

Comprehensive Genomic Profiling (CGP)-Informed Personalized Molecular Residual Disease (MRD) Detection: An Exploratory Analysis from the PREDATOR Study of Metastatic Colorectal Cancer (mCRC) Patients Undergoing Surgical Resection
 , , , ,
, , , , 

Abstract
1. Introduction
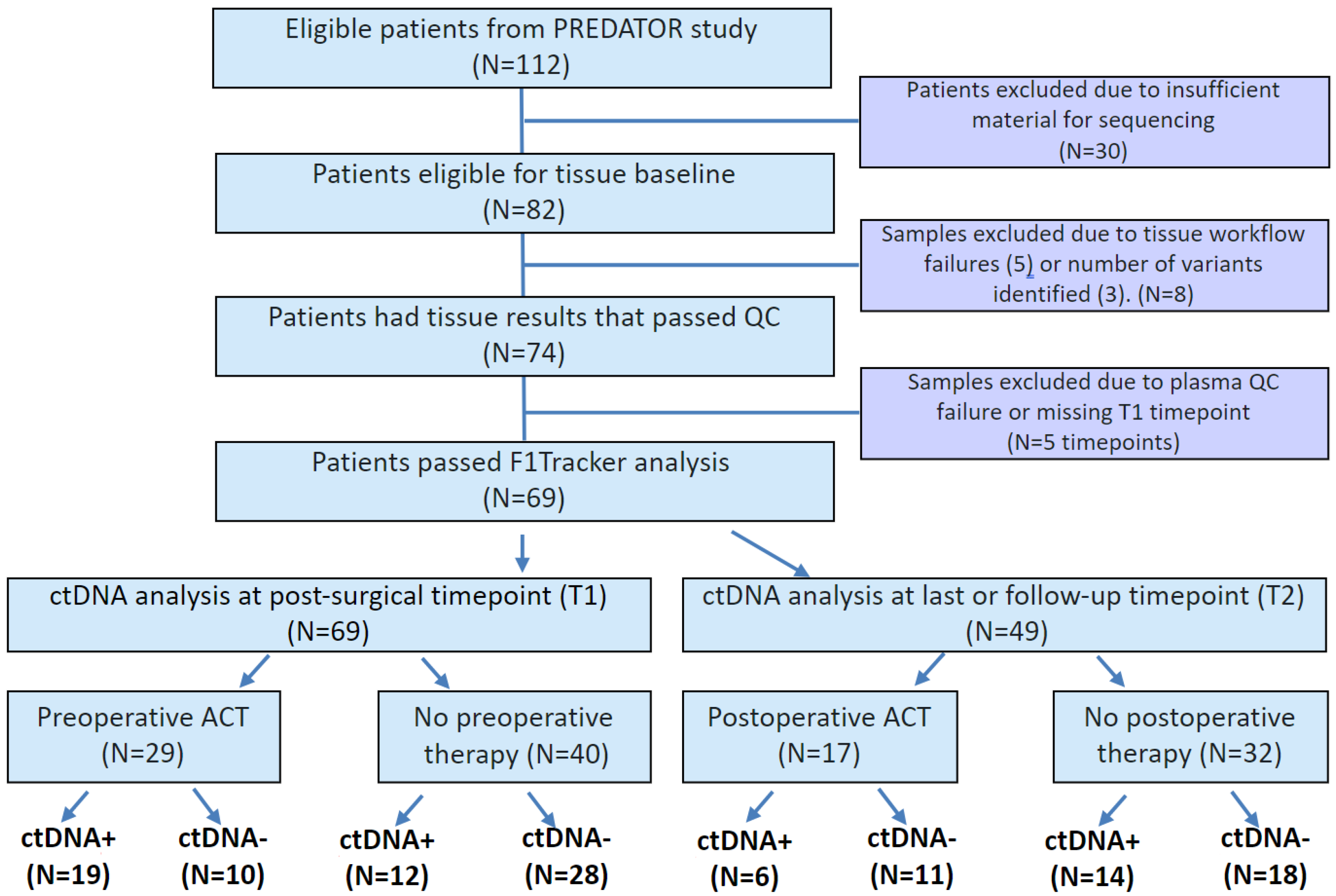
2. Results
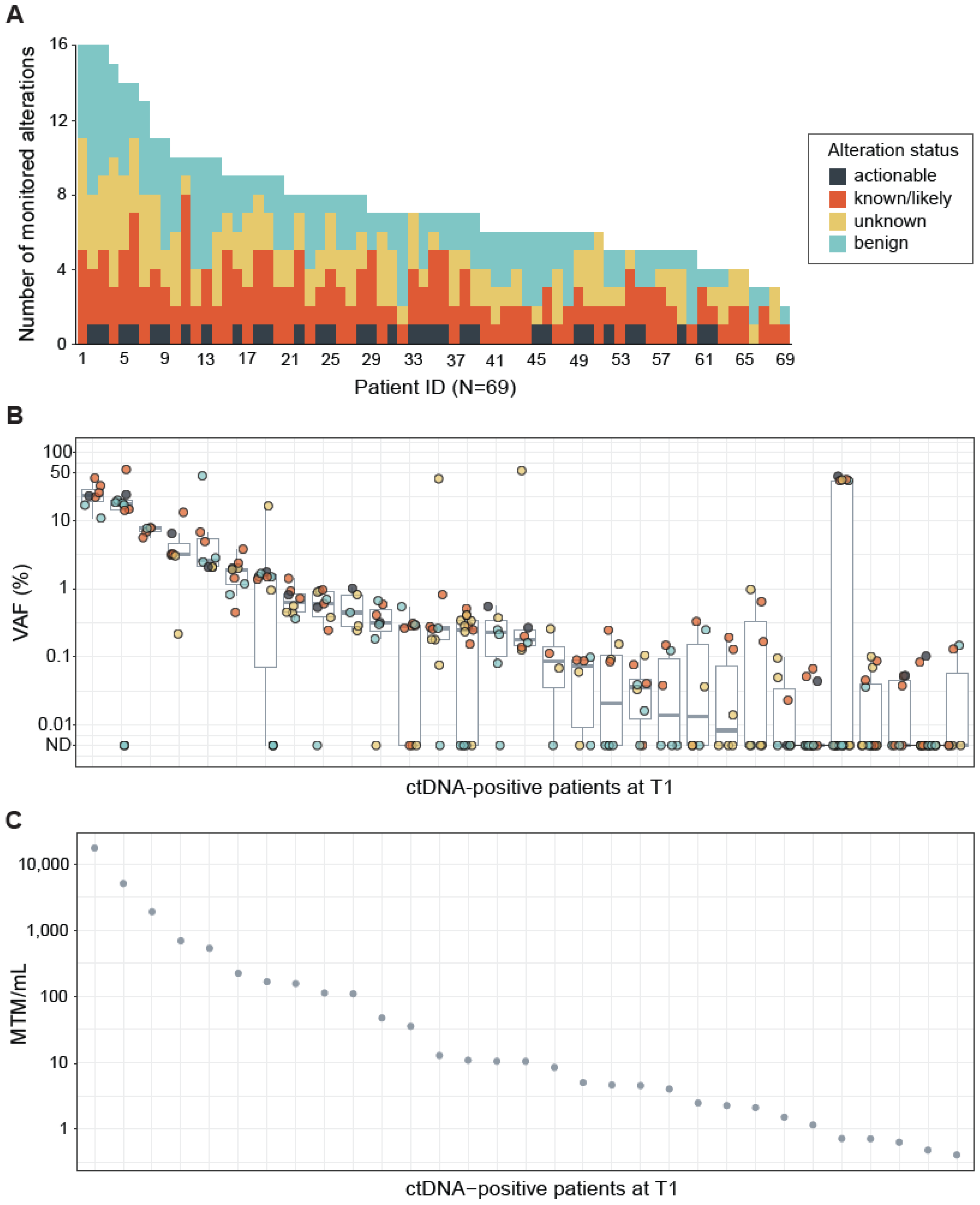
2.1. FoundationOne Tracker Measures Cancer-Associated Alterations across a Large Range of Variant Allele Frequencies
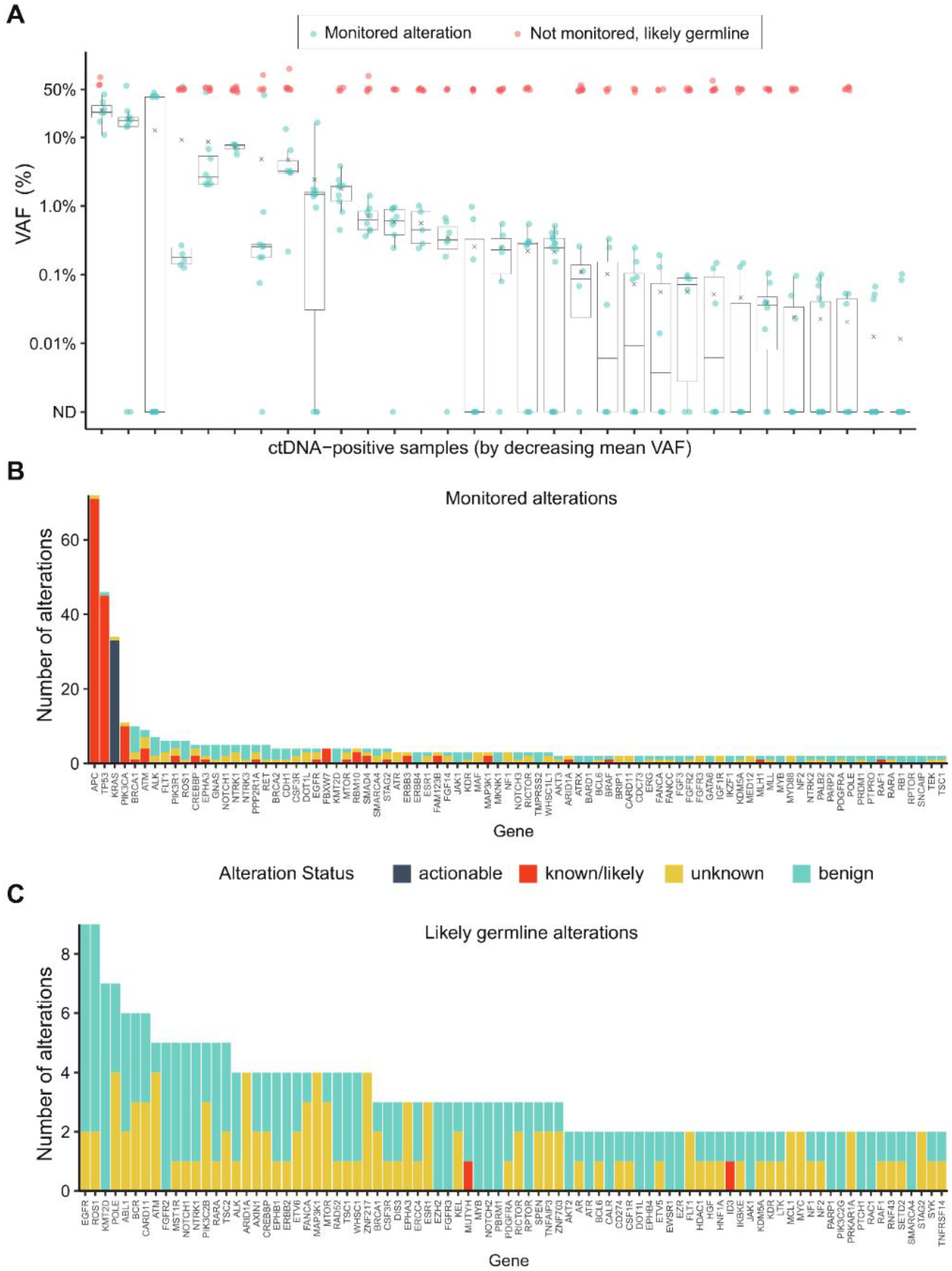
2.2. Landscape of Monitored and Non-Monitored Alterations
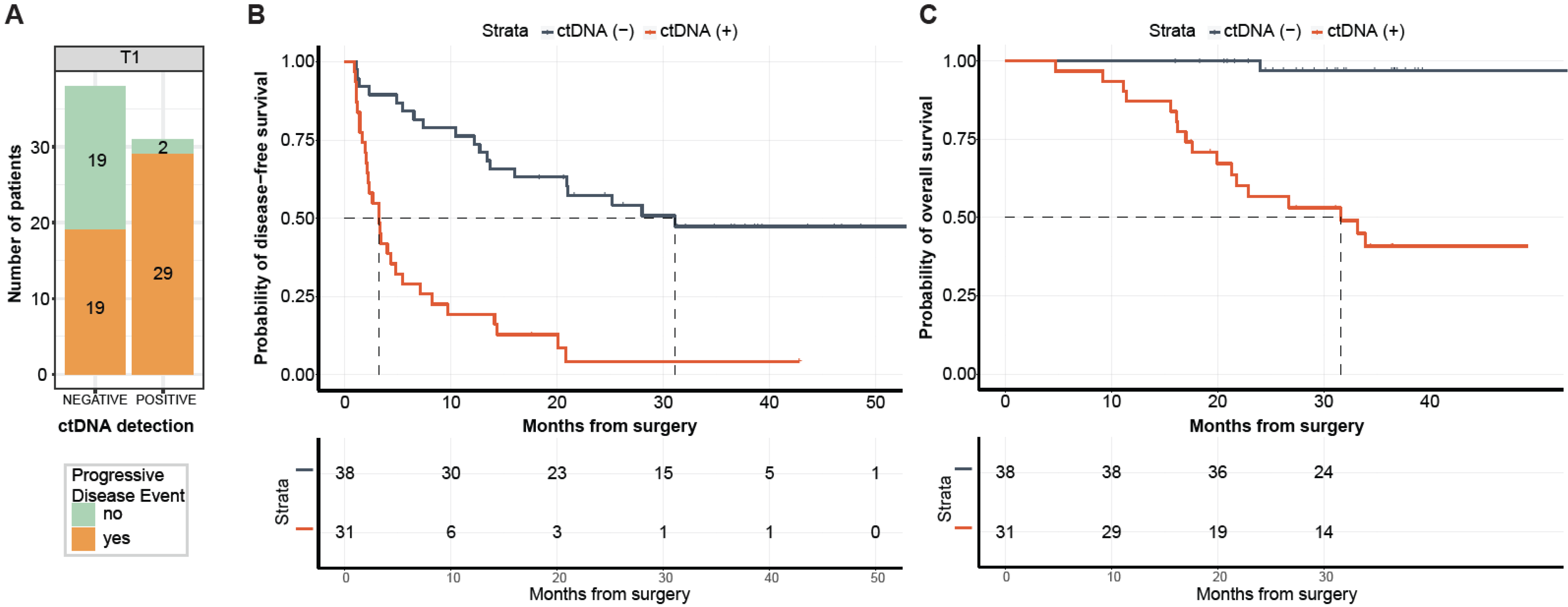
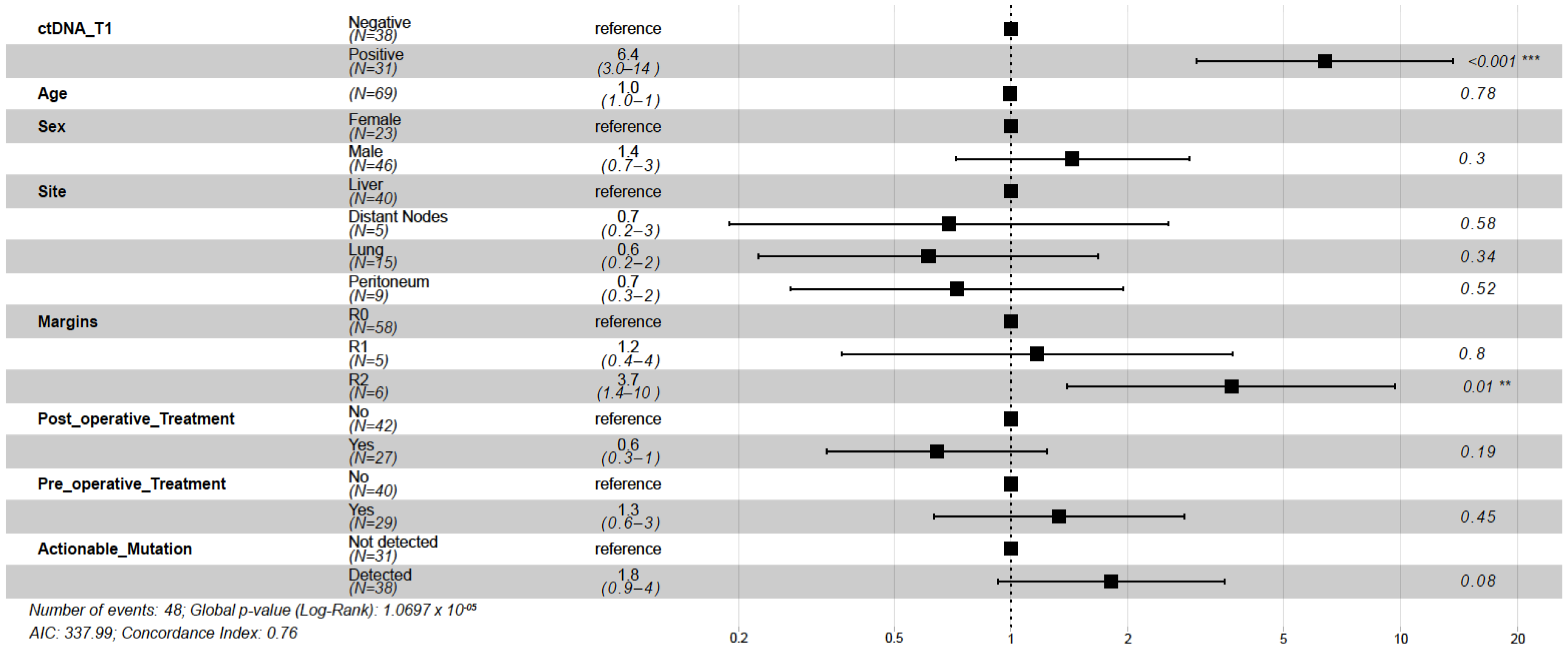
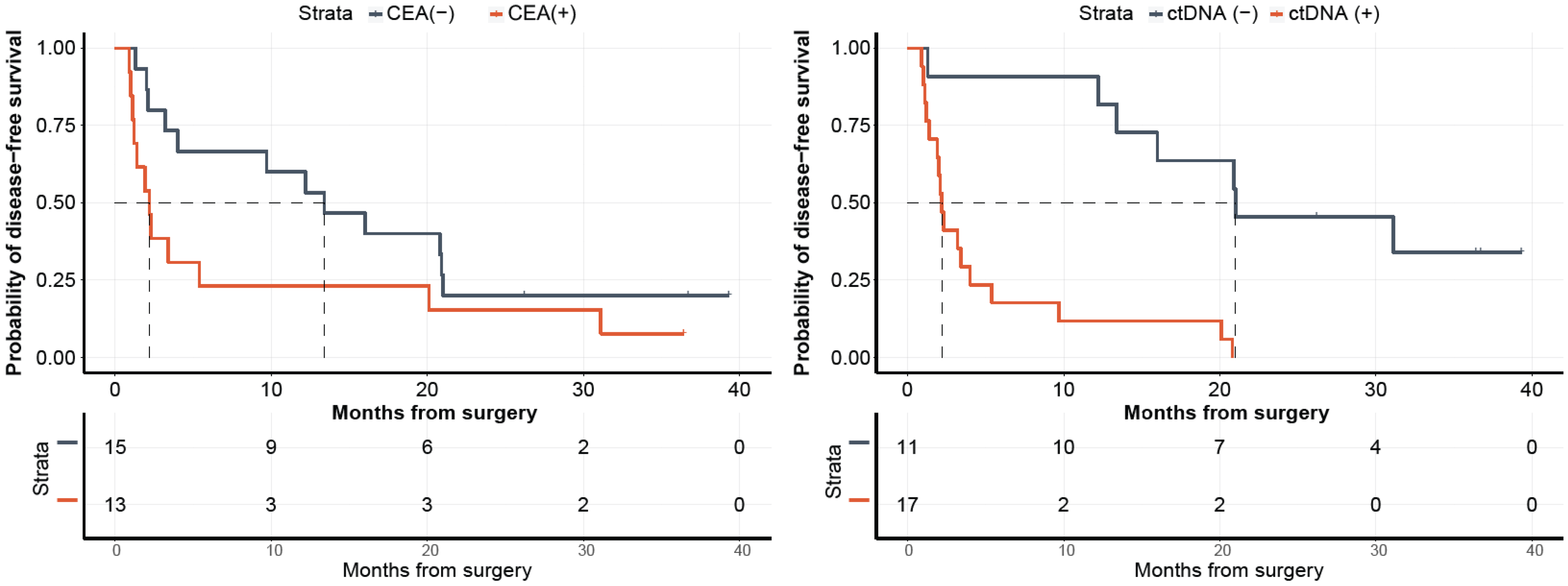
2.3. ctDNA Detection at Postsurgical Time Point Is Predictive of DFS and OS
2.4. ctDNA Detection at Follow-Up Time Point Is Predictive of DFS and OS
2.5. Comparison of ctDNA with CEA and Their Correlation with Disease Progression
3. Discussion
4. Materials and Methods
4.1. Tissue CGP-informed ctDNA assay
4.2. Variant Selection and Primer Design
4.3. Cell-Free DNA Extraction, Library Preparation, and Plasma Multiplex-PCR Next Generation Sequencing Workflow
4.4. Plasma Variant Calling
4.5. Analysis of Monitored and Non-Monitored Variants
4.6. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| mCRC | Metastatic colorectal cancer |
| MRD | Molecular residual disease |
| CGP | Comprehensive genomic profiling |
| ctDNA | Circulating tumor DNA |
| HR | Hazard ratio |
| DFS | Disease-free survival |
| OS | Overall survival |
| CEA | Carcinoembryonic antigen |
| FP | Fluoropyrimidine |
| NCCN | National Comprehensive Cancer Network |
| 5-FU | 5-Fluorouracil |
| IO | Immunotherapy |
| MSI-H | Microsatellite instability high |
| QC | Quality control |
| ACT | Adjuvant chemotherapy |
| VAF | Variant allele frequency |
| MTM/mL | Mean tumor molecules per milliliter |
| ND | Not detected |
| PPV | Positive predictive value |
| CI | Confidence interval |
| NGS | Next generation sequencing |
| FFPE | Formalin-fixed, paraffin-embedded |
| SNP | Single nucleotide polymorphism |
| SNV | Single nucleotide variant |
| WES | Whole exome sequencing |
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Cancer.Net. Colorectal Cancer: Statistics. Available online: https://www.cancer.net/cancer-types/colorectal-cancer/statistics (accessed on 28 February 2022).
- Matsuoka, H.; Morise, Z.; Tanaka, C.; Hayashi, T.; Ikeda, Y.; Maeda, K.; Masumori, K.; Koide, Y.; Katsuno, H.; Tanahashi, Y.; et al. Repeat hepatectomy with systemic chemotherapy might improve survival of recurrent liver metastasis from colorectal cancer-a retrospective observational study. World J. Surg. Oncol. 2019, 17, 33. [Google Scholar] [CrossRef] [PubMed]
- National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology, Colon Cancer, Version 1.2022. Available online: https://www.nccn.org/professionals/physician_gls/pdf/colon.pdf (accessed on 28 February 2022).
- Ciliberto, D.; Prati, U.; Roveda, L.; Barbieri, V.; Staropoli, N.; Abbruzzese, A.; Caraglia, M.; Di Maio, M.; Flotta, D.; Tassone, P.; et al. Role of systemic chemotherapy in the management of resected or resectable colorectal liver metastases: A systematic review and meta-analysis of randomized controlled trials. Oncol. Rep. 2012, 27, 1849–1856. [Google Scholar]
- Wang, Z.M.; Chen, Y.Y.; Chen, F.F.; Wang, S.Y.; Xiong, B. Peri-operative chemotherapy for patients with resectable colorectal hepatic metastasis: A meta-analysis. Eur. J. Surg. Oncol. 2015, 41, 1197–1203. [Google Scholar] [CrossRef]
- Khoo, E.; O’Neill, S.; Brown, E.; Wigmore, S.J.; Harrison, E.M. Systematic review of systemic adjuvant, neoadjuvant and perioperative chemotherapy for resectable colorectal-liver metastases. HPB (Oxford) 2016, 18, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Brandi, G.; De Lorenzo, S.; Nannini, M.; Curti, S.; Ottone, M.; Dall’Olio, F.G.; Barbera, M.A.; Pantaleo, M.A.; Biasco, G. Adjuvant chemotherapy for resected colorectal cancer metastases: Literature review and meta-analysis. World J. Gastroenterol. 2016, 22, 519–533. [Google Scholar] [CrossRef]
- Araujo, R.L.; Gonen, M.; Herman, P. Chemotherapy for patients with colorectal liver metastases who underwent curative resection improves long-term outcomes: Systematic review and meta-analysis. Ann. Surg. Oncol. 2015, 22, 3070–3078. [Google Scholar] [CrossRef]
- Sorensen, C.G.; Karlsson, W.K.; Pommergaard, H.C.; Burcharth, J.; Rosenberg, J. The diagnostic accuracy of carcinoembryonic antigen to detect colorectal cancer recurrence—A systematic review. Int. J. Surg. 2016, 25, 134–144. [Google Scholar] [CrossRef]
- Nicholson, B.D.; Shinkins, B.; Mant, D. Blood Measurement of Carcinoembryonic Antigen Level for Detecting Recurrence of Colorectal Cancer. JAMA 2016, 316, 1310–1311. [Google Scholar] [CrossRef]
- Shinkins, B.; Nicholson, B.D.; Primrose, J.; Perera, R.; James, T.; Pugh, S.; Mant, D. The diagnostic accuracy of a single CEA blood test in detecting colorectal cancer recurrence: Results from the FACS trial. PloS ONE 2017, 12, e0171810. [Google Scholar] [CrossRef]
- Litvak, A.; Cercek, A.; Segal, N.; Reidy-Lagunes, D.; Stadler, Z.K.; Yaeger, R.D.; Kemeny, N.E.; Weiser, M.R.; Pessin, M.S.; Saltz, L. False-positive elevations of carcinoembryonic antigen in patients with a history of resected colorectal cancer. J. Natl. Compr. Cancer Netw. 2014, 12, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Loupakis, F.; Sharma, S.; Derouazi, M.; Murgioni, S.; Biason, P.; Rizzato, M.D.; Rasola, C.; Renner, D.; Shchegrova, S.; Koyen Malashevich, A.; et al. Detection of Molecular Residual Disease Using Personalized Circulating Tumor DNA Assay in Patients with Colorectal Cancer Undergoing Resection of Metastases. JCO Precis. Oncol. 2021, 5, 1166–1177. [Google Scholar] [CrossRef] [PubMed]
- Benesova, L.; Halkova, T.; Ptackova, R.; Semyakina, A.; Menclova, K.; Pudil, J.; Ryska, M.; Levy, M.; Simsa, J.; Pazdirek, F.; et al. Significance of postoperative follow-up of patients with metastatic colorectal cancer using circulating tumor DNA. World J. Gastroenterol. 2019, 25, 6939–6948. [Google Scholar] [CrossRef] [PubMed]
- Norcic, G.; Jelenc, F.; Cerkovnik, P.; Stegel, V.; Novakovic, S. Role of specific DNA mutations in the peripheral blood of colorectal cancer patients for the assessment of tumor stage and residual disease following tumor resection. Oncol. Lett. 2016, 12, 3356–3362. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, B.A.; Winslow, E.R.; Bayasi, M.; Krainock, M.R.; Olshan, P.M.; Billings, P.R.; Aleshin, A. Early Detection of Circulating Tumor DNA Postoperatively Enables Discovery of Resectable Metastatic Disease in a Patient with Colon Cancer. Case Rep. Oncol. 2021, 14, 1748–1753. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, T.V.; Tarazona, N.; Frydendahl, A.; Reinert, T.; Gimeno-Valiente, F.; Carbonell-Asins, J.A.; Sharma, S.; Renner, D.; Hafez, D.; Roda, D.; et al. Circulating Tumor DNA in Stage III Colorectal Cancer, beyond Minimal Residual Disease Detection, toward Assessment of Adjuvant Therapy Efficacy and Clinical Behavior of Recurrences. Clin. Cancer Res. 2022, 28, 507–517. [Google Scholar] [CrossRef]
- Overman, M.J.; Vauthey, J.-N.; Aloia, T.A.; Conrad, C.; Chun, Y.S.; Pereira, A.A.L.; Jiang, Z.; Crosby, S.; Wei, S.; Raghav, K.P.S.; et al. Circulating tumor DNA (ctDNA) utilizing a high-sensitivity panel to detect minimal residual disease post liver hepatectomy and predict disease recurrence. J. Clin. Oncol. 2017, 35, 3522. [Google Scholar] [CrossRef]
- Jones, R.P.; Pugh, S.A.; Graham, J.; Primrose, J.N.; Barriuso, J. Circulating tumour DNA as a biomarker in resectable and irresectable stage IV colorectal cancer; a systematic review and meta-analysis. Eur. J. Cancer 2021, 144, 368–381. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Cervantes, A.; Adam, R.; Sobrero, A.; Van Krieken, J.H.; Aderka, D.; Aranda Aguilar, E.; Bardelli, A.; Benson, A.; Bodoky, G.; et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann. Oncol. 2016, 27, 1386–1422. [Google Scholar] [CrossRef]
- Patelli, G.; Tosi, F.; Amatu, A.; Mauri, G.; Curaba, A.; Patane, D.A.; Pani, A.; Scaglione, F.; Siena, S.; Sartore-Bianchi, A. Strategies to tackle RAS-mutated metastatic colorectal cancer. ESMO Open 2021, 6, 100156. [Google Scholar] [CrossRef]
- Xie, Y.H.; Chen, Y.X.; Fang, J.Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct Target Ther. 2020, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Sorbye, H.; Dahl, O. Carcinoembryonic antigen surge in metastatic colorectal cancer patients responding to oxaliplatin combination chemotherapy: Implications for tumor marker monitoring and guidelines. J. Clin. Oncol. 2003, 21, 4466–4467. [Google Scholar] [CrossRef] [PubMed]
- Siregar, G.A.; Sibarani, H. Comparison of Carcinoembryonic Antigen Levels Among Degree of Differentiation and Colorectal Cancer’s Location in Medan. Open Access Maced J. Med. Sci. 2019, 7, 3447–3450. [Google Scholar] [CrossRef] [PubMed]
- Kasi, P.M.; Kamatham, S.; Shahjehan, F.; Li, Z.; Johnson, P.W.; Merchea, A.; Colibaseanu, D.T. BRAF-V600E and microsatellite instability prediction through CA-19-9/CEA ratio in patients with colorectal cancer. J. Gastrointest Oncol. 2020, 11, 236–241. [Google Scholar] [CrossRef]
- Kasi, P.M.; Budde, G.; Krainock, M.; Aushev, V.N.; Koyen Malashevich, A.; Malhotra, M.; Olshan, P.; Billings, P.R.; Aleshin, A. Circulating tumor DNA (ctDNA) serial analysis during progression on PD-1 blockade and later CTLA-4 rescue in patients with mismatch repair deficient metastatic colorectal cancer. J. Immunother Cancer 2022, 10, e003312. [Google Scholar] [CrossRef]
- Moding, E.J.; Nabet, B.Y.; Alizadeh, A.A.; Diehn, M. Detecting Liquid Remnants of Solid Tumors: Circulating Tumor DNA Minimal Residual Disease. Cancer Discov. 2021, 11, 2968–2986. [Google Scholar] [CrossRef]
- Milbury, C.A.; Creeden, J.; Yip, W.K.; Smith, D.L.; Pattani, V.; Maxwell, K.; Sawchyn, B.; Gjoerup, O.; Meng, W.; Skoletsky, J.; et al. Clinical and analytical validation of FoundationOne(R)CDx, a comprehensive genomic profiling assay for solid tumors. PLoS ONE 2022, 17, e0264138. [Google Scholar] [CrossRef]
- Reinert, T.; Henriksen, T.V.; Christensen, E.; Sharma, S.; Salari, R.; Sethi, H.; Knudsen, M.; Nordentoft, I.; Wu, H.T.; Tin, A.S.; et al. Analysis of Plasma Cell-Free DNA by Ultradeep Sequencing in Patients With Stages I to III Colorectal Cancer. JAMA Oncol. 2019, 5, 1124–1131. [Google Scholar] [CrossRef]
- Abbosh, C.; Birkbak, N.J.; Wilson, G.A.; Jamal-Hanjani, M.; Constantin, T.; Salari, R.; Le Quesne, J.; Moore, D.A.; Veeriah, S.; Rosenthal, R.; et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature 2017, 545, 446–451. [Google Scholar] [CrossRef]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria; Available online: https://cran.r-project.org (accessed on 1 December 2021).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient characteristics (All patients, N = 69) | N | % |
|---|---|---|
| Sex Male Female | 46 23 | 66.7 33.3 |
| Presentation of metastasis Synchronous Metachronous | 35 34 | 50.7 49.3 |
| Median age at first diagnosis, median (range), years | 59.5 | 20.8–82.8 |
| Median age at diagnosis of metastatic disease, median (range), years | 60.1 | 22.1–83.3 |
| Adjuvant therapy administered to metachronous | 26 | 76.5 |
| Site of surgery Liver Lung Peritoneum Other | 40 15 9 5 | 58 21.7 13 7.2 |
| Presurgical treatment Doublet with or w/o biologic, FP monotherapy with or w/o biologic, and triplet with or w/o biologic | 29 | 42 |
| Postsurgical treatment Doublet with or w/o biologic, FP monotherapy, and triplet with or w/o biologic | 27 | 39.1 |
| Event Progressive disease | 48 | 69.6 |
| CEA status: preoperative (N = 28) CEA-positive | 23 | 33.3 |
| CEA status: postoperative (N = 28) CEA-positive | 13 | 18.8 |
| Resection margins R0 R1 R2 | 58 5 6 | 84.1 7.2 8.7 |
| Actionable alterations identified in resected tumor (KRAS = 36, NRAS = 1) | 37 | 54 |
| ctDNA detected at first timepoint (T1) | 31 | 45 |
| ctDNA detection at last or follow-up timepoint (T2) (N = 49) | 20 | 41 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lonardi, S.; Nimeiri, H.; Xu, C.; Zollinger, D.R.; Madison, R.W.; Fine, A.D.; Gjoerup, O.; Rasola, C.; Angerilli, V.; Sharma, S.; et al. Comprehensive Genomic Profiling (CGP)-Informed Personalized Molecular Residual Disease (MRD) Detection: An Exploratory Analysis from the PREDATOR Study of Metastatic Colorectal Cancer (mCRC) Patients Undergoing Surgical Resection. Int. J. Mol. Sci. 2022, 23, 11529. https://doi.org/10.3390/ijms231911529
Lonardi S, Nimeiri H, Xu C, Zollinger DR, Madison RW, Fine AD, Gjoerup O, Rasola C, Angerilli V, Sharma S, et al. Comprehensive Genomic Profiling (CGP)-Informed Personalized Molecular Residual Disease (MRD) Detection: An Exploratory Analysis from the PREDATOR Study of Metastatic Colorectal Cancer (mCRC) Patients Undergoing Surgical Resection. International Journal of Molecular Sciences. 2022; 23(19):11529. https://doi.org/10.3390/ijms231911529
Chicago/Turabian StyleLonardi, Sara, Halla Nimeiri, Chang Xu, Daniel R. Zollinger, Russell W. Madison, Alexander D. Fine, Ole Gjoerup, Cosimo Rasola, Valentina Angerilli, Shruti Sharma, and et al. 2022. "Comprehensive Genomic Profiling (CGP)-Informed Personalized Molecular Residual Disease (MRD) Detection: An Exploratory Analysis from the PREDATOR Study of Metastatic Colorectal Cancer (mCRC) Patients Undergoing Surgical Resection" International Journal of Molecular Sciences 23, no. 19: 11529. https://doi.org/10.3390/ijms231911529
APA StyleLonardi, S., Nimeiri, H., Xu, C., Zollinger, D. R., Madison, R. W., Fine, A. D., Gjoerup, O., Rasola, C., Angerilli, V., Sharma, S., Wu, H.-T., Palsuledesai, C. C., Malhotra, M., Aleshin, A., Loupakis, F., Renkonen, E., Hegde, P., & Fassan, M. (2022). Comprehensive Genomic Profiling (CGP)-Informed Personalized Molecular Residual Disease (MRD) Detection: An Exploratory Analysis from the PREDATOR Study of Metastatic Colorectal Cancer (mCRC) Patients Undergoing Surgical Resection. International Journal of Molecular Sciences, 23(19), 11529. https://doi.org/10.3390/ijms231911529