
Highly Conserved Interaction Profiles between Clinically Relevant Mutants of the Cytomegalovirus CDK-like Kinase pUL97 and Human Cyclins: Functional Significance of Cyclin H
 ,
,  , , and
, , and
Abstract
1. Introduction
2. Results
2.1. Whole Genome Sequencing of Drug-Resistant Clinical Isolates of HCMV
2.2. ORF-UL97 Sequence-Specific Analysis of HCMV Isolates
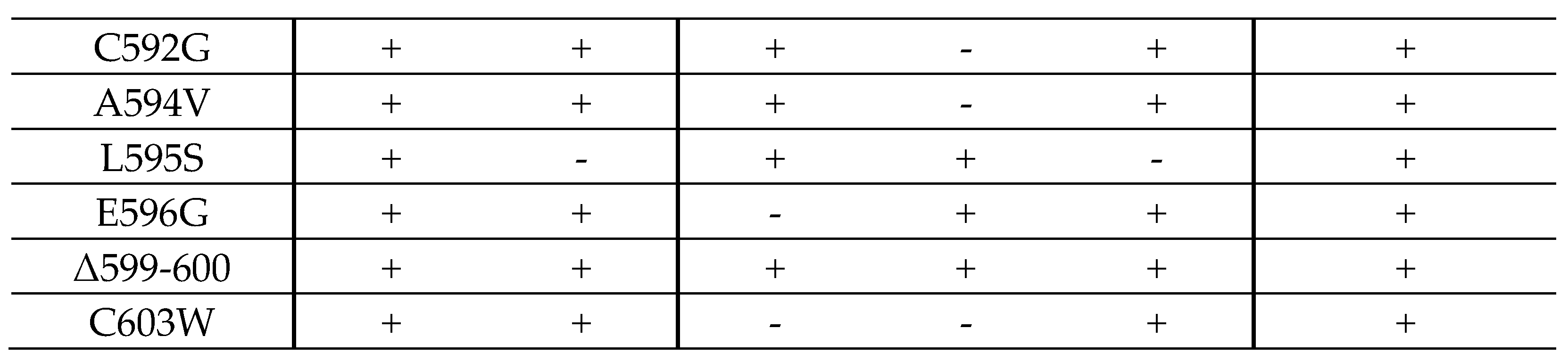
2.3. Resistance-Conferring Mutations of ORF-UL97 in Virus Recombinants Only Show a Minor Impact on Viral Replication Fitness
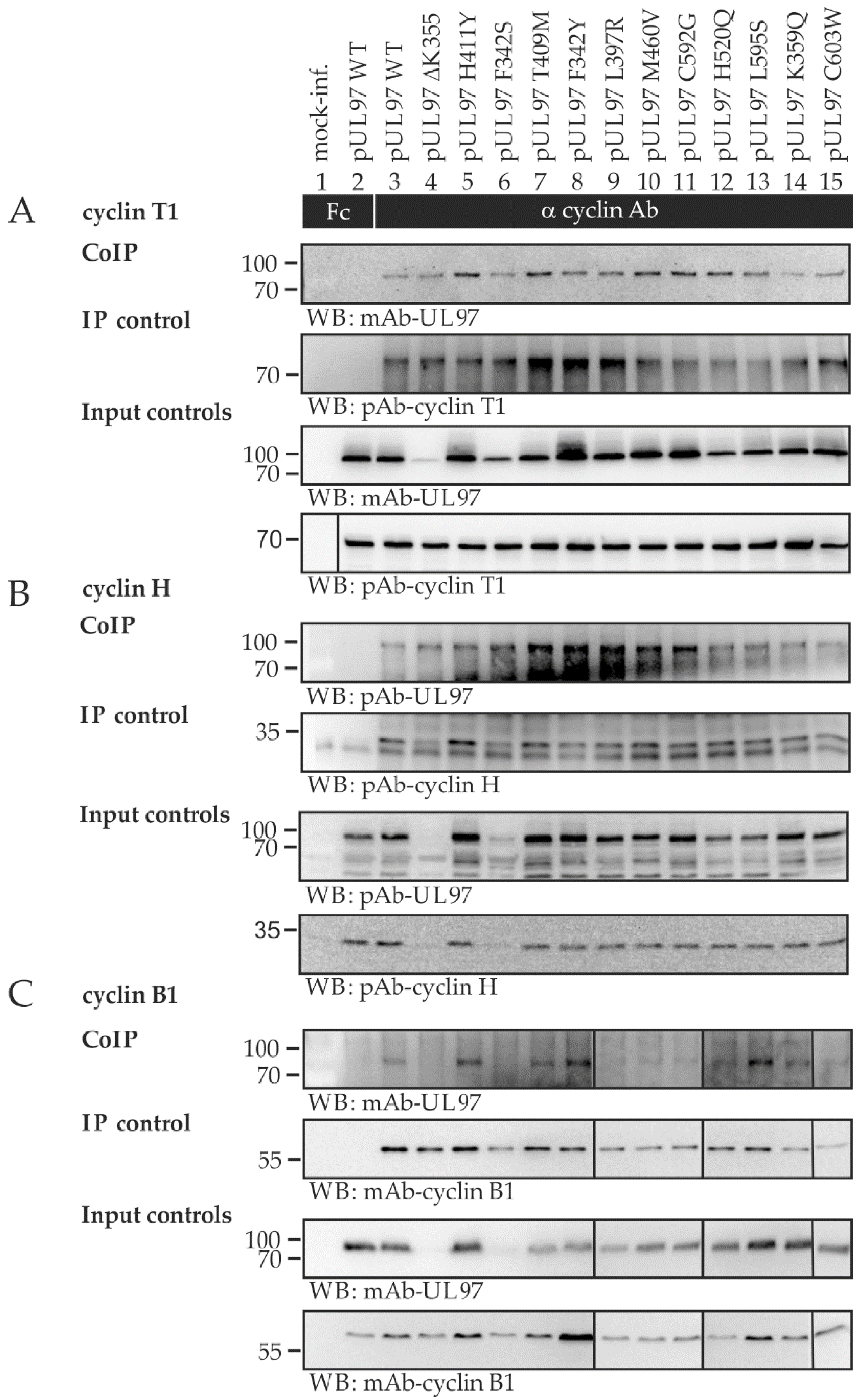
2.4. Interaction Profiles Show a High Conservation of pUL97–Cyclin Complexes for Clinically Relevant Point Mutants in HCMV Strain Backgrounds TB40 and AD169
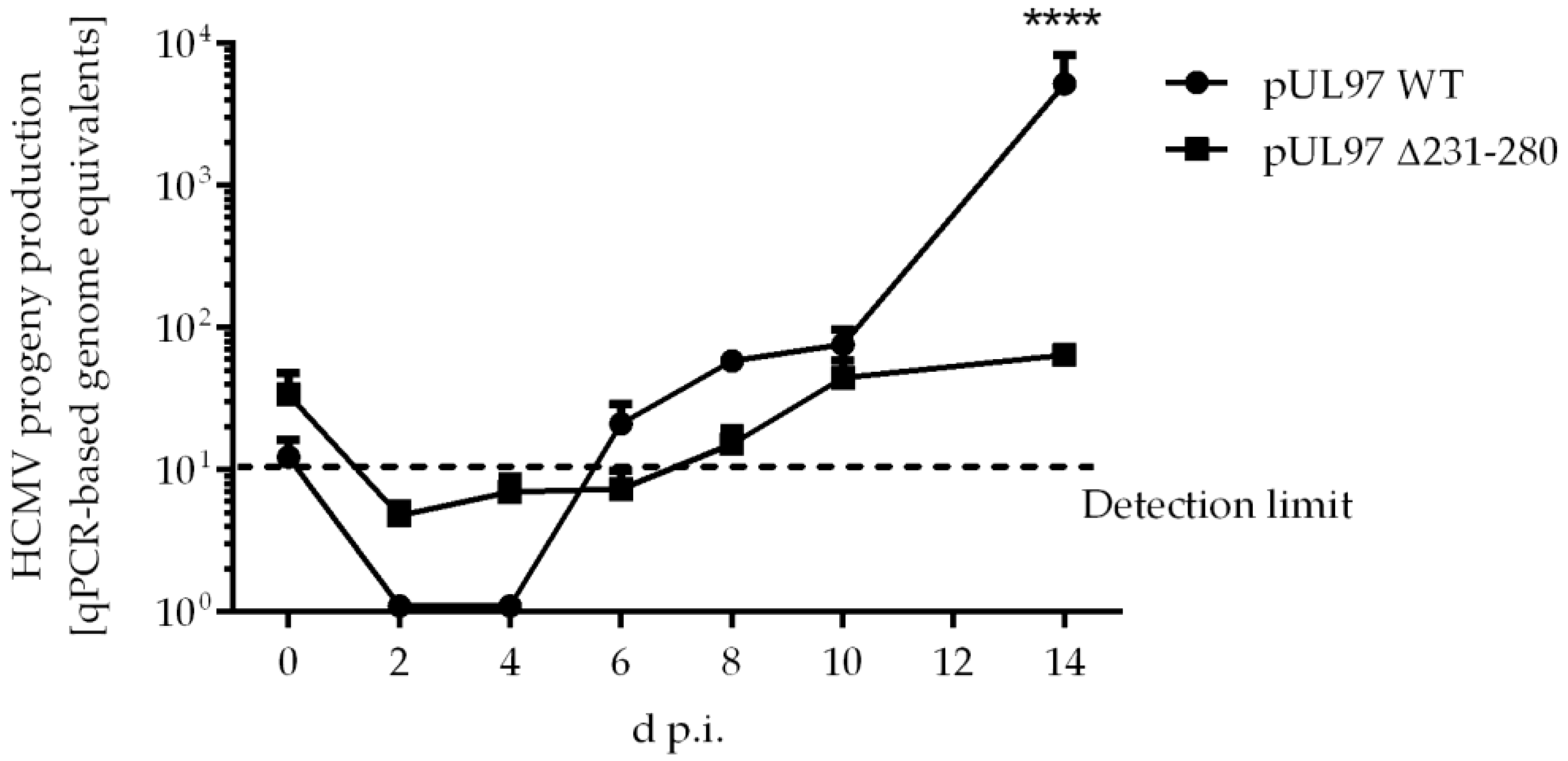
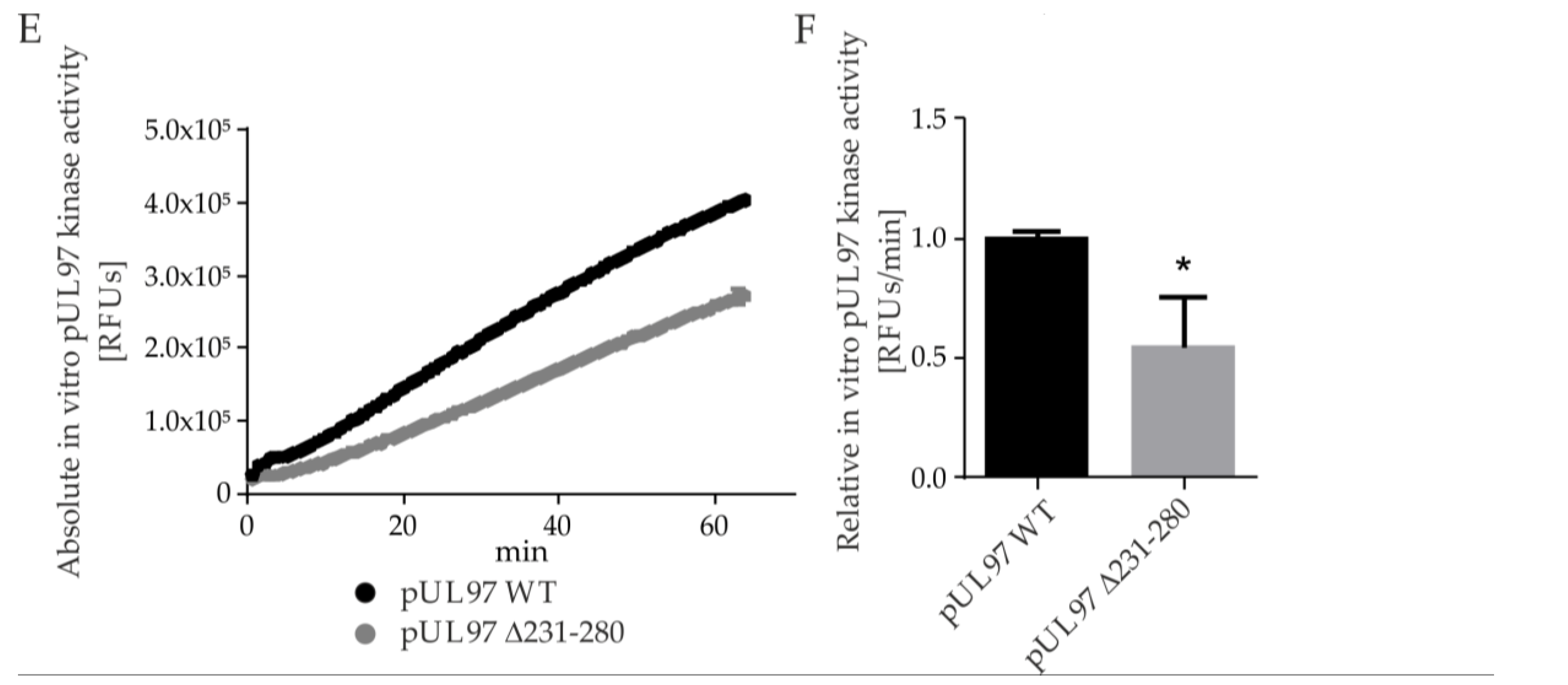
2.5. Amino Acid Region 231-280 of ORF-UL97 Strictly Restricts Cyclin T1/H Interactions and Viral Replication Efficiency
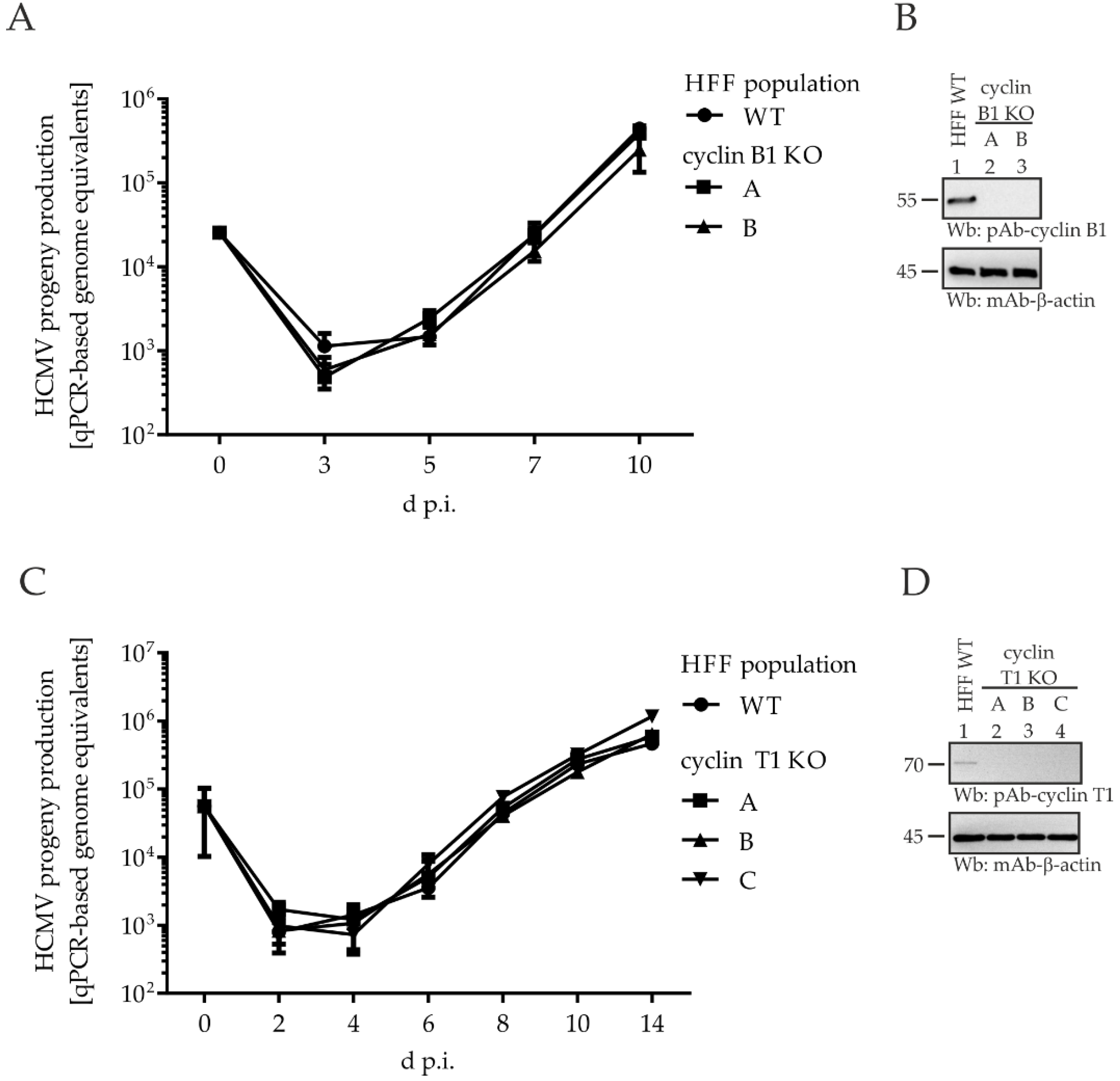
2.6. Comparing HCMV Replication Efficiencies in Cyclin Knock-Out (KO) Cell Populations: Cyclin H KO Cells Populations, but Not Cyclin B1 KO or T1 KO, Determine a Strict Limitation of Viral Replication
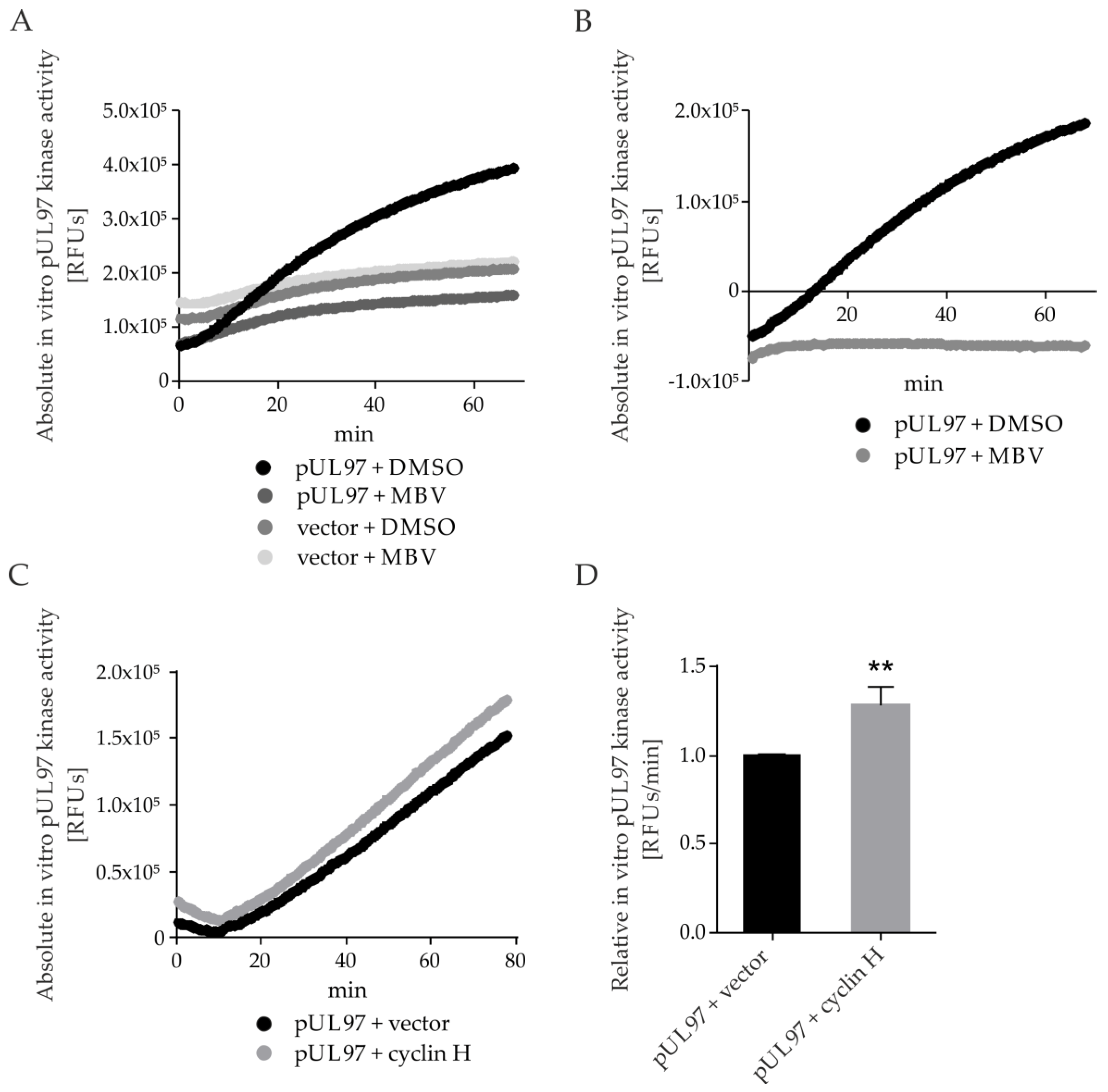
2.7. pUL97 Kinase Activity Is Increased upon co-Transfection of Cyclin H Using a Quantitative Sox Peptide-Based In Vitro Kinase Assay (qSox-IVKA)
3. Discussion
4. Materials and Methods
4.1. Cells and Viruses
4.2. Whole Genome Sequencing
4.3. Generation of Recombinant HCMVs
4.4. Generation of Cyclin KO Cell Lines
4.5. Coimmunoprecipitation (CoIP), SDS-PAGE, and Western Blot (Wb) Analysis
4.6. Antibodies
4.7. Quantitative Polymerase Chain Reaction (qPCR)
4.8. HCMV Fluorescence-Based Replication Assay
4.9. Quantitative Sox Peptide-Based In Vitro Kinase Assay (qSox-IVKA)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ATP | adenosine triphosphate |
| CAK | CDK-activating kinase |
| CDK | cyclin-dependent kinase |
| cCMV | congenital cytomegalovirus infection |
| CoIP | coimmunoprecipitation |
| DMEM | Dulbecco’s modified Eagle’s medium |
| DMSO | dimethyl sulfoxide |
| d p.i. | days post infection |
| GCV | ganciclovir |
| GFP | green fluorescent protein |
| gRNA | guide RNA |
| HCMV | human cytomegalovirus |
| IF | interface |
| HFF | human foreskin fibroblast |
| IVKA | in vitro kinase assay |
| KO | knock-out |
| MBV | maribavir |
| MOI | multiplicity of infection |
| NLS | nuclear localization signal |
| ORF | open reading frame |
| qPCR | quantitative polymerase chain reaction |
| -R | -resistant |
| Rb | retinoblastoma protein |
| vCDK | viral cyclin-dependent kinase ortholog |
| WGS | whole genome sequencing |
| Wb | Western blot |
| WT | wild-type |
| YFP | yellow fluorescent protein |
References
- Griffiths, P.; Baraniak, I.; Reeves, M. The pathogenesis of human cytomegalovirus. J. Pathol. 2015, 235, 288–297. [Google Scholar] [CrossRef]
- Herbein, G. The Human Cytomegalovirus, from Oncomodulation to Oncogenesis. Viruses 2018, 10, 408. [Google Scholar] [CrossRef]
- Goodrum, F.; Britt, W.; Mocarski, E.S. Fields Virology: DNA Viruses, 7th ed.; LWW: Philadelphia, PA, USA, 2021. [Google Scholar]
- Marsico, C.; Kimberlin, D.W. Congenital Cytomegalovirus infection: Advances and challenges in diagnosis, prevention and treatment. Ital. J. Pediatr. 2017, 43, 38. [Google Scholar] [CrossRef] [PubMed]
- Njue, A.; Coyne, C.; Margulis, A.V.; Wang, D.; Marks, M.A.; Russell, K.; Das, R.; Sinha, A. The Role of Congenital Cytomegalovirus Infection in Adverse Birth Outcomes: A Review of the Potential Mechanisms. Viruses 2020, 13, 20. [Google Scholar] [CrossRef] [PubMed]
- Rawlinson, W.D.; Boppana, S.B.; Fowler, K.B.; Kimberlin, D.W.; Lazzarotto, T.; Alain, S.; Daly, K.; Doutre, S.; Gibson, L.; Giles, M.L.; et al. Congenital cytomegalovirus infection in pregnancy and the neonate: Consensus recommendations for prevention, diagnosis, and therapy. Lancet Infect. Dis. 2017, 17, e177–e188. [Google Scholar] [CrossRef]
- Marschall, M.; Häge, S.; Conrad, M.; Alkhashrom, S.; Kicuntod, J.; Schweininger, J.; Kriegel, M.; Lösing, J.; Tillmanns, J.; Neipel, F.; et al. Nuclear Egress Complexes of HCMV and Other Herpesviruses: Solving the Puzzle of Sequence Coevolution, Conserved Structures and Subfamily-Spanning Binding Properties. Viruses 2020, 12, 683. [Google Scholar] [CrossRef] [PubMed]
- Marschall, M.; Muller, Y.A.; Diewald, B.; Sticht, H.; Milbradt, J. The human cytomegalovirus nuclear egress complex unites multiple functions: Recruitment of effectors, nuclear envelope rearrangement, and docking to nuclear capsids. Rev. Med. Virol. 2017, 27, e1934. [Google Scholar] [CrossRef]
- Milbradt, J.; Kraut, A.; Hutterer, C.; Sonntag, E.; Schmeiser, C.; Ferro, M.; Wagner, S.; Lenac, T.; Claus, C.; Pinkert, S.; et al. Proteomic analysis of the multimeric nuclear egress complex of human cytomegalovirus. Mol. Cell. Proteomics 2014, 13, 2132–2146. [Google Scholar] [CrossRef] [PubMed]
- Schütz, M.; Steingruber, M.; Socher, E.; Müller, R.; Wagner, S.; Kögel, M.; Sticht, H.; Marschall, M. Functional Relevance of the Interaction between Human Cyclins and the Cytomegalovirus-Encoded CDK-Like Protein Kinase pUL97. Viruses 2021, 13, 1248. [Google Scholar] [CrossRef]
- Steingruber, M.; Marschall, M. The Cytomegalovirus Protein Kinase pUL97:Host Interactions, Regulatory Mechanisms and Antiviral Drug Targeting. Microorganisms 2020, 8, 515. [Google Scholar] [CrossRef] [PubMed]
- Iwahori, S.; Umaña, A.C.; VanDeusen, H.R.; Kalejta, R.F. Human cytomegalovirus-encoded viral cyclin-dependent kinase (v-CDK) UL97 phosphorylates and inactivates the retinoblastoma protein-related p107 and p130 proteins. J. Biol. Chem. 2017, 292, 6583–6599. [Google Scholar] [CrossRef] [PubMed]
- Iwahori, S.; Hakki, M.; Chou, S.; Kalejta, R.F. Molecular Determinants for the Inactivation of the Retinoblastoma Tumor Suppressor by the Viral Cyclin-dependent Kinase UL97. J. Biol. Chem. 2015, 290, 19666–19680. [Google Scholar] [CrossRef] [PubMed]
- Hume, A.J.; Finkel, J.S.; Kamil, J.P.; Coen, D.M.; Culbertson, M.R.; Kalejta, R.F. Phosphorylation of retinoblastoma protein by viral protein with cyclin-dependent kinase function. Science 2008, 320, 797–799. [Google Scholar] [CrossRef] [PubMed]
- Hertel, L.; Chou, S.; Mocarski, E.S. Viral and cell cycle-regulated kinases in cytomegalovirus-induced pseudomitosis and replication. PLoS Pathog. 2007, 3, e6. [Google Scholar] [CrossRef]
- Chou, S. Advances in the genotypic diagnosis of cytomegalovirus antiviral drug resistance. Antiviral Res. 2020, 176, 104711. [Google Scholar] [CrossRef]
- Prichard, M.N. Function of human cytomegalovirus UL97 kinase in viral infection and its inhibition by maribavir. Rev. Med. Virol. 2009, 19, 215–229. [Google Scholar] [CrossRef]
- Bogdanow, B.; Schmidt, M.; Weisbach, H.; Gruska, I.; Vetter, B.; Imami, K.; Ostermann, E.; Brune, W.; Selbach, M.; Hagemeier, C.; et al. Cross-regulation of viral kinases with cyclin A secures shutoff of host DNA synthesis. Nat. Commun. 2020, 11, 4845. [Google Scholar] [CrossRef]
- Graf, L.; Feichtinger, S.; Naing, Z.; Hutterer, C.; Milbradt, J.; Webel, R.; Wagner, S.; Scott, G.M.; Hamilton, S.T.; Rawlinson, W.D.; et al. New insight into the phosphorylation-regulated intranuclear localization of human cytomegalovirus pUL69 mediated by cyclin-dependent kinases (CDKs) and viral CDK orthologue pUL97. J. Gen. Virol. 2016, 97, 144–151. [Google Scholar] [CrossRef]
- Graf, L.; Webel, R.; Wagner, S.; Hamilton, S.T.; Rawlinson, W.D.; Sticht, H.; Marschall, M. The cyclin-dependent kinase ortholog pUL97 of human cytomegalovirus interacts with cyclins. Viruses 2013, 5, 3213–3230. [Google Scholar] [CrossRef]
- König, P.; Büscher, N.; Steingruber, M.; Socher, E.; Sticht, H.; Tenzer, S.; Plachter, B.; Marschall, M. Dynamic regulatory interaction between cytomegalovirus major tegument protein pp65 and protein kinase pUL97 in intracellular compartments, dense bodies and virions. J. Gen. Virol. 2017, 98, 2850–2863. [Google Scholar] [CrossRef]
- Steingruber, M.; Keller, L.; Socher, E.; Ferre, S.; Hesse, A.M.; Couté, Y.; Hahn, F.; Büscher, N.; Plachter, B.; Sticht, H.; et al. Cyclins B1, T1, and H differ in their molecular mode of interaction with cytomegalovirus protein kinase pUL97. J. Biol. Chem. 2019, 294, 6188–6203. [Google Scholar] [CrossRef] [PubMed]
- Steingruber, M.; Kraut, A.; Socher, E.; Sticht, H.; Reichel, A.; Stamminger, T.; Amin, B.; Couté, Y.; Hutterer, C.; Marschall, M. Proteomic Interaction Patterns between Human Cyclins, the Cyclin-Dependent Kinase Ortholog pUL97 and Additional Cytomegalovirus Proteins. Viruses 2016, 8, 219. [Google Scholar] [CrossRef] [PubMed]
- Steingruber, M.; Socher, E.; Hutterer, C.; Webel, R.; Bergbrede, T.; Lenac, T.; Sticht, H.; Marschall, M. The Interaction between Cyclin B1 and Cytomegalovirus Protein Kinase pUL97 is Determined by an Active Kinase Domain. Viruses 2015, 7, 4582–4601. [Google Scholar] [CrossRef]
- Malumbres, M. Cyclin-dependent kinases. Genome Biol. 2014, 15, 122. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Alonso, D.; Malumbres, M. Mammalian cell cycle cyclins. Semin. Cell Dev. Biol. 2020, 107, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Hutterer, C.; Hamilton, S.; Steingruber, M.; Zeitträger, I.; Bahsi, H.; Thuma, N.; Naing, Z.; Örfi, Z.; Örfi, L.; Socher, E.; et al. The chemical class of quinazoline compounds provides a core structure for the design of anticytomegaloviral kinase inhibitors. Antiviral Res. 2016, 134, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Romaker, D.; Schregel, V.; Maurer, K.; Auerochs, S.; Marzi, A.; Sticht, H.; Marschall, M. Analysis of the structure-activity relationship of four herpesviral UL97 subfamily protein kinases reveals partial but not full functional conservation. J. Med. Chem. 2006, 49, 7044–7053. [Google Scholar] [CrossRef]
- Webel, R.; Milbradt, J.; Auerochs, S.; Schregel, V.; Held, C.; Nöbauer, K.; Razzazi-Fazeli, E.; Jardin, C.; Wittenberg, T.; Sticht, H.; et al. Two isoforms of the protein kinase pUL97 of human cytomegalovirus are differentially regulated in their nuclear translocation. J. Gen. Virol. 2011, 92, 638–649. [Google Scholar] [CrossRef]
- Webel, R.; Solbak, S.; Held, C.; Milbradt, J.; Groß, A.; Eichler, J.; Wittenberg, T.; Jardin, C.; Sticht, H.; Fossen, T.; et al. Nuclear import of isoforms of the cytomegalovirus kinase pUL97 is mediated by differential activity of NLS1 and NLS2 both acting through classical importin-α binding. J. Gen. Virol. 2012, 93, 1756–1768. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.; Yu, D.; Grimwood, J.; Schmutz, J.; Dickson, M.; Jarvis, M.A.; Hahn, G.; Nelson, J.A.; Myers, R.M.; Shenk, T.E. Coding potential of laboratory and clinical strains of human cytomegalovirus. Proc. Natl. Acad. Sci. USA 2003, 100, 14976–14981. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Silva, M.C.; Shenk, T. Functional map of human cytomegalovirus AD169 defined by global mutational analysis. Proc. Natl. Acad. Sci. USA 2003, 100, 12396–12401. [Google Scholar] [CrossRef]
- Dunn, W.; Chou, C.; Li, H.; Hai, R.; Patterson, D.; Stolc, V.; Zhu, H.; Liu, F. Functional profiling of a human cytomegalovirus genome. Proc. Natl. Acad. Sci. USA 2003, 100, 14223–14228. [Google Scholar] [CrossRef]
- Murphy, E.; Rigoutsos, I.; Shibuya, T.; Shenk, T.E. Reevaluation of human cytomegalovirus coding potential. Proc. Natl. Acad. Sci. USA 2003, 100, 13585–13590. [Google Scholar] [CrossRef]
- Wilkinson, G.W.; Davison, A.J.; Tomasec, P.; Fielding, C.A.; Aicheler, R.; Murrell, I.; Seirafian, S.; Wang, E.C.; Weekes, M.; Lehner, P.J.; et al. Human cytomegalovirus: Taking the strain. Med. Microbiol. Immunol. 2015, 204, 273–284. [Google Scholar] [CrossRef]
- Cagliani, R.; Forni, D.; Mozzi, A.; Sironi, M. Evolution and Genetic Diversity of Primate Cytomegaloviruses. Microorganisms 2020, 8, 624. [Google Scholar] [CrossRef]
- Suárez, N.M.; Wilkie, G.S.; Hage, E.; Camiolo, S.; Holton, M.; Hughes, J.; Maabar, M.; Vattipally, S.B.; Dhingra, A.; Gompels, U.A.; et al. Human Cytomegalovirus Genomes Sequenced Directly From Clinical Material: Variation, Multiple-Strain Infection, Recombination, and Gene Loss. J. Infect. Dis. 2019, 220, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Wagenknecht, N.; Reuter, N.; Scherer, M.; Reichel, A.; Müller, R.; Stamminger, T. Contribution of the Major ND10 Proteins PML, hDaxx and Sp100 to the Regulation of Human Cytomegalovirus Latency and Lytic Replication in the Monocytic Cell Line THP-1. Viruses 2015, 7, 2884–2907. [Google Scholar] [CrossRef]
- Houldcroft, C.J.; Bryant, J.M.; Depledge, D.P.; Margetts, B.K.; Simmonds, J.; Nicolaou, S.; Tutill, H.J.; Williams, R.; Worth, A.J.; Marks, S.D.; et al. Detection of Low Frequency Multi-Drug Resistance and Novel Putative Maribavir Resistance in Immunocompromised Pediatric Patients with Cytomegalovirus. Front. Microbiol. 2016, 7, 1317. [Google Scholar] [CrossRef]
- Chen, J.; Larochelle, S.; Li, X.; Suter, B. Xpd/Ercc2 regulates CAK activity and mitotic progression. Nature 2003, 424, 228–232. [Google Scholar] [CrossRef]
- Lolli, G.; Johnson, L.N. CAK-Cyclin-dependent Activating Kinase: A key kinase in cell cycle control and a target for drugs? Cell Cycle 2005, 4, 572–577. [Google Scholar] [CrossRef]
- Wild, M.; Kicuntod, J.; Seyler, L.; Wangen, C.; Bertzbach, L.D.; Conradie, A.M.; Kaufer, B.B.; Wagner, S.; Michel, D.; Eickhoff, J.; et al. Combinatorial Drug Treatments Reveal Promising Anticytomegaloviral Profiles for Clinically Relevant Pharmaceutical Kinase Inhibitors (PKIs). Int. J. Mol. Sci. 2021, 22, 575. [Google Scholar] [CrossRef]
- Wild, M.; Hahn, F.; Brückner, N.; Schütz, M.; Wangen, C.; Wagner, S.; Sommerer, M.; Strobl, S.; Marschall, M. Cyclin-Dependent Kinases (CDKs) and the Human Cytomegalovirus-Encoded CDK Ortholog pUL97 Represent Highly Attractive Targets for Synergistic Drug Combinations. Int. J. Mol. Sci. 2022, 23, 2493. [Google Scholar] [CrossRef]
- Chou, S.; Bowlin, T.L. Cytomegalovirus UL97 mutations affecting cyclopropavir and ganciclovir susceptibility. Antimicrob. Agents Chemother. 2011, 55, 382–384. [Google Scholar] [CrossRef]
- Chou, S.; Ercolani, R.J.; Derakhchan, K. Antiviral activity of maribavir in combination with other drugs active against human cytomegalovirus. Antiviral Res. 2018, 157, 128–133. [Google Scholar] [CrossRef]
- Shults, M.D.; Carrico-Moniz, D.; Imperiali, B. Optimal Sox-based fluorescent chemosensor design for serine/threonine protein kinases. Anal. Biochem. 2006, 352, 198–207. [Google Scholar] [CrossRef]
- Shults, M.D.; Imperiali, B. Versatile fluorescence probes of protein kinase activity. J. Am. Chem. Soc. 2003, 125, 14248–14249. [Google Scholar] [CrossRef]
- Chou, S.; Watters, M.; Sinha, R.; Kleiboeker, S. Ganciclovir and maribavir susceptibility phenotypes of cytomegalovirus UL97 ATP binding region mutations detected by expanded genotypic testing. Antiviral Res. 2021, 193, 105139. [Google Scholar] [CrossRef]
- Marschall, M.; Freitag, M.; Weiler, S.; Sorg, G.; Stamminger, T. Recombinant green fluorescent protein-expressing human cytomegalovirus as a tool for screening antiviral agents. Antimicrob. Agents Chemother. 2000, 44, 1588–1597. [Google Scholar] [CrossRef]
- Rowe, W.P.; Hartley, J.W.; Waterman, S.; Turner, H.C.; Huebner, R.J. Cytopathogenic agent resembling human salivary gland virus recovered from tissue cultures of human adenoids. Proc. Soc. Exp. Biol. Med. 1956, 92, 418–424. [Google Scholar]
- Sinzger, C.; Hahn, G.; Digel, M.; Katona, R.; Sampaio, K.L.; Messerle, M.; Hengel, H.; Koszinowski, U.; Brune, W.; Adler, B. Cloning and sequencing of a highly productive, endotheliotropic virus strain derived from human cytomegalovirus TB40/E. J. Gen. Virol. 2008, 89, 359–368. [Google Scholar] [CrossRef]
- Stanton, R.J.; Baluchova, K.; Dargan, D.J.; Cunningham, C.; Sheehy, O.; Seirafian, S.; McSharry, B.P.; Neale, M.L.; Davies, J.A.; Tomasec, P.; et al. Reconstruction of the complete human cytomegalovirus genome in a BAC reveals RL13 to be a potent inhibitor of replication. J. Clin. Investig. 2010, 120, 3191–3208. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.D.; van Zuylen, W.J.; Hamilton, S.T.; Steingruber, M.; Sonntag, E.; Marschall, M.; Rawlinson, W.D. Patient-Derived Cytomegaloviruses with Different Ganciclovir Sensitivities from UL97 Mutation Retain Their Replication Efficiency and Some Kinase Activity In Vitro. Antimicrob. Agents Chemother. 2019, 63, e02425-18. [Google Scholar] [CrossRef]
- Kapasi, A.J.; Spector, D.H. Inhibition of the cyclin-dependent kinases at the beginning of human cytomegalovirus infection specifically alters the levels and localization of the RNA polymerase II carboxyl-terminal domain kinases cdk9 and cdk7 at the viral transcriptosome. J. Virol. 2008, 82, 394–407. [Google Scholar] [CrossRef] [PubMed]
- Oduro, J.D.; Uecker, R.; Hagemeier, C.; Wiebusch, L. Inhibition of human cytomegalovirus immediate-early gene expression by cyclin A2-dependent kinase activity. J. Virol. 2012, 86, 9369–9383. [Google Scholar] [CrossRef]
- Spector, D.H. Human cytomegalovirus riding the cell cycle. Med. Microbiol. Immunol. 2015, 204, 409–419. [Google Scholar] [CrossRef]
- Couté, Y.; Kraut, A.; Zimmermann, C.; Büscher, N.; Hesse, A.M.; Bruley, C.; De Andrea, M.; Wangen, C.; Hahn, F.; Marschall, M.; et al. Mass Spectrometry-Based Characterization of the Virion Proteome, Phosphoproteome, and Associated Kinase Activity of Human Cytomegalovirus. Microorganisms 2020, 8, 820. [Google Scholar] [CrossRef]
- Kang, C. Maribavir: First Approval. Drugs 2022, 82, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Breen, M.E.; Soellner, M.B. Small molecule substrate phosphorylation site inhibitors of protein kinases: Approaches and challenges. ACS Chem. Biol. 2015, 10, 175–189. [Google Scholar] [CrossRef] [PubMed]
- Alkhashrom, S.; Kicuntod, J.; Häge, S.; Schweininger, J.; Muller, Y.A.; Lischka, P.; Marschall, M.; Eichler, J. Exploring the Human Cytomegalovirus Core Nuclear Egress Complex as a Novel Antiviral Target: A New Type of Small Molecule Inhibitors. Viruses 2021, 13, 471. [Google Scholar] [CrossRef] [PubMed]
- Kicuntod, J.; Alkhashrom, S.; Häge, S.; Diewald, B.; Müller, R.; Hahn, F.; Lischka, P.; Sticht, H.; Eichler, J.; Marschall, M. Properties of Oligomeric Interaction of the Cytomegalovirus Core Nuclear Egress Complex (NEC) and Its Sensitivity to an NEC Inhibitory Small Molecule. Viruses 2021, 13, 462. [Google Scholar] [CrossRef] [PubMed]
- Häge, S.; Sonntag, E.; Svrlanska, A.; Borst, E.M.; Stilp, A.C.; Horsch, D.; Müller, R.; Kropff, B.; Milbradt, J.; Stamminger, T.; et al. Phenotypical Characterization of the Nuclear Egress of Recombinant Cytomegaloviruses Reveals Defective Replication upon ORF-UL50 Deletion but Not pUL50 Phosphosite Mutation. Viruses 2021, 13, 165. [Google Scholar] [CrossRef]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef]
- Lassalle, F.; Depledge, D.P.; Reeves, M.B.; Brown, A.C.; Christiansen, M.T.; Tutill, H.J.; Williams, R.J.; Einer-Jensen, K.; Holdstock, J.; Atkinson, C.; et al. Islands of linkage in an ocean of pervasive recombination reveals two-speed evolution of human cytomegalovirus genomes. Virus Evol 2016, 2, vew017. [Google Scholar] [CrossRef] [PubMed]
- Tischer, B.K.; Smith, G.A.; Osterrieder, N. En passant mutagenesis: A two step markerless red recombination system. In In Vitro Mutagenesis Protocols; Springer: Berlin/Heidelberg, Germany, 2010; pp. 421–430. [Google Scholar]
- Stanton, R.J.; McSharry, B.P.; Armstrong, M.; Tomasec, P.; Wilkinson, G.W. Re-engineering adenovirus vector systems to enable high-throughput analyses of gene function. BioTechniques 2008, 45, 659–662. [Google Scholar] [CrossRef] [PubMed]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef] [PubMed]
- Sonntag, E.; Milbradt, J.; Svrlanska, A.; Strojan, H.; Häge, S.; Kraut, A.; Hesse, A.M.; Amin, B.; Sonnewald, U.; Couté, Y.; et al. Protein kinases responsible for the phosphorylation of the nuclear egress core complex of human cytomegalovirus. J. Gen. Virol. 2017, 98, 2569–2581. [Google Scholar] [CrossRef] [PubMed]
- Hutterer, C.; Wandinger, S.K.; Wagner, S.; Müller, R.; Stamminger, T.; Zeitträger, I.; Godl, K.; Baumgartner, R.; Strobl, S.; Marschall, M. Profiling of the kinome of cytomegalovirus-infected cells reveals the functional importance of host kinases Aurora A, ABL and AMPK. Antiviral Res. 2013, 99, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Rechter, S.; Milbradt, J.; Auerochs, S.; Müller, R.; Stamminger, T.; Marschall, M. Cytomegaloviral protein kinase pUL97 interacts with the nuclear mRNA export factor pUL69 to modulate its intranuclear localization and activity. J. Gen. Virol. 2009, 90, 567–578. [Google Scholar] [CrossRef]
- Milbradt, J.; Sonntag, E.; Wagner, S.; Strojan, H.; Wangen, C.; Lenac Rovis, T.; Lisnic, B.; Jonjic, S.; Sticht, H.; Britt, W.J.; et al. Human Cytomegalovirus Nuclear Capsids Associate with the Core Nuclear Egress Complex and the Viral Protein Kinase pUL97. Viruses 2018, 10, 35. [Google Scholar] [CrossRef] [PubMed]
- Hutterer, C.; Eickhoff, J.; Milbradt, J.; Korn, K.; Zeitträger, I.; Bahsi, H.; Wagner, S.; Zischinsky, G.; Wolf, A.; Degenhart, C.; et al. A novel CDK7 inhibitor of the Pyrazolotriazine class exerts broad-spectrum antiviral activity at nanomolar concentrations. Antimicrob. Agents Chemother. 2015, 59, 2062–2071. [Google Scholar] [CrossRef] [PubMed]
- Schregel, V.; Auerochs, S.; Jochmann, R.; Maurer, K.; Stamminger, T.; Marschall, M. Mapping of a self-interaction domain of the cytomegalovirus protein kinase pUL97. J. Gen. Virol. 2007, 88, 395–404. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Isolate | Mode of Viral Reproduction a | Numbers of Passages | GenBank Accession Number |
|---|---|---|---|
| E16662 | cell-bound mainly (cell-free 5.03 × 106 copies/µL) | <5 | ON119190 |
| E18800 | cell-bound mainly (cell-free 1.10 × 106 copies/µL) | <5 | ON119191 |
| E20744 | cell-bound mainly (cell-free 2.77 × 106 copies/µL) | <5 | ON119192 |
| E20749 | cell-bound mainly (cell-free 3.99 × 105 copies/µL) | <5 | ON119193 |
| E9045 | cell-bound mainly (cell-free 4.11 × 106 copies/µL) | <5 | ON119194 |
| E9361 | cell-bound mainly (cell-free 2.74 × 106 copies/µL) | <5 | ON119195 |
| E31354 | cell-bound mainly (cell-free 9.38 × 105 copies/µL) | <5 | ON119196 |
| E83151 | cell-bound mainly (cell-free 1.86 × 106 copies/µL) | <5 | ON119197 |
| E79446 | cell-bound mainly (cell-free 1.11 × 106 copies/µL) | <5 | ON119198 |
| E29747 | cell-bound mainly (cell-free 2.36 × 106 copies/µL) | <5 | ON119199 |
| Mutant pUL97 | Viral Replication Efficiency | Replication in Cyclin KO Cells | CoIP pUL97-Cyclin | Activity of pUL97 | Cyclin Type with Main Impact on pUL97 and HCMV Replication | Source/Origin of Virus Strains and Recombinants | |
|---|---|---|---|---|---|---|---|
| Laboratory strains (AD169, TB40, Merlin) | no | normal | cycB1/T1: normal cycH KO: impaired | cyclins B1/T1/H | normal | cyclin H | [16,38,44,45,48,49,50,51,52,53] |
| Deletion mutant pUL97 Δ231-280 | yes | defective | n.d. | cyclin B1 only | drastic changes | cyclin H | [10] |
| Drug resistance pUL97 point mutants | yes | Normal | n.d. | cyclins B1/T1/H | t.b.d. | cyclin H | [present study] |
| Isolate | Collected by | Collection Date | Country | Isolation Source | Host |
|---|---|---|---|---|---|
| E16662 | T. Stamminger lab | 28 January 1997 | Germany | human clinical specimens | H. sapiens |
| E18800 | T. Stamminger lab | 7 January 1999 | Germany | human clinical specimens | H. sapiens |
| E20744 | T. Stamminger lab | 5 March 1999 | Germany | human clinical specimens | H. sapiens |
| E20749 | T. Stamminger lab | 15 March 1999 | Germany | human clinical specimens | H. sapiens |
| E9045 | T. Stamminger lab | 7 December 2000 | Germany | human clinical specimens | H. sapiens |
| E9361 | T. Stamminger lab | 7 December 2000 | Germany | human clinical specimens | H. sapiens |
| E31354 | T. Stamminger lab | 21 August 2003 | Germany | human clinical specimens | H. sapiens |
| E83151 | T. Stamminger lab | 25 November 2014 | Germany | human clinical specimens | H. sapiens |
| E79446 | T. Stamminger lab | 14 October 2014 | Germany | human clinical specimens | H. sapiens |
| E29747 | T. Stamminger lab | 15 June 2011 | Germany | human clinical specimens | H. sapiens |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schütz, M.; Müller, R.; Socher, E.; Wangen, C.; Full, F.; Wyler, E.; Wong, D.; Scherer, M.; Stamminger, T.; Chou, S.; et al. Highly Conserved Interaction Profiles between Clinically Relevant Mutants of the Cytomegalovirus CDK-like Kinase pUL97 and Human Cyclins: Functional Significance of Cyclin H. Int. J. Mol. Sci. 2022, 23, 11814. https://doi.org/10.3390/ijms231911814
Schütz M, Müller R, Socher E, Wangen C, Full F, Wyler E, Wong D, Scherer M, Stamminger T, Chou S, et al. Highly Conserved Interaction Profiles between Clinically Relevant Mutants of the Cytomegalovirus CDK-like Kinase pUL97 and Human Cyclins: Functional Significance of Cyclin H. International Journal of Molecular Sciences. 2022; 23(19):11814. https://doi.org/10.3390/ijms231911814
Chicago/Turabian StyleSchütz, Martin, Regina Müller, Eileen Socher, Christina Wangen, Florian Full, Emanuel Wyler, Diana Wong, Myriam Scherer, Thomas Stamminger, Sunwen Chou, and et al. 2022. "Highly Conserved Interaction Profiles between Clinically Relevant Mutants of the Cytomegalovirus CDK-like Kinase pUL97 and Human Cyclins: Functional Significance of Cyclin H" International Journal of Molecular Sciences 23, no. 19: 11814. https://doi.org/10.3390/ijms231911814
APA StyleSchütz, M., Müller, R., Socher, E., Wangen, C., Full, F., Wyler, E., Wong, D., Scherer, M., Stamminger, T., Chou, S., Rawlinson, W. D., Hamilton, S. T., Sticht, H., & Marschall, M. (2022). Highly Conserved Interaction Profiles between Clinically Relevant Mutants of the Cytomegalovirus CDK-like Kinase pUL97 and Human Cyclins: Functional Significance of Cyclin H. International Journal of Molecular Sciences, 23(19), 11814. https://doi.org/10.3390/ijms231911814