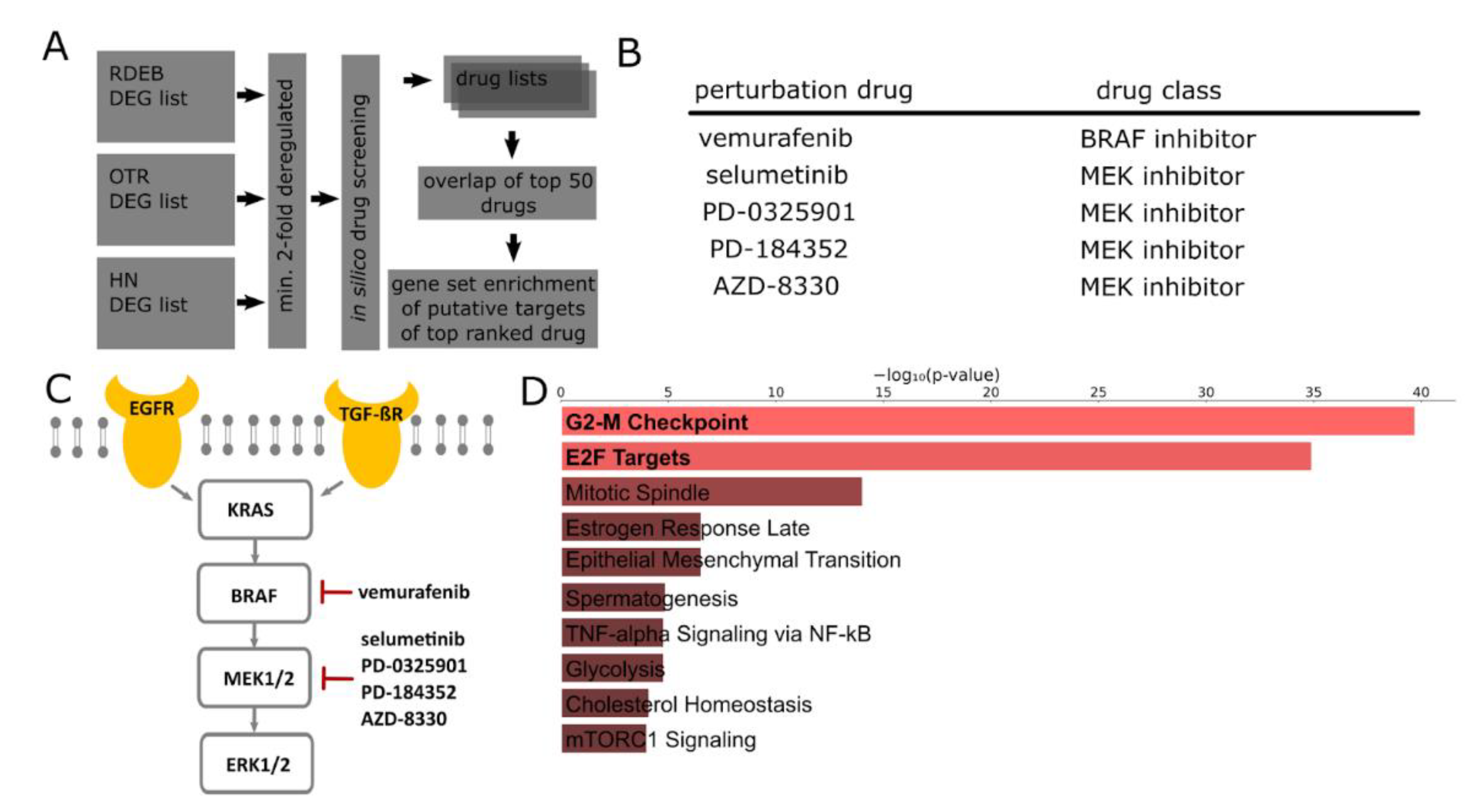
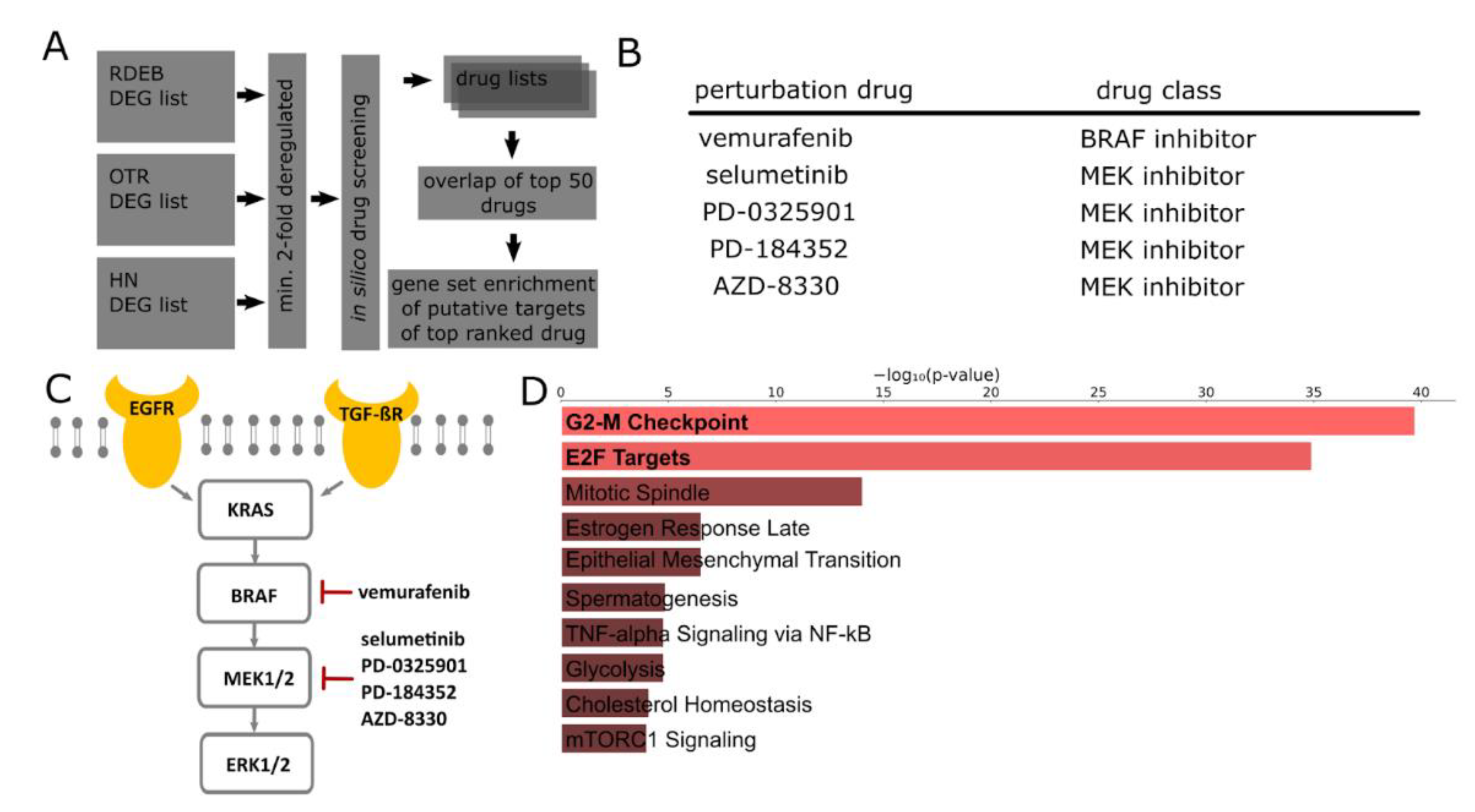
2.2. In Silico Identification of Drugs for the Treatment of SCCs
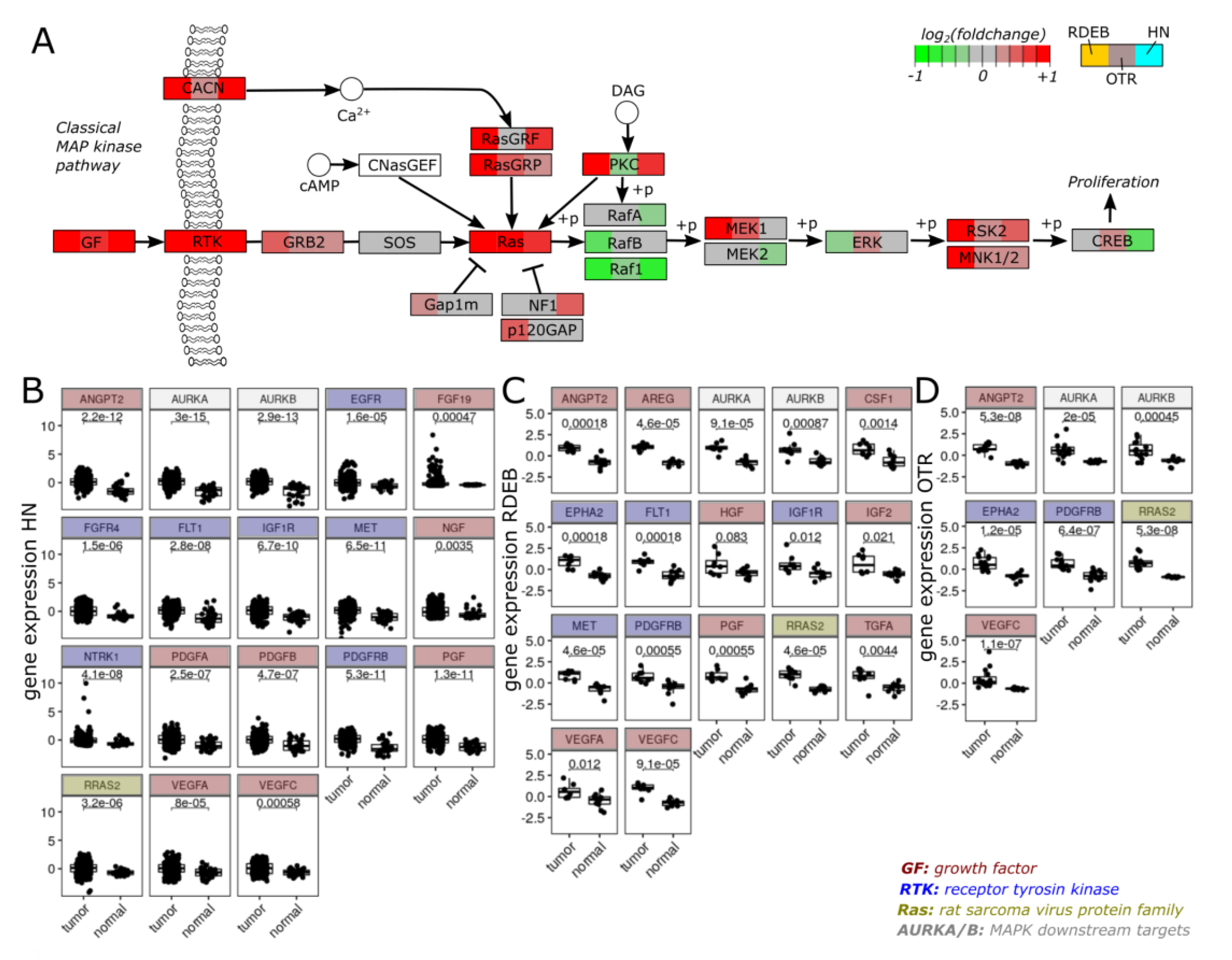
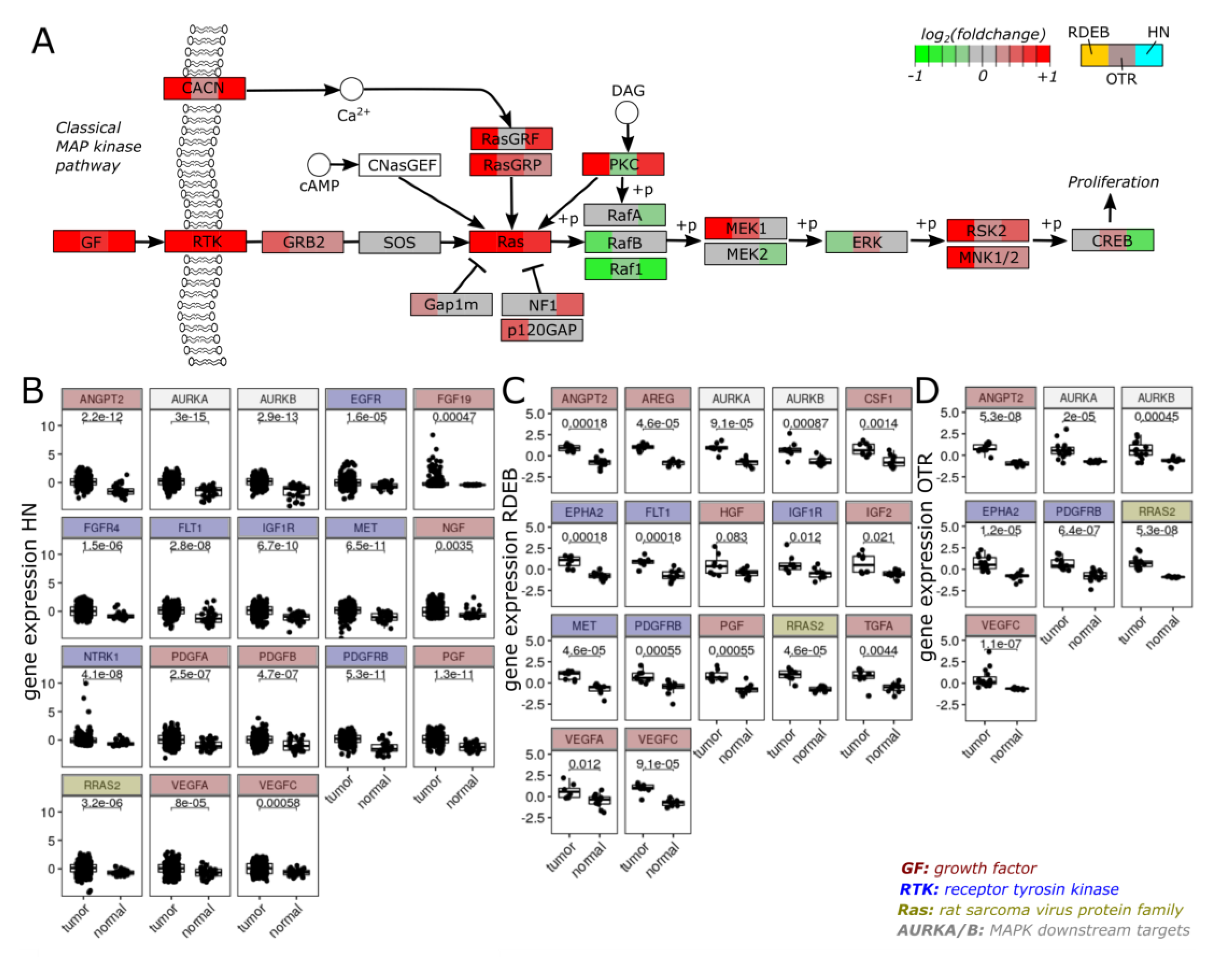
2.3. Transcriptome Profiles Highlight Activation of MAPK Signaling
3. Discussion
4. Materials and Methods
4.1. Data Sets and Extraction of Differentially Expressed Genes
4.2. Evaluation of Similarities between Data Sets
4.3. Gene Set Enrichment Analysis of Overlapping DEGs
4.4. Computational Drug Screening
4.5. Gene Set Enrichment Analysis of Putative Drug Targets
4.6. MAPK Signaling Pathway Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mittal, A.; Colegio, O.R. Skin Cancers in Organ Transplant Recipients. Am. J. Transpl. 2017, 17, 2509–2530. [Google Scholar] [CrossRef]
- Chockalingam, R.; Downing, C.; Tyring, S.K. Cutaneous Squamous Cell Carcinomas in Organ Transplant Recipients. J. Clin. Med. 2015, 4, 1229–1239. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.; Hansen, S.; Møller, B.; Leivestad, T.; Pfeffer, P.; Geiran, O.; Fauchald, P.; Simonsen, S. Skin cancer in kidney and heart transplant recipients and different long-term immunosuppressive therapy regimens. J. Am. Acad. Dermatol. 1999, 40 Pt 1, 177–186. [Google Scholar] [CrossRef]
- Connolly, K.; Manders, P.; Earls, P.; Epstein, R.J. Papillomavirus-associated squamous skin cancers following transplant immunosuppression: One Notch closer to control. Cancer Treat. Rev. 2014, 40, 205–214. [Google Scholar] [CrossRef]
- Montaudié, H.; Chiaverini, C.; Sbidian, E.; Charlesworth, A.; Lacour, J.-P. Inherited epidermolysis bullosa and squamous cell carcinoma: A systematic review of 117 cases. Orphanet J. Rare Dis. 2016, 11, 117. [Google Scholar] [CrossRef] [Green Version]
- Dayal, J.H.S.; Mason, S.M.; Salas-Alanis, J.C.; McGrath, J.A.; Taylor, R.G.; Mellerio, J.E.; Blyth, K.; South, A.P.; Inman, G.J. Heterogeneous addiction to transforming growth factor-beta signalling in recessive dystrophic epidermolysis bull-osa-associated cutaneous squamous cell carcinoma. Br. J. Derm. 2021, 184, 697–708. [Google Scholar] [CrossRef]
- Knaup, J.; Gruber, C.; Krammer, B.; Ziegler, V.; Bauer, J.; Verwanger, T. TGFβ-signaling in squamous cell carcinoma occur-ring in recessive dystrophic epidermolysis bullosa. Anal. Cell. Pathol. 2011, 34, 339–353. [Google Scholar] [CrossRef]
- Hojo, M.; Morimoto, T.; Maluccio, M.; Asano, T.; Morimoto, K.; Lagman, M.; Shimbo, T.; Suthanthiran, M. Cyclosporine in-duces cancer progression by a cell-autonomous mechanism. Nature 1999, 397, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Walsh, S.B.; Xu, J.; Xu, H.; Kurundkar, A.R.; Maheshwari, A.; Grizzle, W.E.; Timares, L.; Huang, C.C.; Kopelovich, L.; Elmets, C.A.; et al. Cyclosporine a mediates pathogenesis of aggressive cutaneous squamous cell carcinoma by augmenting epitheli-al-mesenchymal transition: Role of TGFβ signaling pathway. Mol. Carcinog. 2011, 50, 516–527. [Google Scholar] [CrossRef] [Green Version]
- Condorelli, A.G.; Dellambra, E.; Logli, E.; Zambruno, G.; Castiglia, D. Epidermolysis Bullosa-Associated Squamous Cell Carcinoma: From Pathogenesis to Therapeutic Perspectives. Int. J. Mol. Sci. 2019, 20, 5707. [Google Scholar] [CrossRef] [Green Version]
- Martins, V.L.; Caley, M.P.; Moore, K.; Szentpetery, Z.; Marsh, S.T.; Murrell, D.F.; Kim, M.H.; Avari, M.; McGrath, J.A.; Cerio, R.; et al. Suppression of TGFβ and Angiogenesis by Type VII Collagen in Cutaneous SCC. J. Natl. Cancer Inst. 2016, 108, djv293. [Google Scholar] [CrossRef] [Green Version]
- Que, S.; Zwald, F.; Schmults, C. Cutaneous squamous cell carcinoma: Incidence, risk factors, diagnosis, and staging. J. Am. Acad. Derm. 2018, 78, 237–247. [Google Scholar] [CrossRef] [Green Version]
- Persechino, F.; Ranieri, D.; Guttieri, L.; Nanni, M.; Torrisi, M.; Belleudi, F. Expression Profile of Fibroblast Growth Factor Receptors, Keratinocyte Differentiation Markers, and Epithelial Mesenchymal Transition-Related Genes in Actinic Keratosis: A Possible Predictive Factor for Malignant Progression? Biology 2021, 10, 331. [Google Scholar] [CrossRef]
- Purdie, K.J.; Pourreyron, C.; Fassihi, H.; Cepeda-Valdes, R.; Frew, J.W.; Volz, A.; Weissenborn, S.J.; Pfister, H.; Proby, C.M.; Bruckner-Tuderman, L.; et al. No Evidence That Human Papillomavirus Is Responsible for the Aggressive Nature of Recessive Dystrophic Epidermolysis Bullosa–Associated Squamous Cell Carcinoma. J. Investig. Dermatol. 2010, 130, 2853–2855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuentes, I.; Guttmann-Gruber, C.; Tay, A.S.L.; Hofbauer, J.P.; Denil, S.L.J.; Reichelt, J.; Palisson, F.; Common, J.E.; South, A.P. Reduced Microbial Diversity Is a Feature of Recessive Dystrophic Epidermolysis Bullosa-Involved Skin and Wounds. J. Investig. Dermatol. 2018, 138, 2492–2495. [Google Scholar] [CrossRef] [Green Version]
- Cho, R.J.; Alexandrov, L.B.; den Breems, N.Y.; Atanasova, V.S.; Farshchian, M.; Purdom, E.; Nguyen, T.N.; Coarfa, C.; Rajapakshe, K.; Prisco, M.; et al. APOBEC mutation drives early-onset squamous cell carcinomas in recessive dystrophic epi-dermolysis bullosa. Sci. Transl. Med. 2018, 10, eaas9668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paver, E.C.; Currie, A.M.; Gupta, R.; Dahlstrom, J.E. Human papilloma virus related squamous cell carcinomas of the head and neck: Diagnosis, clinical implications and detection of HPV. Pathology 2019, 52, 179–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannataro, V.L.; Gaffney, S.G.; Sasaki, T.; Issaeva, N.; Grewal, N.; Grandis, J.R.; Yarbrough, W.G.; Burtness, B.; Anderson, K.S.; Townsend, J.P. APOBEC-induced mutations and their cancer effect size in head and neck squamous cell carcinoma. Oncogene 2019, 38, 3475–3487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watt, S.A.; Pourreyron, C.; Purdie, K.; Hogan, C.; Cole, C.L.; Foster, N.; Pratt, N.; Bourdon, J.C.; Appleyard, V.; Murray, K.; et al. Integrative mRNA profiling comparing cultured primary cells with clinical samples reveals PLK1 and C20orf20 as therapeutic targets in cutaneous squamous cell carcinoma. Oncogene 2011, 30, 4666–4677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stathias, V.; Turner, J.; Koleti, A.; Vidovic, D.; Cooper, D.; Fazel-Najafabadi, M.; Pilarczyk, M.; Terryn, R.; Chung, C.; Umeano, A.; et al. LINCS Data Portal 2.0: Next generation access point for perturbation-response signatures. Nucleic Acids Res. 2020, 48, D431–D439. [Google Scholar] [CrossRef] [Green Version]
- Hameetman, L.; Commandeur, S.; Bavinck, J.N.B.; Wisgerhof, H.C.; De Gruijl, F.R.; Willemze, R.; Mullenders, L.; Tensen, C.P.; Vrieling, H. Molecular profiling of cutaneous squamous cell carcinomas and actinic keratoses from organ transplant recipients. BMC Cancer 2013, 13, 58. [Google Scholar] [CrossRef] [Green Version]
- Ramos, M.; Geistlinger, L.; Oh, S.; Schiffer, L.; Azhar, R.; Kodali, H.; de Bruijn, I.; Gao, J.; Carey, V.J.; Morgan, M.; et al. Mul-tiomic Integration of Public Oncology Databases in Bioconductor. JCO Clin. Cancer Inform. 2020, 4, 958–971. [Google Scholar] [CrossRef] [PubMed]
- Duan, Q.; Reid, S.P.; Clark, N.R.; Wang, Z.; Fernandez, N.F.; Rouillard, A.D.; Readhead, B.; Tritsch, S.R.; Hodos, R.; Hafner, M.; et al. L1000CDS2: LINCS L1000 characteristic direction signatures search engine. NPJ Syst. Biol. Appl. 2016, 2, 16015. [Google Scholar] [CrossRef]
- Harder, A. MEK inhibitors—Novel targeted therapies of neurofibromatosis associated benign and malignant lesions. Biomark. Res. 2021, 9, 26. [Google Scholar] [CrossRef]
- Alitalo, A.K.; Proulx, S.; Karaman, S.; Aebischer, D.; Martino, S.; Jost, M.; Schneider, N.; Bry, M.; Detmar, M. VEGF-C and VEGF-D Blockade Inhibits Inflammatory Skin Carcinogenesis. Cancer Res. 2013, 73, 4212–4221. [Google Scholar] [CrossRef] [Green Version]
- Kiss, E.A.; Saharinen, P. Anti-angiogenic Targets: Angiopoietin and Angiopoietin Receptors. Tumor Angiogenesis 2020, 24, 227–250. [Google Scholar] [CrossRef]
- Peguera, A.B.; Simón-Extremera, P.; Da Silva-Diz, V.; De Munain, M.L.; Díaz-Gil, L.; Penin, R.M.; González-Suárez, E.; Sidelnikova, D.P.; Bermejo, O.; Viñals, J.M.; et al. PDGFR-induced autocrine SDF-1 signaling in cancer cells promotes metastasis in advanced skin carcinoma. Oncogene 2019, 38, 5021–5037. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Pyon, J.; Lee, S.; Lee, Y.; Kang, S.; Kim, C.; Kim, D.; Nam, H.; Park, Y.; Jeong, D.; et al. Greater expression of TC21/R-ras2 in highly aggressive malignant skin cancer. Int. J. Derm. 2011, 50, 956–960. [Google Scholar] [CrossRef]
- Furukawa, T.; Kanai, N.; Shiwaku, H.; Soga, N.; Uehara, A.; Horii, A. AURKA is one of the downstream targets of MAPK1/ERK2 in pancreatic cancer. Oncogene 2006, 25, 4831–4839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Zhou, L.; Xie, N.; Nice, E.C.; Zhang, T.; Cui, Y.; Huang, C. Overcoming cancer therapeutic bottleneck by drug repurposing. Signal Transduct. Target. Ther. 2020, 5, 113. [Google Scholar] [CrossRef]
- Roessler, H.I.; Knoers, N.; van Haelst, M.M.; van Haaften, G. Drug Repurposing for Rare Diseases. Trends Pharm. Sci. 2021, 42, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Fania, L.; Didona, D.; Di Pietro, F.; Verkhovskaia, S.; Morese, R.; Paolino, G.; Donati, M.; Ricci, F.; Coco, V.; Ricci, F.; et al. Cutaneous Squamous Cell Carcinoma: From Pathophysiology to Novel Therapeutic Approaches. Biomedicines 2021, 9, 171. [Google Scholar] [CrossRef]
- Hall-Jackson, C.A.; Eyers, P.A.; Cohen, P.; Goedert, M.; Boyle, F.T.; Hewitt, N.; Plant, H.; Hedge, P. Paradoxical activation of Raf by a novel Raf inhibitor. Chem. Biol. 1999, 6, 559–568. [Google Scholar] [CrossRef]
- Escuin-Ordinas, H.; Li, S.; Xie, M.W.; Sun, L.; Hugo, W.; Huang, R.R.; Jiao, J.; De-Faria, F.M.; Realegeno, S.; Krystofinski, P.; et al. Cutaneous wound healing through paradoxical MAPK activation by BRAF inhibitors. Nat. Commun. 2016, 7, 12348. [Google Scholar] [CrossRef]
- Mellerio, J.E.; Robertson, S.; Bernardis, C.; Diem, A.; Fine, J.D.; George, R.S.; Goldberg, D.; Halmos, G.; Harries, M.; Jonkman, M.F.; et al. Management of cutaneous squamous cell carcinoma in patients with epidermolysis bullosa: Best clinical practice guidelines. Br. J. Dermatol. 2015, 174, 56–67. [Google Scholar] [CrossRef] [Green Version]
- Medek, K.; Koelblinger, P.; Koller, J.; Diem, A.; Ude-Schoder, K.; Bauer, J.W.; Laimer, M. Wound healing deficits in severe generalized recessive dystrophic epidermolysis bullosa along anticancer treatment with cetuximab. J. Dtsch. Dermatol. Ges. 2019, 17, 448–450. [Google Scholar] [CrossRef]
- Livingstone, E.; Zimmer, L.; Vaubel, J.; Schadendorf, D. Current advances and perspectives in the treatment of advanced melanoma. J. Dtsch. Dermatol. Ges. 2012, 10, 319–325. [Google Scholar] [CrossRef]
- Athuluri-Divakar, S.K.; Vasquez-Del Carpio, R.; Dutta, K.; Baker, S.J.; Cosenza, S.C.; Basu, I.; Gupta, Y.K.; Reddy, M.V.R.; Ueno, L.; Hart, J.R.; et al. A Small Molecule RAS-Mimetic Disrupts RAS Association with Effector Proteins to Block Signaling. Cell 2016, 165, 643–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atanasova, V.S.; Pourreyron, C.; Farshchian, M.; Lawler, M.; Brown, C.A.; Watt, S.A.; Wright, S.; Warkala, M.; Guttmann-Gruber, C.; Hofbauer, J.P.; et al. Identification of Rigosertib for the Treatment of Recessive Dystrophic Epidermolysis Bullosa-Associated Squamous Cell Carcinoma. Clin. Cancer Res. 2019, 25, 3384–3391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and neck squamous cell carcinoma. Nat. Rev. Dis. Primers 2020, 6, 92. [Google Scholar] [CrossRef]
- Onoufriadis, A.; Proudfoot, L.E.; Ainali, C.; Torre, D.; Papanikolaou, M.; Rayinda, T.; Rashidghamat, E.; Danarti, R.; Mellerio, J.E.; Ma’Ayan, A.; et al. Transcriptomic profiling of recessive dystrophic epidermolysis bullosa wounded skin highlights drug repurposing opportunities to improve wound healing. Exp. Dermatol. 2021, 10, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.-H.; Keam, B.; Ahn, Y.-O.; Park, H.-R.; Kim, M.; Kim, T.M.; Kim, D.-W.; Heo, D.S. Inhibition of MEK with trametinib enhances the efficacy of anti-PD-L1 inhibitor by regulating anti-tumor immunity in head and neck squamous cell carcinoma. Oncoimmunology 2019, 8, e1515057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Feng, X.; Mintz, A.; Petty, W.J.; Hsu, W. Regulation of brachyury by fibroblast growth factor receptor 1 in lung cancer. Oncotarget 2016, 7, 87124–87135. [Google Scholar] [CrossRef]
- Giuliani, N.; Lunghi, P.; Morandi, F.; Colla, S.; Bonomini, S.; Hojden, M.; Rizzoli, V.; Bonati, A. Downmodulation of ERK protein kinase activity inhibits VEGF secrection by human myeloma cells and myeloma-induced angiogenesis. Leukemia 2004, 18, 628–635. [Google Scholar] [CrossRef] [Green Version]
- Xiao, T.; Xiao, Y.; Wang, W.; Tang, Y.Y.; Xiao, Z.; Su, M. Targeting EphA2 in cancer. J. Hematol. Oncol. 2020, 13, 114. [Google Scholar] [CrossRef]
- O’Donovan, S.M.; Imami, A.; Eby, H.; Henkel, N.D.; Creeden, J.F.; Asah, S.; Zhang, X.; Wu, X.; Alnafisah, R.; Taylor, R.T.; et al. Identification of candidate repurposable drugs to combat COVID-19 using a signature-based approach. Sci. Rep. 2021, 11, 4495. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Bai, H. Integrating LINCS Data to Evaluate Cancer Transcriptome Modifying Potential of Small-molecule Compounds for Drug Repositioning. Comb. Chem. High Throughput Screen. 2021, 24, 1340–1350. [Google Scholar] [CrossRef]
- Tippmann, S. Programming tools: Adventures with R. Nature 2015, 517, 109–110. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Drai, D.; Elmer, G.; Kafkafi, N.; Golani, I. Controlling the false discovery rate in behavior genetics research. Behav. Brain Res. 2001, 125, 279–284. [Google Scholar] [CrossRef] [Green Version]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, W.; Brouwer, C. Pathview: An R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics 2013, 29, 1830–1831. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Data Set | Type | Samples Included | Platform | Reference |
|---|---|---|---|---|
| RDEB (GSE111582) | tissue | SCC (n = 8) | RNA-seq, Illumina HiSeq | [16] |
| control (n = 9) | ||||
| OTR (GSE32979) | tissue | SCC (n = 15) | Array, Illumina human-6 v2.0 expression beadchip | [21] |
| control (n = 13) | ||||
| HN (TCGA) | tissue | SCC (n = 263) | RNA-seq | [22] |
| control (n = 31) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zauner, R.; Wimmer, M.; Dorfer, S.; Ablinger, M.; Koller, U.; Piñón Hofbauer, J.; Guttmann-Gruber, C.; Bauer, J.W.; Wally, V. Transcriptome-Guided Drug Repurposing for Aggressive SCCs. Int. J. Mol. Sci. 2022, 23, 1007. https://doi.org/10.3390/ijms23021007
Zauner R, Wimmer M, Dorfer S, Ablinger M, Koller U, Piñón Hofbauer J, Guttmann-Gruber C, Bauer JW, Wally V. Transcriptome-Guided Drug Repurposing for Aggressive SCCs. International Journal of Molecular Sciences. 2022; 23(2):1007. https://doi.org/10.3390/ijms23021007
Chicago/Turabian StyleZauner, Roland, Monika Wimmer, Sonja Dorfer, Michael Ablinger, Ulrich Koller, Josefina Piñón Hofbauer, Christina Guttmann-Gruber, Johann W. Bauer, and Verena Wally. 2022. "Transcriptome-Guided Drug Repurposing for Aggressive SCCs" International Journal of Molecular Sciences 23, no. 2: 1007. https://doi.org/10.3390/ijms23021007
APA StyleZauner, R., Wimmer, M., Dorfer, S., Ablinger, M., Koller, U., Piñón Hofbauer, J., Guttmann-Gruber, C., Bauer, J. W., & Wally, V. (2022). Transcriptome-Guided Drug Repurposing for Aggressive SCCs. International Journal of Molecular Sciences, 23(2), 1007. https://doi.org/10.3390/ijms23021007