
Mutations, Genes, and Phenotypes Related to Movement Disorders and Ataxias
 ,
,  add
Show full author list
add
Show full author list
Abstract
1. Introduction
2. Results
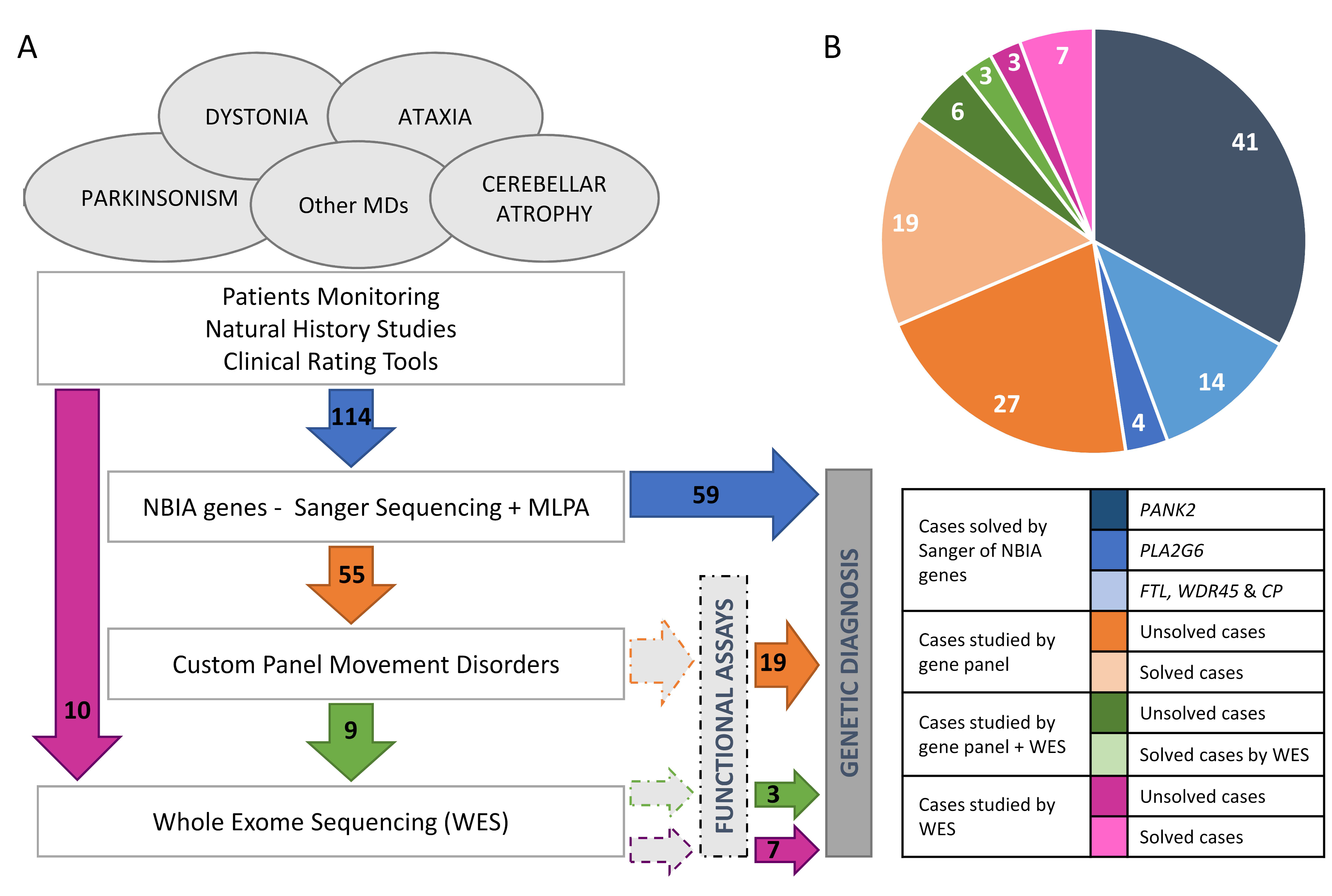
2.1. Genetics
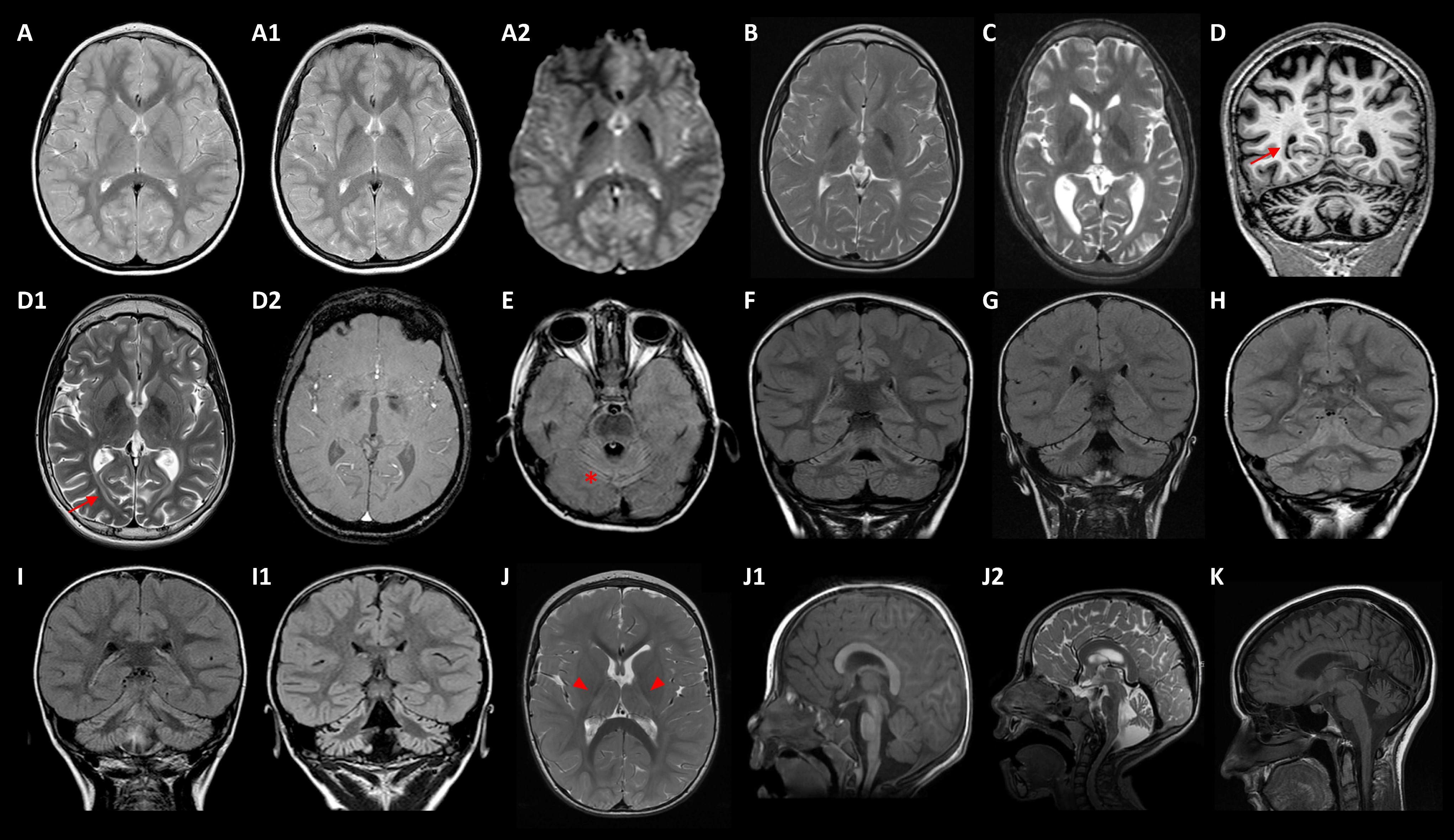
2.2. Genotype-Phenotype Correlation
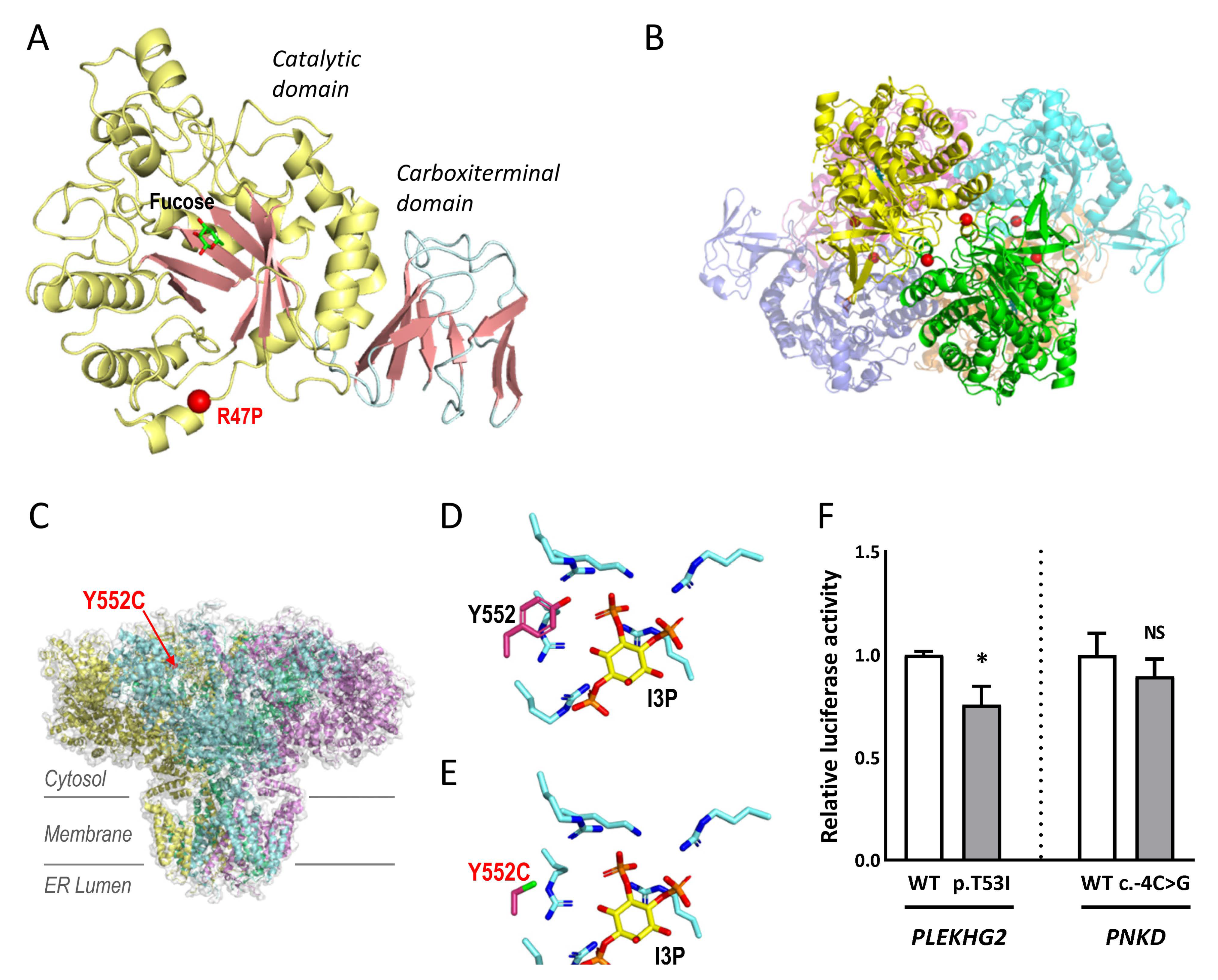
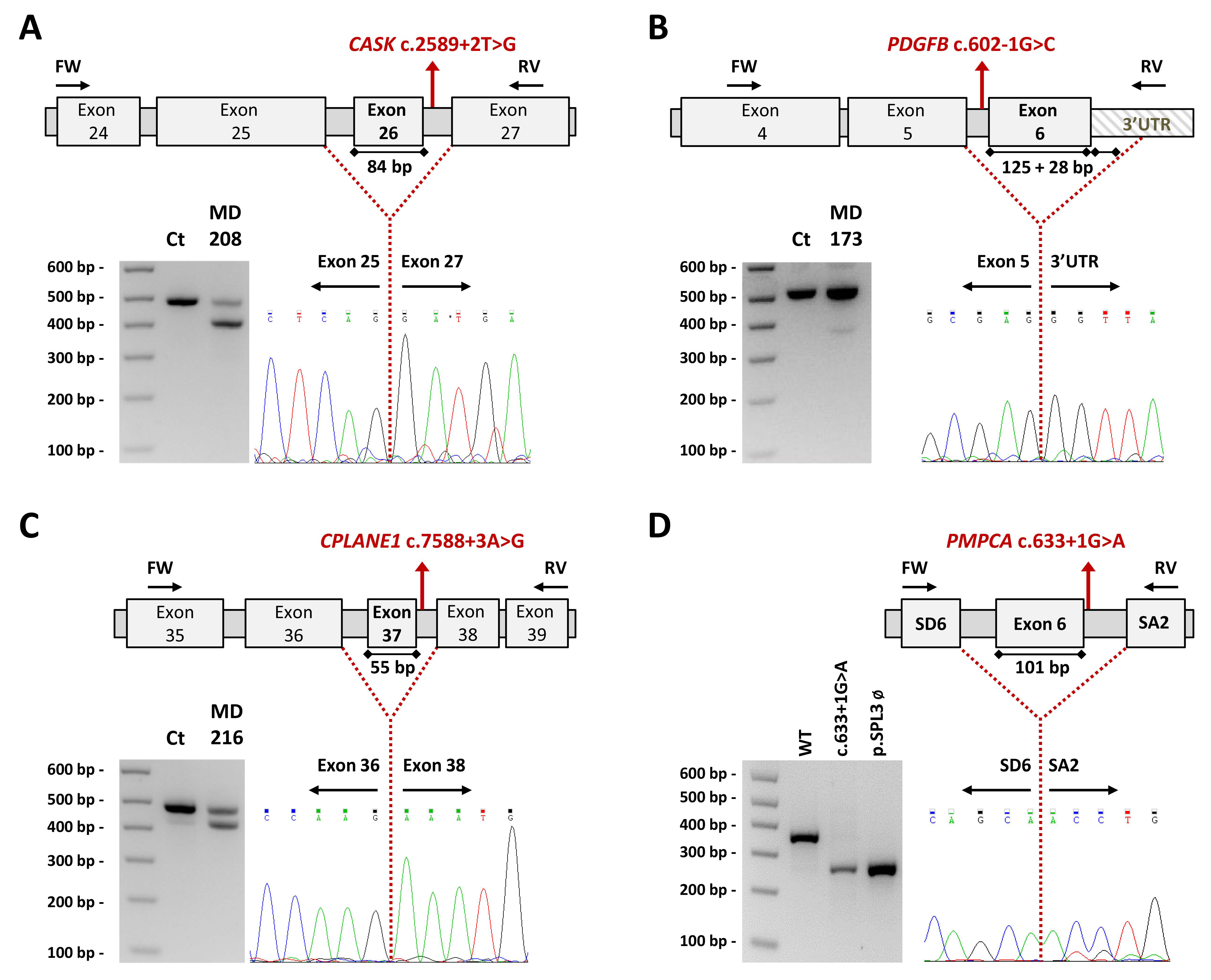
2.3. Studies to Investigate the Pathogenicity of the Novel Variants
3. Discussion
4. Material and Methods
4.1. Patients
4.2. Neuroimaging Approaches
4.3. Genetic and Genomic Studies
4.3.1. Genetic Analysis of NBIA Genes
4.3.2. Gene Panel, WES and Chromosomal Microarray Analysis
4.4. Studies to Investigate the Pathogenicity of Novel Variants
4.4.1. Structural Modelling: ITPR1 and FUCA1
4.4.2. Luciferase Reporter Assays: PLEKHG2 and PNKD
4.4.3. Transcript Analysis and Minigenes: CASK, PDGFB, CPLANE1/C5orf42 and PMPCA
4.4.4. NIPA1 Subcellular Location
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hinarejos, I.; Machuca-Arellano, C.; Sancho, P.; Espinos, C. Mitochondrial Dysfunction, Oxidative Stress and Neuroinflammation in Neurodegeneration with Brain Iron Accumulation (NBIA). Antioxidants 2020, 9, 1020. [Google Scholar] [CrossRef] [PubMed]
- Tello, C.; Darling, A.; Lupo, V.; Pérez-Dueñas, B.; Espinós, C. On the complexity of clinical and molecular bases of neurodegeneration with brain iron accumulation. Clin. Genet. 2018, 93, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Yun, J.Y.; Gregory, A.; Hogarth, P.; Hayflick, S.J. Brain MRI Pattern Recognition in Neurodegeneration with Brain Iron Accumulation. Front. Neurol. 2020, 11, 1024. [Google Scholar] [CrossRef]
- Zhou, B.; Westaway, S.K.; Levinson, B.; Johnson, M.A.; Gitschier, J.; Hayflick, S.J. A novel pantothenate kinase gene (PANK2) is defective in Hallervorden-Spatz syndrome. Nat. Genet. 2001, 28, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.; Aguilera-Albesa, S.; Tello, C.A.; Serrano, M.; Tomas, M.; Camino-Leon, R.; Fernandez-Ramos, J.; Jimenez-Escrig, A.; Poo, P.; O’Callaghan, M.; et al. PLA2G6-associated neurodegeneration: New insights into brain abnormalities and disease progression. Park. Relat. Disord. 2019, 61, 179–186. [Google Scholar] [CrossRef]
- Montaut, S.; Tranchant, C.; Drouot, N.; Rudolf, G.; Guissart, C.; Tarabeux, J.; Stemmelen, T.; Velt, A.; Fourrage, C.; Nitschke, P.; et al. Assessment of a Targeted Gene Panel for Identification of Genes Associated with Movement Disorders. JAMA Neurol. 2018, 75, 1234–1245. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Johnson, A.K.; Nelakuditi, V.; Guidugli, L.; Fischer, D.; Arndt, K.; Ma, L.; Sandford, E.; Shakkottai, V.; Boycott, K.; et al. Targeted exome analysis identifies the genetic basis of disease in over 50% of patients with a wide range of ataxia-related phenotypes. Genet. Med. 2018, 21, 195–206. [Google Scholar] [CrossRef]
- Coutelier, M.; Hammer, M.B.; Stevanin, G.; Monin, M.L.; Davoine, C.S.; Mochel, F.; Labauge, P.; Ewenczyk, C.; Ding, J.; Gibbs, J.R.; et al. Efficacy of Exome-Targeted Capture Sequencing to Detect Mutations in Known Cerebellar Ataxia Genes. JAMA Neurol. 2018, 75, 591–599. [Google Scholar] [CrossRef]
- Elert-Dobkowska, E.; Stepniak, I.; Krysa, W.; Ziora-Jakutowicz, K.; Rakowicz, M.; Sobanska, A.; Pilch, J.; Antczak-Marach, D.; Zaremba, J.; Sulek, A. Next-generation sequencing study reveals the broader variant spectrum of hereditary spastic paraplegia and related phenotypes. Neurogenetics 2019, 20, 27–38. [Google Scholar] [CrossRef]
- D’Amore, A.; Tessa, A.; Casali, C.; Dotti, M.T.; Filla, A.; Silvestri, G.; Antenora, A.; Astrea, G.; Barghigiani, M.; Battini, R.; et al. Next Generation Molecular Diagnosis of Hereditary Spastic Paraplegias: An Italian Cross-Sectional Study. Front. Neurol. 2018, 9, 981. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Jesús, S.; Hinarejos, I.; Carrillo, F.; Martínez-Rubio, M.D.; Macías-García, D.; Sánchez-Monteagudo, A.; Adarmes, A.; Lupo, V.; Pérez-Dueñas, B.; Mir, P.; et al. NR4A2 is involved in dystonia-parkinsonism with intellectual disability, language impairment and motor tics. Neurol. Genet. 2021, 7, e543. [Google Scholar] [CrossRef]
- Sancho, P.; Andrés-Bordería, A.; Gorría-Redondo, N.; Llano, K.; Martínez-Rubio, D.; Yoldi-Petri, M.E.; Blumkin, L.; Rodríguez de la Fuente, P.; Gil-Ortiz, F.; Fernández-Murga, L.; et al. Expanding the β-III spectrin-associated phenotypes toward non-progressive congenital ataxias with neurodegeneration. Int. J. Mol. Sci. 2021, 22, 2505. [Google Scholar] [CrossRef] [PubMed]
- Correa-Vela, M.; Lupo, V.; Montpeyo, M.; Sancho, P.; Marce-Grau, A.; Hernandez-Vara, J.; Darling, A.; Jenkins, A.; Fernandez-Rodriguez, S.; Tello, C.; et al. Impaired proteasome activity and neurodegeneration with brain iron accumulation in FBXO7 defect. Ann. Clin. Transl. Neurol. 2020, 7, 1436–1442. [Google Scholar] [CrossRef]
- Campins-Romeu, M.; Baviera-Munoz, R.; Sastre-Bataller, I.; Bataller, L.; Jaijo, T.; Martinez-Torres, I. Hereditary Spastic Paraplegia 7 Presenting as Multifocal Dystonia with Prominent Cranio-Cervical Involvement. Mov. Disord. Clin. Pract. 2021, 8, 966–968. [Google Scholar] [CrossRef]
- Baviera-Munoz, R.; Martinez-Rubio, D.; Sastre-Bataller, I.; Campins-Romeu, M.; Losada-Lopez, M.; Perez-Garcia, J.; Novella-Maestre, E.; Martinez-Torres, I.; Espinos, C. A 3.9-Mb Deletion on 2p11.2 Comprising the REEP1 Gene Causes Early-Onset Atypical Parkinsonism. Neurol. Genet. 2021, 7, e642. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Rubio, D.; Rodriguez-Prieto, A.; Sancho, P.; Navarro-Gonzalez, C.; Gorria-Redondo, N.; Miquel-Leal, J.; Marco-Marin, C.; Jenkins, A.; Soriano-Navarro, M.; Hernandez, A.; et al. Protein misfolding and clearance in the pathogenesis of a new infantile onset ataxia caused by mutations in PRDX3. Hum. Mol. Genet. 2022, ddac146. [Google Scholar] [CrossRef] [PubMed]
- Bertini, E.; Zanni, G.; Boltshauser, E. Nonprogressive congenital ataxias. Handb Clin. Neurol. 2018, 155, 91–103. [Google Scholar] [PubMed]
- Baviera-Munoz, R.; Campins-Romeu, M.; Carretero-Vilarroig, L.; Sastre-Bataller, I.; Martinez-Torres, I.; Vazquez-Costa, J.F.; Muelas, N.; Sevilla, T.; Vilchez, J.J.; Aller, E.; et al. Clinical and genetic characteristics of 21 Spanish patients with biallelic pathogenic SPG7 mutations. J. Neurol. Sci. 2021, 429, 118062. [Google Scholar] [CrossRef] [PubMed]
- Jonch, A.E.; Douard, E.; Moreau, C.; Van Dijck, A.; Passeggeri, M.; Kooy, F.; Puechberty, J.; Campbell, C.; Sanlaville, D.; Lefroy, H.; et al. Estimating the effect size of the 15q11.2 BP1-BP2 deletion and its contribution to neurodevelopmental symptoms: Recommendations for practice. J. Med. Genet. 2019, 56, 701–710. [Google Scholar] [CrossRef]
- Sulzenbacher, G.; Bignon, C.; Nishimura, T.; Tarling, C.A.; Withers, S.G.; Henrissat, B.; Bourne, Y. Crystal structure of Thermotoga maritima alpha-L-fucosidase. Insights into the catalytic mechanism and the molecular basis for fucosidosis. J. Biol. Chem. 2004, 279, 13119–13128. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.; Baker, M.L.; Wang, Z.; Baker, M.R.; Sinyagovskiy, P.A.; Chiu, W.; Ludtke, S.J.; Serysheva, I.I. Gating machinery of InsP3R channels revealed by electron cryomicroscopy. Nature 2015, 527, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Bosanac, I.; Alattia, J.R.; Mal, T.K.; Chan, J.; Talarico, S.; Tong, F.K.; Tong, K.I.; Yoshikawa, F.; Furuichi, T.; Iwai, M.; et al. Structure of the inositol 1,4,5-trisphosphate receptor binding core in complex with its ligand. Nature 2002, 420, 696–700. [Google Scholar] [CrossRef] [PubMed]
- Edvardson, S.; Wang, H.; Dor, T.; Atawneh, O.; Yaacov, B.; Gartner, J.; Cinnamon, Y.; Chen, S.; Elpeleg, O. Microcephaly-dystonia due to mutated PLEKHG2 with impaired actin polymerization. Neurogenetics 2016, 17, 25–30. [Google Scholar] [CrossRef]
- Zhao, J.; Matthies, D.S.; Botzolakis, E.J.; Macdonald, R.L.; Blakely, R.D.; Hedera, P. Hereditary spastic paraplegia-associated mutations in the NIPA1 gene and its Caenorhabditis elegans homolog trigger neural degeneration in vitro and in vivo through a gain-of-function mechanism. J. Neurosci. 2008, 28, 13938–13951. [Google Scholar] [CrossRef]
- Goytain, A.; Hines, R.M.; El-Husseini, A.; Quamme, G.A. NIPA1(SPG6), the basis for autosomal dominant form of hereditary spastic paraplegia, encodes a functional Mg2+ transporter. J. Biol. Chem. 2007, 282, 8060–8088. [Google Scholar] [CrossRef]
- Sevilla, T.; Martinez-Rubio, D.; Marquez, C.; Paradas, C.; Colomer, J.; Jaijo, T.; Millan, J.M.; Palau, F.; Espinos, C. Genetics of the Charcot-Marie-Tooth disease in the Spanish Gypsy population: The hereditary motor and sensory neuropathy-Russe in depth. Clin. Genet. 2013, 83, 565–570. [Google Scholar] [CrossRef]
- Casana, P.; Martinez, F.; Haya, S.; Lorenzo, J.I.; Espinos, C.; Aznar, J.A. Q1311X: A novel nonsense mutation of putative ancient origin in the von Willebrand factor gene. Br. J. Haematol. 2000, 111, 552–555. [Google Scholar]
- Gil-Pena, H.; Coto, E.; Santos, F.; Espino, M.; Cea Crespo, J.M.; Chantzopoulos, G.; Komianou, F.; Gomez, J.; Alonso, B.; Iglesias, S.; et al. A new SLC12A3 founder mutation (p.Val647Met) in Gitelman’s syndrome patients of Roma ancestry. Nefrologia 2017, 37, 423–428. [Google Scholar] [CrossRef]
- Wu, Y.W.; Hess, C.P.; Singhal, N.S.; Groden, C.; Toro, C. Idiopathic basal ganglia calcifications: An atypical presentation of PKAN. Pediatr. Neurol. 2013, 49, 351–354. [Google Scholar] [CrossRef]
- Dusek, P.; Tovar Martinez, E.M.; Madai, V.I.; Jech, R.; Sobesky, J.; Paul, F.; Niendorf, T.; Wuerfel, J.; Schneider, S.A. 7-Tesla Magnetic Resonance Imaging for Brain Iron Quantification in Homozygous and Heterozygous PANK2 Mutation Carriers. Mov. Disord. Clin. Pract. 2014, 1, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Akcakaya, N.H.; Iseri, S.U.; Bilir, B.; Battaloglu, E.; Tekturk, P.; Gultekin, M.; Akar, G.; Yigiter, R.; Hanagasi, H.; Alp, R.; et al. Clinical and genetic features of PKAN patients in a tertiary centre in Turkey. Clin. Neurol. Neurosurg. 2017, 154, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Yapici, Z.; Akcakaya, N.H.; Tekturk, P.; Iseri, S.A.; Ozbek, U. A novel gene mutation in PANK2 in a patient with severe jaw-opening dystonia. Brain Dev. 2016, 38, 755–758. [Google Scholar] [CrossRef]
- Darling, A.; Tello, C.; Martí, M.J.; Garrido, C.; Aguilera-Albesa, S.; Tomás Vila, M.; Gastón, I.; Madruga, M.; González Gutiérrez, L.; Ramos Lizana, J.; et al. Clinical rating scale for pantothenate kinase-associated neurodegeneration: A pilot study. Mov. Disord. 2017, 32, 1620–1630. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.F.; FitzPatrick, D.R.; Firth, H.V. Paediatric genomics: Diagnosing rare disease in children. Nat. Rev. Genet. 2018, 19, 325. [Google Scholar] [CrossRef]
- Ngo, K.J.; Rexach, J.E.; Lee, H.; Petty, L.E.; Perlman, S.; Valera, J.M.; Deignan, J.L.; Mao, Y.; Aker, M.; Posey, J.E.; et al. A diagnostic ceiling for exome sequencing in cerebellar ataxia and related neurological disorders. Hum. Mutat. 2020, 41, 487–501. [Google Scholar] [CrossRef] [PubMed]
- Djarmati, A.; Svetel, M.; Momcilovic, D.; Kostic, V.; Klein, C. Significance of recurrent mutations in the myofibrillogenesis regulator 1 gene. Arch. Neurol. 2005, 62, 1641. [Google Scholar] [CrossRef] [PubMed]
- Panedey, S.; Ramkumarsingh, L.; Mahadevan, L. Progressive nonparoxysmal chorea and dystonia due to myofibrillogenesis regulator-1 gene mutation. Park. Relat. Disord. 2019, 60, 186–187. [Google Scholar] [CrossRef]
- Bartels, E.; Draaken, B.; Kazmierczak, B.; Spranger, S.; Schramm, C.; Baudisch, F.; Nöthen, M.M.; Schmiedeke, E.; Ludwig, M.; Reutter, H. De novo partial trisomy 18p and partial monosomy 18q in a patient with anorectal malformation. Cytogenet. Genome Res. 2011, 134, 243–248. [Google Scholar]
- Hu, H.; Hao, J.; Yao, H.; Chang, Q.; Li, R.; Zhang, X.; Liang, Z. Prenatal diagnosis of de novo partial trisomy 18p and partial monosomy 18q recurrent in a family with fatal aortic coarctation. Gene 2013, 517, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Gordon, M.F.; Bressman, S.; Brin, M.F.; de Leon, D.; Warburton, D.; Yeboa, K.; Fahn, S. Dystonia in a patient with deletion of 18q. Mov. Disord. 1995, 10, 496–499. [Google Scholar] [CrossRef]
- Gautschi, M.; Merlini, L.; Calza, A.M.; Hayflick, S.; Nuoffer, J.M.; Fluss, J. Late diagnosis of fucosidosis in a child with progressive fixed dystonia, bilateral pallidal lesions and red spots on the skin. Eur. J. Paediatr. Neurol. 2014, 18, 516–519. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, R.; Momoi, T.; Yoshida, A.; Okumura, M.; Yamakura, S.; Takasaki, Y.; Kiyomasu, T.; Yamanaka, C. Type 3 GM1 gangliosidosis: Clinical and neuroradiological findings in an 11-year-old girl. J. Neurol. 1995, 242, 299–303. [Google Scholar] [CrossRef] [PubMed]
- De Grandis, E.; Di Rocco, M.; Pessagno, A.; Veneselli, E.; Rossi, A. MR imaging findings in 2 cases of late infantile GM1 gangliosidosis. AJNR Am. J. Neuroradiol. 2009, 30, 1325–1327. [Google Scholar] [CrossRef]
- Vieira, J.P.; Conceicao, C.; Scortenschi, E. GM1 gangliosidosis, late infantile onset dystonia, and T2 Hypointensity in the globus pallidus and substantia Nigra. Pediatr. Neurol. 2013, 49, 195–197. [Google Scholar] [CrossRef] [PubMed]
- Takenouchi, T.; Kosaki, R.; Nakabayashi, K.; Hata, K.; Takahashi, T.; Kosaki, K. Paramagnetic signals in the globus pallidus as late radiographic sign of juvenile-onset GM1 gangliosidosis. Pediatr. Neurol. 2015, 52, 226–229. [Google Scholar] [CrossRef] [PubMed]
- Zoons, E.; de Koning, T.J.; Abeling, N.G.; Tijssen, M.A. Neurodegeneration with Brain Iron Accumulation on MRI: An Adult Case of alpha-Mannosidosis. JIMD Rep. 2012, 4, 99–102. [Google Scholar] [PubMed]
- Hajirnis, O.; Udwadia-Hegde, A. Chronic GM1 gangliosidosis with characteristic “wish bone sign” on brain MRI. Another type of neurodegeneration with brain iron accumulation? Mov. Disord.Clin. Pract. 2015, 2, 323–325. [Google Scholar] [CrossRef]
- Drecourt, A.; Babdor, J.; Dussiot, M.; Petit, F.; Goudin, N.; Garfa-Traoré, M.; Habarou, F.; Bole-Feysot, C.; Nitschke, P.; Ottolenghi, C.; et al. Impaired transferrin receptor palmitoylation and recycling in neurodegeneration with brain iron accumulation. Am. J. Hum. Genet. 2018, 102, 266–277. [Google Scholar] [CrossRef]
- Kolarova, H.; Tan, J.; Strom, T.M.; Meitinger, T.; Wagner, M.; Klopstock, T. Lifetime risk of autosomal recessive neurodegeneration with brain iron accumulation (NBIA) disorders calculated from genetic databases. EBioMedicine 2022, 77, 103869. [Google Scholar] [CrossRef]
- Dusek, P.; Hofer, T.; Alexander, J.; Roos, P.M.; Aaseth, J.O. Cerebral iron deposition in neurodegeneration. Biomolecules 2022, 12, 714. [Google Scholar] [CrossRef]
- Bolthauser, E.; Schmahmann, J. Cerebellar Disorders in Children. In Clinics in Developmental Medicine, 1st ed.; John Wiley & Sons, I.: Hoboken, NJ, USA, 2012; Volume 191–192, p. 415. [Google Scholar]
- Jobling, R.K.; Assoum, M.; Gakh, O.; Blaser, S.; Raiman, J.A.; Mignot, C.; Roze, E.; Durr, A.; Brice, A.; Levy, N.; et al. PMPCA mutations cause abnormal mitochondrial protein processing in patients with non-progressive cerebellar ataxia. Brain 2015, 138, 1505–1517. [Google Scholar] [CrossRef]
- Romaniello, R.; Pasca, L.; Panzeri, E.; D’Abrusco, F.; Travaglini, L.; Serpieri, V.; Signorini, S.; Aiello, C.; Bertini, E.; Bassi, M.T.; et al. Superior Cerebellar Atrophy: An Imaging Clue to Diagnose ITPR1-Related Disorders. Int. J. Mol. Sci. 2022, 23, 6723. [Google Scholar] [CrossRef]
- Vanegas, M.I.; Marce-Grau, A.; Marti-Sanchez, L.; Mellid, S.; Baide-Mairena, H.; Correa-Vela, M.; Cazurro, A.; Rodriguez, C.; Toledo, L.; Fernandez-Ramos, J.A.; et al. Delineating the motor phenotype of SGCE-myoclonus dystonia syndrome. Park. Relat. Disord. 2020, 80, 165–174. [Google Scholar] [CrossRef]
- Bienert, S.; Waterhouse, A.; de Beer, T.A.; Tauriello, G.; Studer, G.; Bordoli, L.; Schwede, T. The SWISS-MODEL Repository-new features and functionality. Nucleic Acids Res. 2017, 45, D313–D319. [Google Scholar] [CrossRef] [PubMed]
- Runne, C.; Chen, S. PLEKHG2 promotes heterotrimeric G protein beta gamma-stimulated lymphocyte migration via Rac and Cdc42 activation and actin polymerization. Mol. Cell. Biol. 2013, 33, 4294–4307. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sanchez-Monteagudo, A.; Alvarez-Sauco, M.; Sastre, I.; Martinez-Torres, I.; Lupo, V.; Berenguer, M.; Espinós, C. Genetics of Wilson disease and Wilson-like phenotype in a clinical series from eastern Spain. Clin. Genet. 2020, 97, 758–763. [Google Scholar] [CrossRef]
- Austin, C.P.; Cutillo, C.M.; Lau, L.P.L.; Jonker, A.H.; Rath, A.; Julkowska, D.; Thomson, D.; Terry, S.F.; de Montleau, B.; Ardigo, D.; et al. International Rare Diseases Research, C. Future of Rare Diseases Research 2017–2027: An IRDiRC Perspective. Clin. Transl. Sci. 2018, 11, 21–27. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Gene | RefSeq | Position | DNA Change | Protein Change | Prediction ◊ | rs number (MAF) ǂ | References PMID § | Method |
|---|---|---|---|---|---|---|---|---|---|
| MD-208 | CASK | NM_003688.3 | X:41383202 | c.2589+2T>G | ― | P | NA | None | Gene Panel |
| MD-216 | CPLANE1 | NM_023073.3 | 5:37201821 | c.3379T>G | p.S1127A | LP | rs776423792 (1.193 × 10−5) | None | Gene Panel |
| 5:37164372 | c.7588+3A>G | ― | P | NA | None | ||||
| MD-122 | EXOSC3 | NM_013042.3 | 9:37783990 | c.395A>C | p.D132A | P | rs141138948 (4.067 × 10−4) | 22544365, 23564332, 23564332, 23975261, 24524299, 25533962, 27777260, 28687512, 30950035, 31692161 | Gene Panel |
| HOMOZYGOSIS (No Consanguinity) | |||||||||
| MD-012 | EXOSC3 | NM_013042.3 | 9:37783990 | c.395A>C | p.D132A | P | rs141138948 (4.067 × 10−4) | 22544365, 23564332, 23564332, 23975261, 24524299, 25533962, 27777260, 28687512, 30950035, 31692161 | Gene Panel |
| HOMOZYGOSIS (No Consanguinity) | |||||||||
| MD-018 # | FBXO7 | NM_012179.3 | 22:32875213 | c.368C>G | p.S123 * | P | NA | 32767480 | Gene Panel |
| HOMOZYGOSIS (Consanguinity) | |||||||||
| MD-137 | FUCA1 | NM_000147.4 | 1:24194637 | c.140G>C | p.R47P | LP | NA | None | Gene Panel |
| HOMOZYGOSIS (Consanguinity) | |||||||||
| MD-020 | GLB1 | NM_000404.3 | 3:33114105 | c.176G>A | p.R59H | P | rs72555392 (3.607 × 10−5) | 10338095, 17664528, 28939701, 31761138 | Gene Panel |
| 3:33114174 | c.107A>G | p.Y36C | P | rs748345527 (4.809 × 10−5) | None | ||||
| MD-270 | HEXA | NM_000520.5 | 15:72646027 | c.459+5G>A | ― | P | rs762060470 (2.829 × 10−5) | 1837283, 25525159 | Gene Panel |
| 15:72638893 | c.1305C>T | ― | P | rs587779406 (8.488 × 10−5) | 20363167, 25606403 | ||||
| MD-320 | ITPR1 | NM_00116872.1 | 3:4687357 | c.800C>T | p.T267M | P | rs797044955 (NA) | 24091540, 28659154, 29878067, 29925855, 29925855, 30842224, 31632679, 32695065 | WES-proband |
| MD-106 | ITPR1 | NM_00116872.1 | 3:4687362 | c.805C>T | p.R269W | P | NA | 27062503, 25533962, 28826917, 28191890, 28135719, 28659154, 29925855 | Gene Panel |
| MD-041 | ITPR1 | NM_00116872.1 | 3:4706967 | c.1655A>G | p.Y552C | LP | NA | None | Gene Panel |
| MD-200 | KIF1A | NM_001244008.1 | 2:241725854 | c.506C>G | p.R169T | LP | NA | 34121983 | Gene Panel |
| MD-189 | KIF1A | NM_001244008.1 | 2:241715280 | c.946C>T | p.R316W | P | NA | 25265257, 26354034, 28554332 | Gene Panel |
| MD-299 | KIF1A | NM_001244008.1 | 2:241715280 | c.946C>T | p.R316W | P | NA | 25265257, 26354034, 28554332 | WES-proband |
| MD-319 | LRRK2 | NM_198578.4 | 12:40734202 | c.6055G>A | p.G2019S | P | rs34637584 (4.884 × 10−4) | 15726496, 16102999, 16333314, 16750377, 16966501, 17060589, 17116211, 17210620, 17200152, 19072560, 20008657, 19283415, 21686713, 19302196, 19741132 σ | WES-proband |
| HOMOZYGOSIS (Consanguinity) | |||||||||
| MD-277 # | NR4A2 | NM_006186.4 | 2:157184954 | c.956G>A | p.R319Q | LP | NA | 33585677 | WES-trio |
| MD-173 | PDGFB | NM_002608.3 | 22:39621853 | c.602-1G>C | ― | P | NA | None | Gene Panel |
| MD-252 | PNKD | NM_015488.4 | 2:219187987 | c.-4C>G | ― | LP | rs1461115674 (8.152 × 10−6) | None | Gene Panel |
| MD-341 | PLA2G6 | NM_003560 | 22:38519251 | c.1442T>A | p.L481Q | LP | rs587784330 (5.922 × 10−6) | 16783378, 24870368, 25164370 | WES-proband |
| HOMOZYGOSIS (Consanguinity) | |||||||||
| MD-181 | PLEKHG2 | NM_022835.2 | 19:39905680 | c.158C>T | p.T53I | P | NA | None | WES-trio |
| HOMOZYGOSIS (Consanguinity) | |||||||||
| MD-323 | PMPCA | NM_015160.2 | 9:139310844 | c.633+1G>A | --- | P | rs1442110087 (7.961 × 10−6) | None | WES-proband |
| 9:139306513 | c.136T>C | p.S46P | LP | NA | None | ||||
| MD-174 # | PRDX3 | NM_006793.5 | 10:120931956 | c.489C>G | p.R163E | P | NA | 35766882 | WES-trio |
| HOMOZYGOSIS (Consanguinity) | |||||||||
| MD-296 | QARS1 | NM_005051 | 3:49138870 | c.794G>A | p.R265H | LP | rs916890735 (6.575 × 10−6) | None | WES-proband |
| HOMOZYGOSIS (Consanguinity) | |||||||||
| MD-126 # | REEP1 | arr[hg19]2p11.2 (chr2:83,335,425-87,271,924; Hg 19) | P | ― | 22062632, 24986827 | Gene Panel | |||
| MD-307 | RPGRIP1L | NM_015272 | 16:53720424 | c.697A>T | p.K233* | P | rs121918197 (2.788 × 10−5) | 17558409, 25525159 | WES-proband |
| 16:53686828 | c.1769_1770delCT | p.S590Cfs* | P | NA | None | ||||
| MD-159 # | SPG7 | NM_003119.3 | 16:89613145 | c.1529C>T | p.A510V | LP | rs61755320 (2.899 × 10−5) | 18799786, 20981092, 20186691, 21623769, 22995991, 23269439, 22571692, 25133958, 25525159, 26626314, 29057857, 27957547, 29026558, 28362824, 28832565, 29482223, 29913018, 30369941, 31433872, 30098094, 31316545, 29915382, 30537300, 31692161 | Gene Panel |
| 16:89616953 | c.1715C>T | p.A572V | P | rs72547551 (3.537 × 10−5) | 14985266, 25681447, 29482223 | ||||
| MD-219 # | SPTBN2 | NM_006946.3 | 11:66483417 | c.193A>G | p.K65E | P | NA | 33801522 | Gene Panel |
| MD-207 # | SPTBN2 | NM_006946.3 | 11:66481110 | c.764A>G | p.D255G | P | NA | 33801522 | Gene Panel |
| MD-153 | TPP1 | NM_000391.3 | 11:6636487 | c.1340G>A | p.R447H | P | rs119455956 (7.953 × 10−5) | 10330339, 19038966, 20340139, 26143525, 29655203 | Gene Panel |
| 11:6640007 | c.229G>C | p.G77R | P | rs121908195 (7.969 × 10−5) | 26633542, 31741823 | ||||
| Patient Sex | Gene | Origin | Presentation Inheritance | Disease (OMIM) | Age of Onset Age at Testing | Early Symptoms | Brain MRI | Additional Clinical Features |
|---|---|---|---|---|---|---|---|---|
| MD-208 Female | CASK | Spain | de novo XLD | Mental retardation and microcephaly with pontine and cerebellar hypoplasia (300749) | 1 mo 3 yo | Hypotonia Microcephaly DD | Severe PCH | Mild limb dystonia, choreoathetosis, progressive scoliosis from 3 yo |
| MD-216 Male | CPLANE1 | Morocco | Familial AR | Joubert syndrome 17 (614615) | 1 yo 10 yo | DD Hypotonia, ataxia OMA | Molar tooth sign Cerebellar hypoplasia | ID, ataxia and OMA improve with age |
| MD-122 Female | EXOSC3 | Spain | Sporadic AR | Pontocerebellar hypoplasia type 1B (614678) | 3 mo 3 yo | Hypotonia, axial hyperextension, strabismus | Progressive vermis atrophy | Spasticity progressing to flaccid paralysis from 3-yo, g-tube feeding, neurogenic EMG pattern, exitus at 4 yo |
| MD-012 Male | EXOSC3 | Spain | Sporadic AR | 4 mo 8 yo | Hypotonia, DD | Progressive vermis atrophy | Spasticity from 4 yo, axonal neuropathy with loss of tendon reflexes from 5 yo, g-tube feeding, limb dystonia from 6 yo | |
| MD-018 # Female | FBXO7 | Morocco | Sporadic AR | Parkinson disease 15 (260300) | 2 yo 21 yo | Mild DD, infection-triggered acute ataxia at 2 yo | CA from 10 yo Brain iron deposits from 15 yo | Absence epilepsy from 5 yo, acute encephalopathy at 12 yo, progressive deterioration, spastic paraparesis, drug-resistant seizures, optic neuropathy, parkinsonism from 15 yo |
| MD-137 Male | FUCA1 | Greece | Sporadic AR | Fucosidosis (230000) | 3 yo 10 yo | Speech difficulties, gait disturbances | GP T2 hypointensity from 10 yo | Static encephalopathy, progression of gait disorder with dystonia, ID, dysmorphic features, ADHD symptoms, motor stereotypes |
| MD-020 Female | GLB1 | Spain | Sporadic AR | GM1-gangliosidosis (230500, 230600, 230650) | 3 yo 12 yo | Speech difficulties, motor deterioration | Brain iron deposits from 10 yo | Oromandibular dystonia, dysarthria, neuropathy, spasticity, scoliosis |
| MD-270 Female | HEXA | Spain | NA AR | GM2-gangliosidosis (272800) | 26 yo 23 yo | DD, hypotonia | CA | Cerebellar syndrome, stereotyped behaviour, progressive cognitive decline |
| MD-320 Male | ITPR1 | Senegal | de novo AD | SCA15 (606658) SCA29 congenital nonprogressive (117360) | 3 mo 5 yo | DD, hypotonia, gaze-evoked nystagmus | CA Cerebellar cortical hyperintensity from 3 yo (upper > lower hemispheres) | Ataxia, NPCA, strabismus, ID, mild lower limbs spasticity |
| MD-106 Male | ITPR1 | Morocco | de novo AD | <1 yo 6 yo | DD, hypotonia | CA Cerebellar cortical hyperintensity from 3 yo (upper > lower hemispheres) | Ataxia, NPCA, strabismus, gaze-evoked nystagmus, ID, lower limbs spasticity | |
| MD-041 Male | ITPR1 | Spain | de novo AD | 2 mo 3 yo | Gaze-evoked nystagmus, hypotonia | CA Cerebellar cortical FLAIR hyperintensity at age of 4 (upper > lower hemispheres) | Ataxia, NPCA, strabismus, abnormal ocular movements, ID | |
| MD-200 Male | KIF1A | Spain | de novo AD | NESCAV syndrome (614255) | <1 yo 12 yo | DD, microcephaly, nystagmus | CA | Ataxia, NPCA, lower limbs spasticity, seizures (4 yo), dysphagia, intellectual disability, axonal neuropathy, optic atrophy (9 yo) |
| MD-189 Male | KIF1A | Portugal | de novo AD | SPG30 (610357) | 12 mo 22 yo | DD, visual deficit | CA Brain iron deposits from 17 yo | Ataxia, NPCA, optic atrophy, stuttering (2 yo), lower limbs spasticity (3 yo), cognitive regression (4 yo), peripheral neuropathy, seizures (14 yo) |
| MD-299 Male | KIF1A | Spain | de novo AD | SPG30 (610357) | 11 mo 9 yo | DD, hypotonia | CA Cerebellar cortical and deep WM FLAIR hyperintensity | Ataxia (2 yo), NPCA, acquired microcephaly, lower limbs spasticity (3 yo), cognitive impairment, no regression |
| MD-319 Male | LRRK2 | Morocco | Sporadic AR | Parkinson disease (607060) | 5 yo 14 yo | Tremor | Normal | Hands tremor, cervical tremor, mild bradykinesia, hoarseness voice (13 yo) |
| MD-277 # Male | NR4A2 | Spain | de novo AD | AD early-onset dystonia-parkinsonism with intellectual disability (601828) | 12 yo 29 yo | Mild DD from 1 yo | Normal | Borderline IQ (77; 7 yo), motor tics from 16-yo, dystonia and parkinsonism from 28 yo |
| MD-173 Female | PDGFB | Spain | Familial AD | Basal ganglia calcification idiopathic 5 (615483) | 19 yo 19 yo | Postural and intentional tremor | Calcifications | Tremor |
| MD-252 Male | PNKD | Spain | Familial? AD | Paroxysmal nonkinesigenic dyskinesia 1 (118800) | 49 yo 49 yo | Paroxysmal dystonia | Normal | Dystonia, parkinsonism |
| MD-341 Female | PLA2G6 | Morocco | Sporadic AR | Infantile neuroaxonal dystrophy 1 (256600) NBIA2B (610217) Parkinson disease 14 (612953) | 12 mo 5 yo | Ataxic gait, DD | CA Cerebellar cortical FLAIR hyperintensity | Ataxia, dysmetria, hypotonia, spasticity, axonal motor impairment, optic atrophy, intellectual disability |
| MD-181 Male | PLEKHG2 | Spain | Sporadic AR | Leukodystrophy and acquired microcephaly with or without dystonia (616763) | 1 mo 6 yo | Hypotonia, DD | Thalamic lesions CA | Spastic-dystonic tetraparesia from 6 mo, bilateral cataracts, OMA, neuropathy |
| MD-323 Male | PMPCA | Spain | Sporadic AR | SCAR2 (213200) | 21 mo 9 yo | Motor development delay, ataxia, nystagmus | CA Cerebellar cortical hyperintensity | Ataxia, NPCA, mild cognitive impairment |
| MD-174 # Male | PRDX3 | Morocco | Sporadic AR | NA | 2 yo 3 yo | Non-triggered acute cerebellar syndrome | Rapid progression of CA | Ataxia, chronic cerebellar syndrome following acute onset, demyelinating neuropathy from 5 yo |
| MD-296 Male | QARS1 | Morocco | Sporadic AR | Microcephaly, progressive seizures, and cerebral and cerebellar atrophy (615760) | 18 mo 13 yo | Febrile seizures, DD, atypical absences | CA | ID, language impairment, drug-resistant epilepsy with non-motor and motor seizures |
| MD-126 # Female | REEP1 | Spain | de novo AD | SPG31 (610250) | 33 yo 35 yo | Parkinsonism | Calcifications | Parkinsonism, tremor, spasticity, dystonia, slow saccades, dysarthria |
| MD-307 Male | RPGRIP1L | Spain | Sporadic AR | COACH syndrome (216360) Joubert syndrome 7 (611560) Meckel syndrome 5 (611561) | 1 mo 1 yo | Hypotonia, nystagmus, strabismus | PCH | Profound DD, g-tube feeding, awake apneas, seizures, dyskinesia, exitus at 24 mo |
| MD-159 # Male | SPG7 | Spain | Sporadic AR | SPG7 (607259) | 17 yo 26 yo | Dystonia, postural instability, dysarthria | CA | Mild dystonic gait with mild spastic-ataxia gait, dysmetric saccades, gaze-evoked nystagmus, intention tremor |
| MD-219 # Male | SPTBN2 | Spain | de novo AD | SCA5 (600224) | 4 mo 11 yo | Hypotonia, transient upgaze deviation | Severe CA Cerebellar cortical hyperintensity | Ataxia, NPCA, moderate ID |
| MD-207 # Male | SPTBN2 | Spain | de novo AD | SCA5 (600224) | 12 mo 8 yo | Motor delay, DD | Severe CA Cerebellar cortical hyperintensity | Ataxia, NPCA, ADHD, borderline IQ |
| MD-153 Male | TPP1 | Cuba | Sporadic AR | Ceroid lipofuscinosis neuronal 2 (204500) SCA7 (609270) | 5 yo 13 yo | Speech delay, stuttering, motor clumsiness | GP FLAIR low signal, cerebral and CA, white matter T2-high signal | Limb and orofacial dystonia from 11 yo, spasticity, cognitive decline, no seizures |
| Patient Sex | Gene | Candidate Mutation rs Number (MAF) § | Onset Status Inheritance | Disease (OMIM) Inheritance | Main Clinical Features | References PMID § | Observations |
|---|---|---|---|---|---|---|---|
| MD-168 Male | NIPA1 | arr[hg19] 15q11.2(22,770,421–23,277,436)×1 507 Kb deletion that includes seven genes: TUBGCP5, CYFIP1, NIPA2, NIPA1, LOC283683, WHAMMP3, GOLGA8I | 2 yo Sporadic case Heterozygosis AD | The 15q11.2 BP1-BP2 microdeletion syndrome (615656) AD | Autism, obsessive phobic disorder, language delay, dysarthria, severe hypermetropia, generalised dystonia | 31451536, 25689425, 30909440, 30542208, 19328872, 21359847, 28387067, 32117010 | The 15q11.2 BP1-BP2, although associated with neurodevelopmental disorders in multiple papers, should not be considered as a disease causing mutation, since this microdeletion or reported microduplication may explain only a small fraction of the clinical phenotype |
| MD-143 Male | NIPA1 | arr[hg19] 15q11.2(22,770,421–23,214,655)×1 444 Kb deletion that includes six genes: TUBGCP5, CYFIP1, NIPA2, NIPA1, LOC283683, WHAMMP3 | 73 yo Sporadic case Heterozygosis AD | Chorea, parkinsonism | |||
| MD-232 Male | NIPA1 | c.272G>C (p.P91R) novel | 12 yo Sporadic case Heterozygosis AD | Spastic paraplegia 6 (600363) AD | GP T2 hypointensity, lower limb spasticity-dystonia, intellectual disability, sensorineural deafness | novel | The proband also had a 18p duplication and a 18q deletion with unclear clinical implications. |
| MD-179 Female | GCH1 | c.671A>G (p.K224R) rs41298442(MAF: 3.709 × 10−4) | 20 yo Sporadic case Heterozygosis AD | Dystonia DOPA-responsive (128230) AD | GP T2 hypointensity, dystonia, dysarthria, upper limbs tremor | 8852666, 12391354, 15303002, 25497597, 30314816 | Her two asymptomatic children carried the mutation in heterozygosis |
| MD-342 Female | PARK2 PRKN | c.220_221dupTG (p.W74Cfs*) rs746646126(MAF: 3.191 × 10−5) | 9 yo Sporadic case Homozygosis AR | Parkinson disease juvenile type 2 (600116) AR | Ataxia, motor delay, intention tremor, dysarthria, hypotonia, spasticity | 10072423 | The proband’s phenotype does not fit with the PARK2-associated parkinsonism |
| MD-347 Female | SACS | c.4016C>A (p.Q1143K) rs144267558(MAF: 1.066 × 10−3) | 2 mo Sporadic case Compound Heterozygosis AR | ARSACS (270550) AR | Cerebellar vermis hypoplasia, oculomotor apraxia, improving with age | 29915382 | MD-347 seems to be a carrier of a deleterious mutation (p.Q1143K), whereas p.N4573H (VUS) likely does not contribute to the clinical outcome. |
| c.14306A>C (p.N4573H) rs34382952(MAF: 3.124 × 10−3) | None |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Rubio, D.; Hinarejos, I.; Sancho, P.; Gorría-Redondo, N.; Bernadó-Fonz, R.; Tello, C.; Marco-Marín, C.; Martí-Carrera, I.; Martínez-González, M.J.; García-Ribes, A.; et al. Mutations, Genes, and Phenotypes Related to Movement Disorders and Ataxias. Int. J. Mol. Sci. 2022, 23, 11847. https://doi.org/10.3390/ijms231911847
Martínez-Rubio D, Hinarejos I, Sancho P, Gorría-Redondo N, Bernadó-Fonz R, Tello C, Marco-Marín C, Martí-Carrera I, Martínez-González MJ, García-Ribes A, et al. Mutations, Genes, and Phenotypes Related to Movement Disorders and Ataxias. International Journal of Molecular Sciences. 2022; 23(19):11847. https://doi.org/10.3390/ijms231911847
Chicago/Turabian StyleMartínez-Rubio, Dolores, Isabel Hinarejos, Paula Sancho, Nerea Gorría-Redondo, Raquel Bernadó-Fonz, Cristina Tello, Clara Marco-Marín, Itxaso Martí-Carrera, María Jesús Martínez-González, Ainhoa García-Ribes, and et al. 2022. "Mutations, Genes, and Phenotypes Related to Movement Disorders and Ataxias" International Journal of Molecular Sciences 23, no. 19: 11847. https://doi.org/10.3390/ijms231911847
APA StyleMartínez-Rubio, D., Hinarejos, I., Sancho, P., Gorría-Redondo, N., Bernadó-Fonz, R., Tello, C., Marco-Marín, C., Martí-Carrera, I., Martínez-González, M. J., García-Ribes, A., Baviera-Muñoz, R., Sastre-Bataller, I., Martínez-Torres, I., Duat-Rodríguez, A., Janeiro, P., Moreno, E., Pías-Peleteiro, L., Gordo, M. O., Ruiz-Gómez, Á., ... Espinós, C. (2022). Mutations, Genes, and Phenotypes Related to Movement Disorders and Ataxias. International Journal of Molecular Sciences, 23(19), 11847. https://doi.org/10.3390/ijms231911847