
Mammary-Enriched Transcription Factors Synergize to Activate the Wap Super-Enhancer for Mammary Gland Development


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
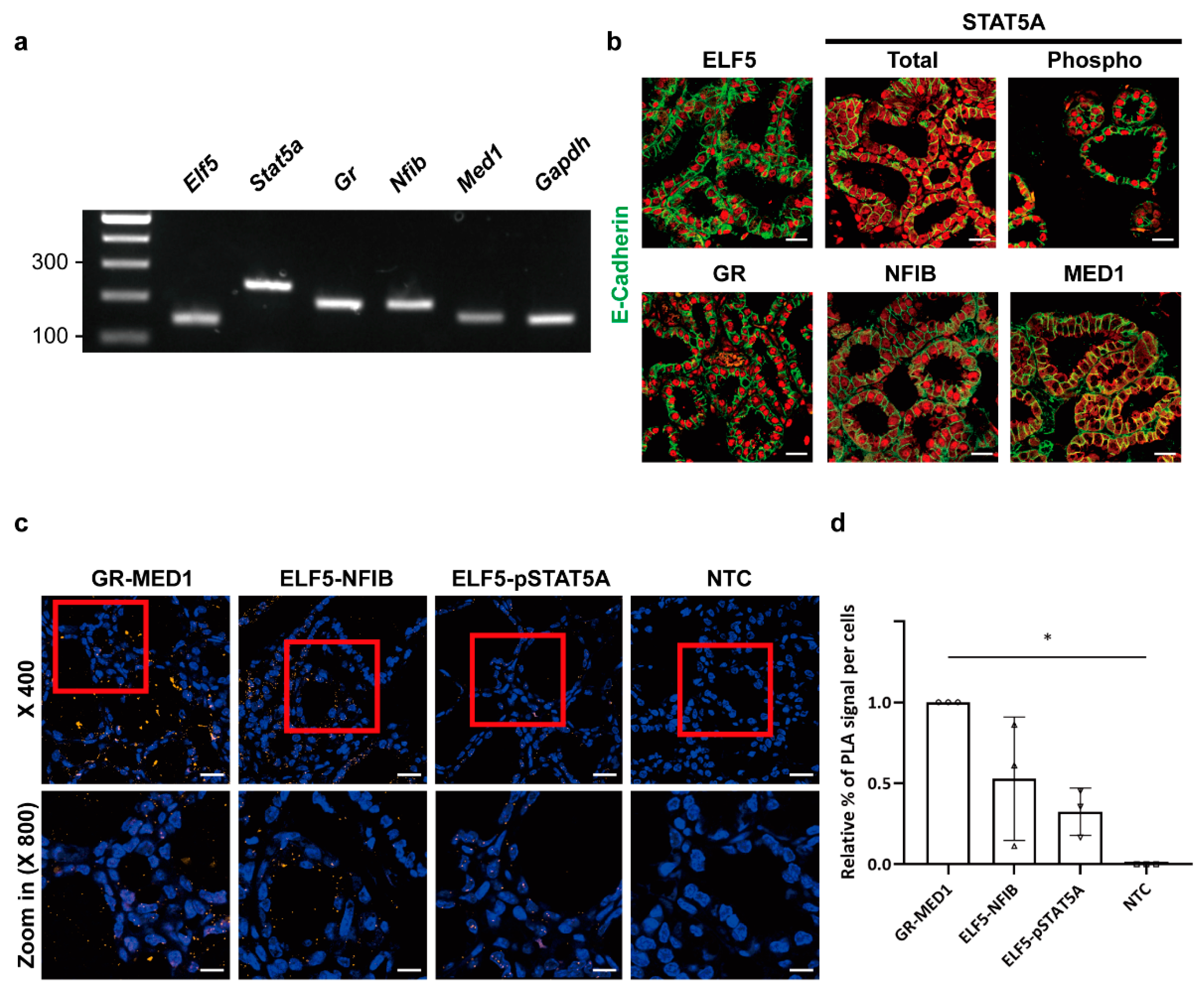
2.1. Cellular Localization of Mammary-Enriched Transcription Factors Constituting the Wap Super-Enhancer in Mammary Epithelial Cells
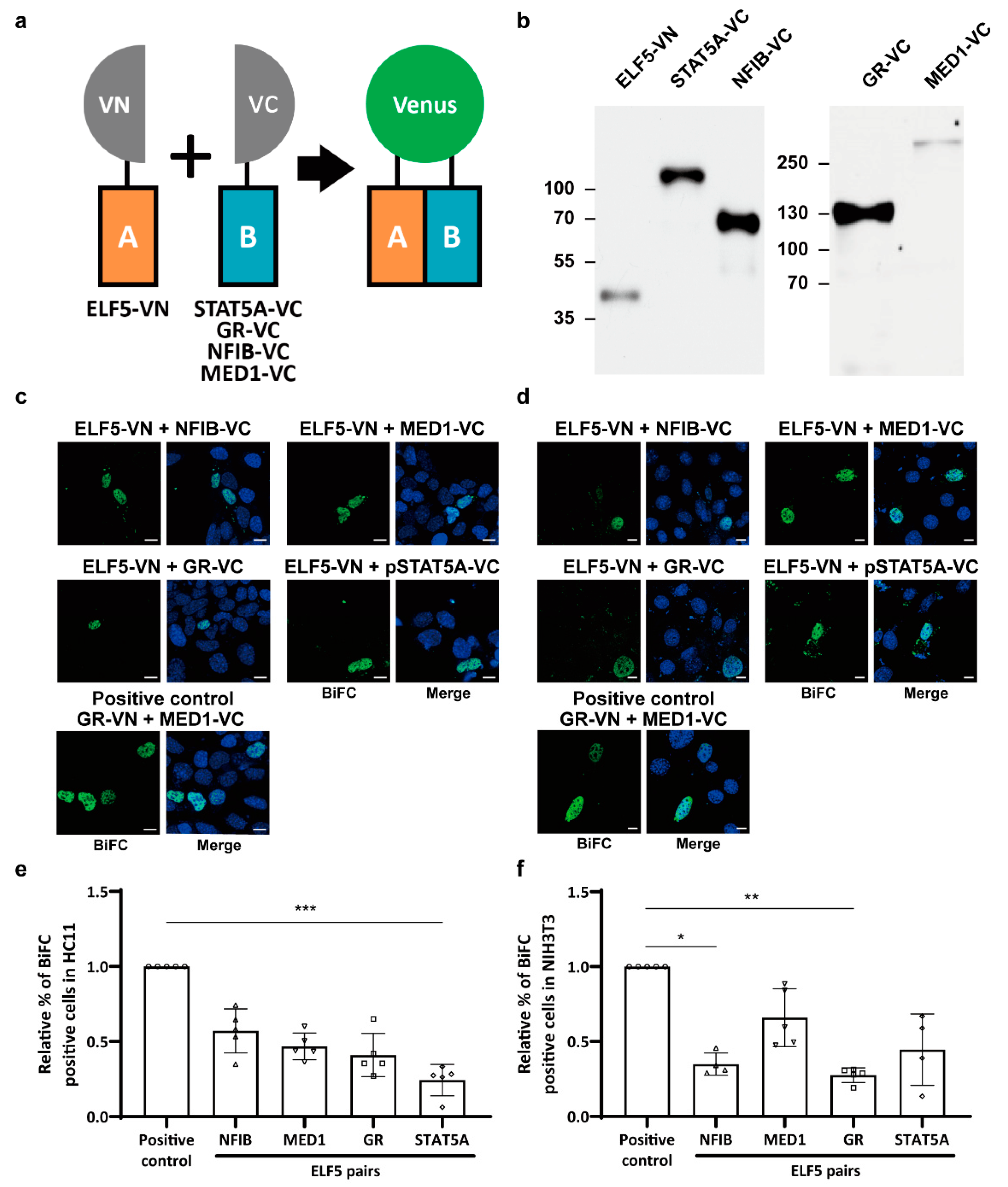
2.2. Protein Interactions between Mammary-Enriched Transcription Factors in Mammary Epithelial Cells Differ from Those in Non-Mammary Cells
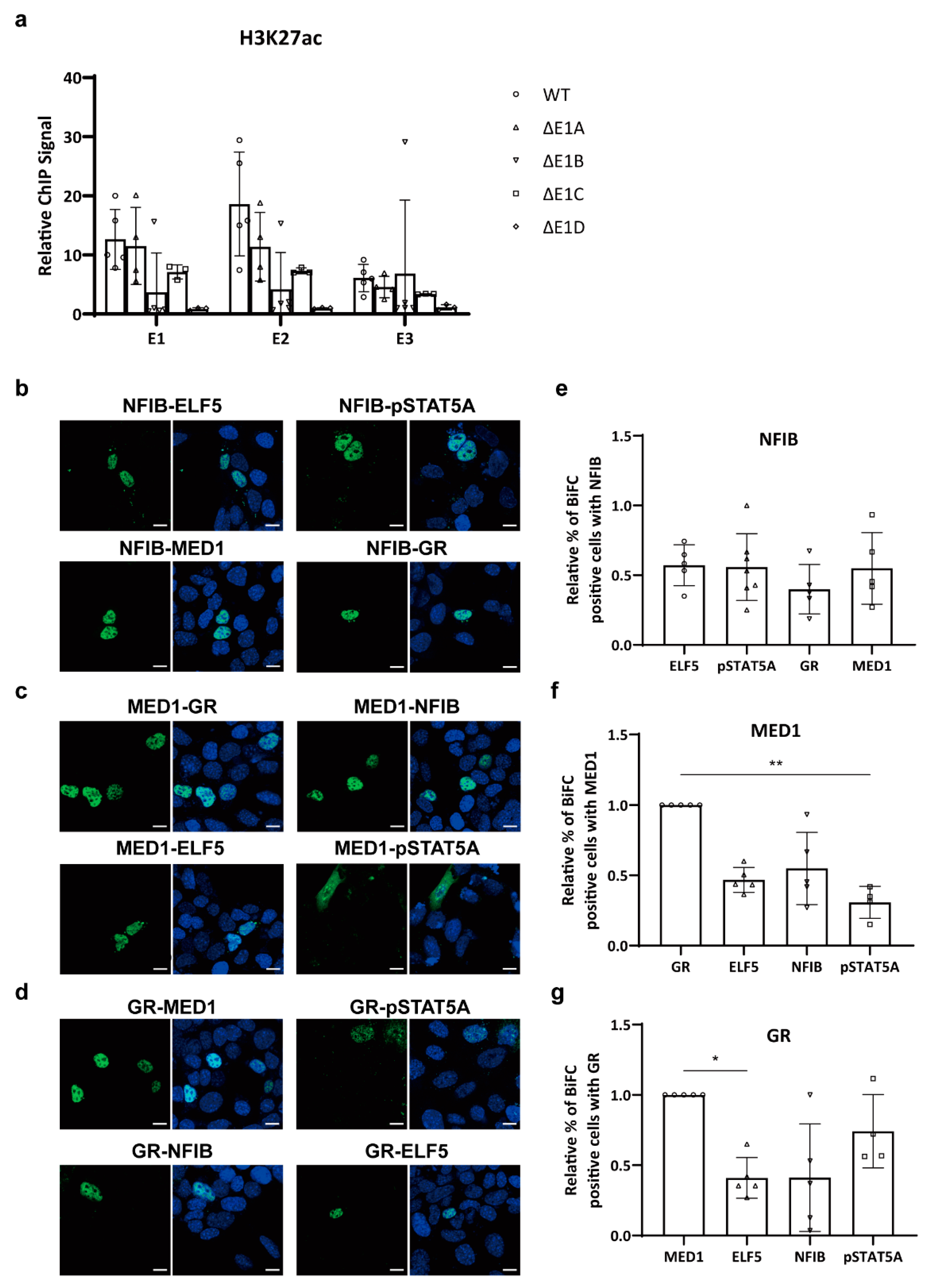
2.3. Mammary-Enriched Transcription Factors Synergize to Activate the Wap Super-Enhancer for Mammary Gland Differentiation
2.4. Protein–Protein Interactions within a Mammary-Enriched Transcription Factor Complex Are Essential for Activation of Mammary-Specific Wap Super-Enhancers
3. Discussion
4. Materials and Methods
4.1. ChIP-Seq Data
4.2. Plasmids Construction
4.3. Cell Culture
4.4. Western Blot Analysis
4.5. Bimolecular Fluorescence Complementation (BiFC) Assay
4.6. Mice
4.7. Generation of Homozygous Mice and Genotyping
4.8. Histological Analysis
4.9. In Situ Proximity Ligation Assay (PLA)
4.10. Quantitative (RT-qPCR) and Conventional RT-PCR
4.11. Chromatin Immunoprecipitation (ChIP) Analysis
4.12. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hennighausen, L.; Robinson, G.W. Information networks in the mammary gland. Nat. Rev. Mol. Cell Biol. 2005, 6, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.J.; Khaled, W.T. Mammary development in the embryo and adult: A journey of morphogenesis and commitment. Development 2008, 135, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Horseman, N.D.; Zhao, W.; Montecino-Rodriguez, E.; Tanaka, M.; Nakashima, K.; Engle, S.J.; Smith, F.; Markoff, E.; Dorshkind, K. Defective mammopoiesis, but normal hematopoiesis, in mice with a targeted disruption of the prolactin gene. EMBO J. 1997, 16, 6926–6935. [Google Scholar] [CrossRef] [PubMed]
- Ormandy, C.J.; Camus, A.; Barra, J.; Damotte, D.; Lucas, B.; Buteau, H.; Edery, M.; Brousse, N.; Babinet, C.; Binart, N.; et al. Null mutation of the prolactin receptor gene produces multiple reproductive defects in the mouse. Genes Dev. 1997, 11, 167–178. [Google Scholar] [CrossRef]
- Neville, M.C.; McFadden, T.B.; Forsyth, I. Hormonal regulation of mammary differentiation and milk secretion. J. Mammary Gland Biol. Neoplasia 2002, 7, 49–66. [Google Scholar] [CrossRef]
- Shin, H.Y.; Willi, M.; HyunYoo, K.; Zeng, X.; Wang, C.; Metser, G.; Hennighausen, L. Hierarchy within the mammary STAT5-driven Wap super-enhancer. Nat. Genet. 2016, 48, 904–911. [Google Scholar] [CrossRef]
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-Andre, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-enhancers in the control of cell identity and disease. Cell 2013, 155, 934–947. [Google Scholar] [CrossRef]
- Whyte, W.A.; Orlando, D.A.; Hnisz, D.; Abraham, B.J.; Lin, C.Y.; Kagey, M.H.; Rahl, P.B.; Lee, T.I.; Young, R.A. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013, 153, 307–319. [Google Scholar] [CrossRef]
- Yoo, K.H.; Hennighausen, L.; Shin, H.Y. Dissecting Tissue-Specific Super-Enhancers by Integrating Genome-Wide Analyses and CRISPR/Cas9 Genome Editing. J. Mammary Gland Biol. Neoplasia 2019, 24, 47–59. [Google Scholar] [CrossRef]
- Hnisz, D.; Schuijers, J.; Lin, C.Y.; Weintraub, A.S.; Abraham, B.J.; Lee, T.I.; Bradner, J.E.; Young, R.A. Convergence of developmental and oncogenic signaling pathways at transcriptional super-enhancers. Mol. Cell 2015, 58, 362–370. [Google Scholar] [CrossRef]
- Loven, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef] [PubMed]
- Parker, S.C.; Stitzel, M.L.; Taylor, D.L.; Orozco, J.M.; Erdos, M.R.; Akiyama, J.A.; van Bueren, K.L.; Chines, P.S.; Narisu, N.; Program, N.C.S.; et al. Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants. Proc. Natl. Acad. Sci. USA 2013, 110, 17921–17926. [Google Scholar] [CrossRef] [PubMed]
- Vahedi, G.; Kanno, Y.; Furumoto, Y.; Jiang, K.; Parker, S.C.; Erdos, M.R.; Davis, S.R.; Roychoudhuri, R.; Restifo, N.P.; Gadina, M.; et al. Super-enhancers delineate disease-associated regulatory nodes in T cells. Nature 2015, 520, 558–562. [Google Scholar] [CrossRef]
- Shin, H.Y. Targeting Super-Enhancers for Disease Treatment and Diagnosis. Mol. Cells 2018, 41, 506–514. [Google Scholar] [CrossRef]
- Pittius, C.W.; Sankaran, L.; Topper, Y.J.; Hennighausen, L. Comparison of the regulation of the whey acidic protein gene with that of a hybrid gene containing the whey acidic protein gene promoter in transgenic mice. Mol. Endocrinol. 1988, 2, 1027–1032. [Google Scholar] [CrossRef]
- Robinson, G.W.; Kang, K.; Yoo, K.H.; Tang, Y.; Zhu, B.M.; Yamaji, D.; Colditz, V.; Jang, S.J.; Gronostajski, R.M.; Hennighausen, L. Coregulation of genetic programs by the transcription factors NFIB and STAT5. Mol. Endocrinol. 2014, 28, 758–767. [Google Scholar] [CrossRef]
- Zhou, J.; Chehab, R.; Tkalcevic, J.; Naylor, M.J.; Harris, J.; Wilson, T.J.; Tsao, S.; Tellis, I.; Zavarsek, S.; Xu, D.; et al. Elf5 is essential for early embryogenesis and mammary gland development during pregnancy and lactation. EMBO J. 2005, 24, 635–644. [Google Scholar] [CrossRef]
- Liu, X.; Robinson, G.W.; Wagner, K.U.; Garrett, L.; Wynshaw-Boris, A.; Hennighausen, L. Stat5a is mandatory for adult mammary gland development and lactogenesis. Genes Dev. 1997, 11, 179–186. [Google Scholar] [CrossRef]
- Shyu, Y.J.; Liu, H.; Deng, X.; Hu, C.D. Identification of new fluorescent protein fragments for bimolecular fluorescence complementation analysis under physiological conditions. Biotechniques 2006, 40, 61–66. [Google Scholar] [CrossRef]
- Kerppola, T.K. Design and implementation of bimolecular fluorescence complementation (BiFC) assays for the visualization of protein interactions in living cells. Nat. Protoc. 2006, 1, 1278–1286. [Google Scholar] [CrossRef]
- Miller, K.E.; Kim, Y.; Huh, W.K.; Park, H.O. Bimolecular Fluorescence Complementation (BiFC) Analysis: Advances and Recent Applications for Genome-Wide Interaction Studies. J. Mol. Biol. 2015, 427, 2039–2055. [Google Scholar] [CrossRef] [PubMed]
- Soderberg, O.; Gullberg, M.; Jarvius, M.; Ridderstrale, K.; Leuchowius, K.J.; Jarvius, J.; Wester, K.; Hydbring, P.; Bahram, F.; Larsson, L.G.; et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 2006, 3, 995–1000. [Google Scholar] [CrossRef] [PubMed]
- Gustafsdottir, S.M.; Schallmeiner, E.; Fredriksson, S.; Gullberg, M.; Soderberg, O.; Jarvius, M.; Jarvius, J.; Howell, M.; Landegren, U. Proximity ligation assays for sensitive and specific protein analyses. Anal. Biochem. 2005, 345, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Bellucci, A.; Fiorentini, C.; Zaltieri, M.; Missale, C.; Spano, P. The “in situ” proximity ligation assay to probe protein-protein interactions in intact tissues. Methods Mol. Biol. 2014, 1174, 397–405. [Google Scholar] [CrossRef]
- Willi, M.; Yoo, K.H.; Reinisch, F.; Kuhns, T.M.; Lee, H.K.; Wang, C.; Hennighausen, L. Facultative CTCF sites moderate mammary super-enhancer activity and regulate juxtaposed gene in non-mammary cells. Nat. Commun. 2017, 8, 16069. [Google Scholar] [CrossRef]
- Shin, H.Y. The structural and functional roles of CTCF in the regulation of cell type-specific and human disease-associated super-enhancers. Genes Genom. 2019, 41, 257–265. [Google Scholar] [CrossRef]
- Chen, W.; Roeder, R.G. The Mediator subunit MED1/TRAP220 is required for optimal glucocorticoid receptor-mediated transcription activation. Nucleic Acids Res. 2007, 35, 6161–6169. [Google Scholar] [CrossRef]
- Siersbaek, R.; Rabiee, A.; Nielsen, R.; Sidoli, S.; Traynor, S.; Loft, A.; Poulsen, L.C.; Rogowska-Wrzesinska, A.; Jensen, O.N.; Mandrup, S. Transcription factor cooperativity in early adipogenic hotspots and super-enhancers. Cell Rep. 2014, 7, 1443–1455. [Google Scholar] [CrossRef]
- Adam, R.C.; Yang, H.; Rockowitz, S.; Larsen, S.B.; Nikolova, M.; Oristian, D.S.; Polak, L.; Kadaja, M.; Asare, A.; Zheng, D.; et al. Pioneer factors govern super-enhancer dynamics in stem cell plasticity and lineage choice. Nature 2015, 521, 366–370. [Google Scholar] [CrossRef]
- Engelen, E.; Brandsma, J.H.; Moen, M.J.; Signorile, L.; Dekkers, D.H.; Demmers, J.; Kockx, C.E.; Ozgur, Z.; van, I.W.F.; van den Berg, D.L.; et al. Proteins that bind regulatory regions identified by histone modification chromatin immunoprecipitations and mass spectrometry. Nat. Commun. 2015, 6, 7155. [Google Scholar] [CrossRef]
- Hnisz, D.; Shrinivas, K.; Young, R.A.; Chakraborty, A.K.; Sharp, P.A. A Phase Separation Model for Transcriptional Control. Cell 2017, 169, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yue, W.; Zhou, Y.; Liao, M.; Chen, X.; Hua, J. Super enhancers-Functional cores under the 3D genome. Cell Prolif. 2021, 54, e12970. [Google Scholar] [CrossRef] [PubMed]
- Piggin, C.L.; Roden, D.L.; Law, A.M.K.; Molloy, M.P.; Krisp, C.; Swarbrick, A.; Naylor, M.J.; Kalyuga, M.; Kaplan, W.; Oakes, S.R.; et al. ELF5 modulates the estrogen receptor cistrome in breast cancer. PLoS Genet. 2020, 16, e1008531. [Google Scholar] [CrossRef] [PubMed]
- Raisner, R.; Bainer, R.; Haverty, P.M.; Benedetti, K.L.; Gascoigne, K.E. Super-enhancer acquisition drives oncogene expression in triple negative breast cancer. PLoS ONE 2020, 15, e0235343. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Hu, J.; Maryam, A.; Huang, Q.; Zhang, Y.; Ramakrishnan, S.; Li, J.; Ma, H.; Ma, V.W.S.; Cheuk, W.; et al. Defining super-enhancer landscape in triple-negative breast cancer by multiomic profiling. Nat. Commun. 2021, 12, 2242. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, U.; Kim, S.; Kim, N.; Shin, H.Y. Mammary-Enriched Transcription Factors Synergize to Activate the Wap Super-Enhancer for Mammary Gland Development. Int. J. Mol. Sci. 2022, 23, 11680. https://doi.org/10.3390/ijms231911680
Kim U, Kim S, Kim N, Shin HY. Mammary-Enriched Transcription Factors Synergize to Activate the Wap Super-Enhancer for Mammary Gland Development. International Journal of Molecular Sciences. 2022; 23(19):11680. https://doi.org/10.3390/ijms231911680
Chicago/Turabian StyleKim, Uijin, Suyeon Kim, Nahyun Kim, and Ha Youn Shin. 2022. "Mammary-Enriched Transcription Factors Synergize to Activate the Wap Super-Enhancer for Mammary Gland Development" International Journal of Molecular Sciences 23, no. 19: 11680. https://doi.org/10.3390/ijms231911680
APA StyleKim, U., Kim, S., Kim, N., & Shin, H. Y. (2022). Mammary-Enriched Transcription Factors Synergize to Activate the Wap Super-Enhancer for Mammary Gland Development. International Journal of Molecular Sciences, 23(19), 11680. https://doi.org/10.3390/ijms231911680